
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSERS detection of microRNA biomarkers for cancer diagnosis using gold-coated paramagnetic nanoparticles to capture SERS-active gold nanoparticles†
Hao Zhang *a,
Yu Yib,
Chunhui Zhoua,
Guoqing Yingb,
Xiangdong Zhoua,
Chaopeng Fua,
Yifeng Zhu*a and
Youqing Shen*c
*a,
Yu Yib,
Chunhui Zhoua,
Guoqing Yingb,
Xiangdong Zhoua,
Chaopeng Fua,
Yifeng Zhu*a and
Youqing Shen*c
aCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: hzhang6@163.com; yifeng@zjut.edu.cn
bCollege of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310014, China
cCollege of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: shenyq@zju.edu.cn
First published on 14th November 2017
Abstract
In this paper, a magnetic-based, surface-enhanced Raman scattering (SERS) assay for detection of a cancer-related microRNA biomarker, miR-141, has been developed. The detection is based on hybridization-dependent recognition, in which the miR-141 target sequences were captured by complementary reporter and capture oligonucleotide probes conjugated to Raman-tagged gold nanoparticles (GNPs) and gold-coated paramagnetic nanoparticles (Au@MNPs) respectively. The resultant hybridization complexes, Raman-tagged GNPs/miR-141/Au@MNPs, are retrieved from solution by magnetic pull-down and concentrated within the focus of laser excitation. A signature spectrum for the Raman tag, 5,5′-dithiobis(succinimidyl-2-nitrobenzoate) (DSNB), was observed in concentrated pellets and specific for the miR-141 sequences. The viability of SERS detection has been demonstrated in a microfluidic platform, in which the hybridizations containing dilutions of the miR-141 sequences yielded a reduction in the DSNB spectrum peaks' intensity. The limit of detection (LOD) is estimated to be 100 fM, which is 100-fold lower than the LOD of 10 pM previously reported in a similar magnetic-capture SERS detection of small oligonucleotides using nonplasmonic MNPs. These results indicate that the addition of Au shells to MNPs facilitates the formation of SERS-active junction regions (“hot spots”) with nearby Au contents within the magnetic concentrates, which substantially improves the SERS signal and, therefore, detection sensitivity.
Introduction
MicroRNAs (miRNA), a class of small (18–24 nucleotides in length) non-protein-coding RNAs, are critical regulators of cellular processes, such as proliferation, division, differentiation and cell death.1,2 Because of these recognized roles, it is not surprising that miRNA expression patterns have been implicated in several diseases, particularly cancers.3 For instance, all tumors analyzed by miRNA profiling have exhibited very distinct miRNA signatures compared to normal cells from the same tissue.4 In addition to tissue samples, tumor-derived miRNAs in serum have exhibited potential as ideal biomarkers for noninvasive detection of human cancers.3,5 For instance, the serum-circulating miR-141 has been recognized as a critical biomarker for various malignant cancers.6,7 Several methods have been developed for profiling miRNA expression, including northern blotting,8 real-time quantitative reverse-transcription polymerase chain reaction methods (qRT-PCR),9 and microarray methods.10 Nevertheless, miRNAs' intrinsic properties, such as short lengths, high-sequence similarity within miRNA families and low abundance in serum, have restrained the accurate measurement of miRNAs by conventional methods.11 Moreover, these sensing processes normally rely on fluorescent-dye signaling, which exhibits sensitivity and multiplex-detection limitations, due to the intrinsic fluorophore photobleaching and the broad fluorescent bands that are easily overlapped.12Recent advances in the fields of nanotechnology and surface-enhanced Raman scattering (SERS) spectroscopy have opened up possibilities for sensing a large number of biomarkers, such as nucleic acids,13 proteins,14 and even cells.15 The scattering enhancement of SERS mainly comes from localized surface plasmon resonance (LSPR) on the nanostructured surfaces of noble metals (mostly gold and silver), under irradiation with light within a certain wavelength range.16 When positioned within an electromagnetic field induced by LSPR, light scattering cross-sections of Raman dye compounds are significantly enlarged, resulting in an enhanced SERS signal. SERS offers a number of advantages over fluorescence-based detection methods, including ultrahigh sensitivity,17 low photobleaching,18 and multiplexing capabilities.19,20
The comprehensive SERS advantages are very valuable in miRNA profiling. In an early study,21 Driskell developed a SERS platform that enabled rapid and specific miRNA detection without enzyme-assisted amplification of target sequences. In a later study, he demonstrated that the reproducibility of SERS spectra was sufficient for quantitative analysis of miRNA profiles.22 Recently, Wang employed an “OFF-to-ON” SERS signal switch for multiplexed detection of breast cancer-related miRNAs, by sensing the spectroscopic “brightness” of Raman labels on plasmonic nanostars.23 Label-free SERS assays have also been developed for the direct detection of miRNA based on single- versus double-stranded oligonucleotide affinity for SERS substrates with limit of detection (LOD) sensitivities in a dynamic range of 100 pM to 100 μM.24–26
To achieve higher detection sensitivity, signal amplification systems have been incorporated in SERS detection of miRNAs. Pang reported a SERS-based detection of miRNA with LOD sensitivity of 0.3 fM, using duplex-specific nuclease (DSN) to trigger the signal-amplifying recycling.27 Recently, enzyme-free nucleic acid amplification methods, including hybridization chain reaction (LOD of 0.3 fM)28 and DNA machines (LOD of 0.17 fM),29 have been used in SERS detection of miRNAs for subfemtomolar sensitivity. Despite the high-level LOD sensitivities, these amplification systems face the challenges of the low miRNA expression efficiency, laborious probes fabrication, time-consuming hybridization reactions and extra washing steps. It has been found that the greatest Raman enhancements are located within nanoscale interstitial spaces or gaps between SERS substrates, named as “hot spots”.30 Fabricating nanogaps for SERS signaling, such as nanowire interstices,31 nanogaps in gold–silver nanomushrooms,13 and DNA-mediated nanogaps,32 has enabled femtomolar and even attomolar miRNA LOD sensitivities. However, the preparation of nanogap-based SERS substrates demands technical expertise and complicated reaction conditions that may be difficult to scale up.
Recently, paramagnetic nanoparticles (MNPs) have been used to enhance SERS signaling in detection of target DNA sequences and proteins by magnetic manipulation and concentration of Raman-active gold nanoparticles (GNPs) conjugated with probes specific to target biomarkers.33,34 For instance, a co-author's previous study showed that the incorporation of silica-coated MNPs (Silica@MNPs) into SERS-based hybridization assay enhanced SERS intensity of GNPs coated with 5,5′-dithiobis(succinimidyl-2-nitrobenzoate) (DSNB) by 200 times and resulted in a LOD sensitivity of 10 pM for small-target DNA sequences (53 nucleotides).35 This assay provides intriguing opportunities for detecting miRNAs of a similar length scale and recognizable by DNA/RNA hybridization reactions. However, there is still room for more research into using MNPs as magnetic substrates to improve SERS signaling even more due to their nonplasmonic surfaces, which would decrease the probability of hot-spot formation between plasmonic GNPs in magnetically concentrated aggregates.36 In the present study, we describe a magnetic-capture SERS assay for detection of target miR-141 sequences, using gold-coated MNPs (Au@MNPs) as magnetic substrates. Fig. 1 illustrates the scheme summarizing capture and detection of miR-141. The miR-141 sequence is first recognized by hybridization reactions with Au@MNPs conjugated with complementary capture oligonucleotides (Capture-Au@MNPs) and GNPs conjugated with complementary reporter oligonucleotides and Raman reporters, DSNB (Reporter-GNPs; Fig. 1A). The resultant hybridization complexes, Capture-Au@MNPs/miR-141/Reporter-GNPs, are then injected into the microfluidic channel and magnetically concentrated (Fig. 1B). Laser interrogation of the sample pellets yields a Raman spectrum profile of DSNB that is restricted to miR-141. The current LOD sensitivity for miR-141 sequence is estimated to be 100 fM, which is improved by a 100-fold compared to the LOD sensitivity (10 pM) previously reported for detecting small oligonucleotides using the same DSNB-coated GNPs as Raman reporters but using Silica@MNPs as magnetic captures.35 These results indicate that the MNP's additional Au shell decreases exposure of nonplasmonic iron oxide surfaces and increases the probability of forming SERS hot spots with nearby nanostructured Au contents in magnetic concentrates (Fig. 1C). This offers improved SERS signal enhancement and detection sensitivity over nonplasmonic MNPs used in a conventional magnetic-based SERS assay (Fig. 1D).
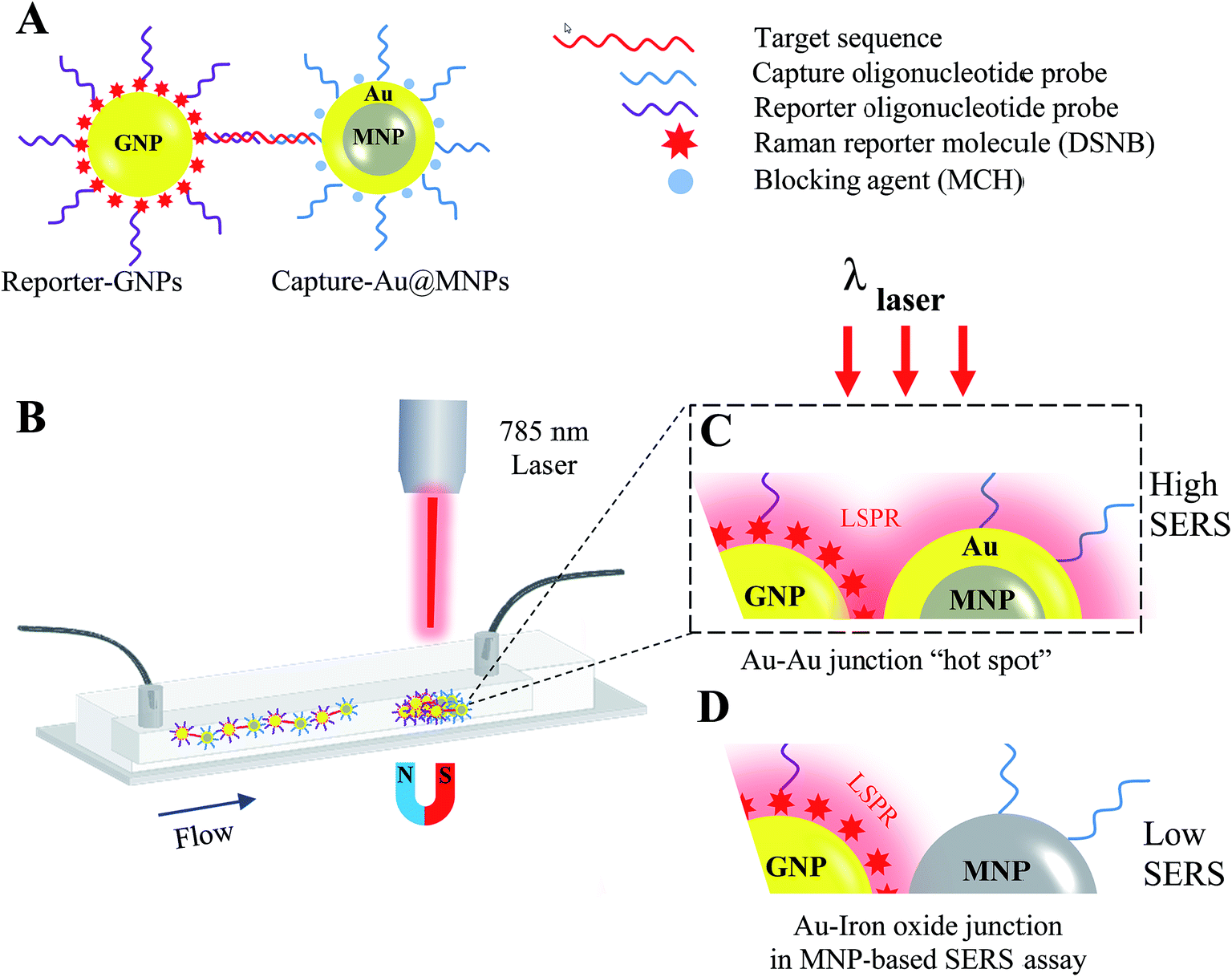 | ||
| Fig. 1 Schematic of magnetic capture and SERS detection of cancer biomarkers, miR-141, using Au@MNPs conjugated with capture probes and SERS-active GNPs conjugated with reporter probes. | ||
Experimental
Materials
Unless indicated otherwise, all other chemicals and materials were purchased from Sigma-Aldrich. GNPs of 30 nm (2 × 1011 particles per mL) were ordered from Ted Pella (USA). All solutions were prepared using RNase-free water obtained from Takara Biotechnology Co., Ltd. (Dalian, China). All HPLC purified oligonucleotides used in this study were purchased from Sangong Biotech Co., Ltd. (Shanghai China), and validated by quality control mass spectroscopy. Their sequences are shown in Table S1.†Synthesis and characterization of Au@MNPs
To synthesize the Au@MNPs, the core MNPs were first synthesized according to previous methods with modification.37 Briefly, 3 mmol of Fe(acac)3, 12 mmol of 1,2-hexadecanediol, 6 mmol of oleic acid, 6 mmol of oleylamine, and 20 mL of benzyl ether (99%) were vigorously stirred under an N2 blanket. The solution was heated to 200 °C for 2 h, followed by heating to 300 °C for 2 h. After reaction, the 13 nm of MNPs were precipitated by addition of ethanol and recovered by centrifugation.Au@MNPs were synthesized via reductions of Au(OOCCH3)3 using 13 nm of MNPs as seeds, similar to previous methods with modification.33,38 A 100 mg portion of MNPs, 5 mmol of Au(OOCCH3)3, 12 mmol of 1,2-hexadecanediol, 1.5 mmol of oleic acid and 6 mmol of oleylamine were added to 30 mL of benzyl ether and vigorously stirred under argon for 2 h at 200 °C, followed by heating to 250 °C for 3 h. Precipitation by ethanol and separation by centrifugation produced 27 nm Au@MNPs with ∼7 nm Au shells. The hydrophobic particles were treated with tetramethylammonium-11-aminoun-decanoate in dichloromethane (2% w/v) to obtain hydrophilic 27 nm Au@MNPs, which were dispersed in phosphate-buffered saline (PBS) solution (10 mM phosphate buffer, 300 mM NaCl, pH 7.4).
The physical properties of synthesized MNPs and Au@MNPs were characterized by transmission electron microscopy (TEM), UV-vis absorption spectroscopy, X-ray diffraction (XRD) and dynamic light scattering (DLS). TEM images were taken on a Hitachi H-7650 electron microscope at an accelerating voltage of 200 kV. The nanoparticle (NP) samples dispersed in hexane solution were deposited on formvar-coated copper grid films, followed by evaporation at room temperature. X-ray powder diffraction patterns of dried NP samples were obtained on a Siemens D-500 diffractometer under Co Ka radiation (λ = 1.788965 Å). UV-vis absorbance spectra were obtained with a Cary 50 UV/vis-NIR spectrophotometer over the range of 300–750 nm. Particle size distributions were determined by DLS using a Zetasizer Nano ZS ZEN 3600 (Malvern, UK).
Fabrication and characterization of SERS NP assemblies
The thiolated capture probe was immobilized onto the gold surface of Au@MNPs, according to previous report.33 Briefly, 1 mL of 0.1 mg mL−1 Au@MNPs (∼5 × 1011 particles per mL) in PBS buffer and 40 μL of 100 μM thiol-modified capture probe in PBS buffer were incubated overnight at room temperature. Excess unbounded reagents were removed by magnetic separation, and the capture-probe-conjugated Au@MNPs were redispersed into 1 mL PBS buffer. The gold surface of the Au@MNPs was then passivated using 0.1 mM 6-mercapto-1-hexanol (MCH) in 1 mL of PBS buffer. The Capture-Au@MNPs were washed with PBS buffer using repeated magnetic separation and finally resuspended in 1 mL PBS buffer.The Raman reporter label DSNB was synthesized and attached to colloidal GNPs according to previous methods.39 Next, 1 mL of DSNB-conjugated GNPs (∼2.5 × 1011 particles per mL) in PBS buffer and 40 μL of 100 μM amino-modified reporter probes were mixed and incubated for 12 h. The amines of reporter probes favored the formation of an amide linkage by reaction with succinimidyl ester of DSNB to produce the Reporter-GNPs. Excess unconjugated reporter probes were removed by centrifugation at 7000 rpm for 20 min. The Reporter-GNP precipitate was washed three times and finally dispersed in 1 mL PBS buffer.
Surface modifications of NP assemblies were analyzed by X-ray photoelectron spectroscopy (XPS) to determine the element and chemical bond compositions. XPS measurements were conducted on a Kratos AXIS Ultra DLD spectrometer using a focused monochromatic Al Kα X-ray (1486.7 eV) source for excitation. Compositional survey and high-resolution spectra of NP assemblies were acquired using pass energy of 80 eV and 20 eV respectively. For the high-resolution spectra, the collected binding energies were referenced to the Au4f peak at 84.0 eV. XPS spectra were deconvoluted into separate peaks using the software program XPS Peak 4.1.
Microfluidic device fabrication
The straight microfluidic channel with cross-sectional dimension of 150 × 150 μm was fabricated using standard PDMS soft lithography techniques.40 Briefly, a master of the microchannel features was created by photoresist methods (MicroChem, USA) on a silicon wafer. The polydimethylsiloxane (PDMS) (Sylgard 184, Dow Corning) solution was cast upon the master wafer and the cured PDMS microchannel replicas were cut and then bonded to clean glass slides following treatment by oxygen plasma. Sample solutions are injected into the PDMS microchannel via syringe pump (NanoJet syringe pump, USA).Procedures of miRNAs detection
All hybridization reactions were conducted in 1.5 mL PBS buffer solution or diluted human serum solution (Sigma, USA), contained ∼2.5 × 1011 Reporter-GNPs per mL, 0.25 mg mL−1 of Capture-Au@MNPs and target miR-141 or control miRNA sequence, miR-429. These hybridization reaction mixtures were incubated in capped glass vials at 37 °C for 2 h, after which the hybridization solutions were pumped through the straight microfluidic channel and accumulated by a small external magnet. Laser excitation of concentrated sample pellets was performed using an Advantage NIR Raman spectrometer (DeltaNu Inc., USA; 60 mW power, 785 nm laser) fitted with right angle input optics and camera with 100× magnification. Spectral data was acquired using NuSpec software and recorded in the 200–2000 vibrational unit range (Raman shift/cm−1), with a integration time of 5 s. Grams/AI software (Thermo Fisher Scientific) performed the data analysis.QCM-D modeling of hybridization complex formation
The formation of Capture probe/miR-141/Reporter-GNPs hybridization complexes on Au-coated quartz crystals (5 MHz AT-cut) was monitored by QCM-D analysis (E4, Biolin Scientific, Sweden) using conditions similar to previous reports.41,42 In brief, sensor crystals were boiled in a cleaning solution (30% H2O2, 28% ammonia, and deionized water in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5) for 5 min. The freshly cleaned crystals were immersed in 100 mM thiolated capture probe in PBS buffer overnight, and then passivated using 2 mM MCH in PBS buffer. The immobilization of capture probes generated a surface that supports the hybridization reaction with target miRNA and subsequent hybridization with Reporter-GNP while resisting nonspecific adsorption. A nonspecific surface saturated with MCH was also produced by immersing cleaned sensor crystal in 2 mM MCH PBS buffer solution. After rinsing with RNAnase-free water and drying with nitrogen, the treated sensor crystals were ready to be used for the measurements.
:1:5) for 5 min. The freshly cleaned crystals were immersed in 100 mM thiolated capture probe in PBS buffer overnight, and then passivated using 2 mM MCH in PBS buffer. The immobilization of capture probes generated a surface that supports the hybridization reaction with target miRNA and subsequent hybridization with Reporter-GNP while resisting nonspecific adsorption. A nonspecific surface saturated with MCH was also produced by immersing cleaned sensor crystal in 2 mM MCH PBS buffer solution. After rinsing with RNAnase-free water and drying with nitrogen, the treated sensor crystals were ready to be used for the measurements.
After mounting into QCM chambers, the frequency baselines of the treated sensor crystals were recorded using PBS buffer solution before 1 mM PBS solution of target or nonspecific control miRNA sequence was injected. Once the changes in resonant frequency (Δf) and dissipation (ΔD) plateaued, the crystals were rinsed with PBS buffer as a step to wash off excess unbounded agents. The baseline-stabilized surfaces were then washed with ∼2 × 1010 particles per mL PBS solutions of either specific Reporter-GNPs or nonspecific DSNB-GNPs as control particles, until the signal stabilized, and then rinsed again with PBS buffer. Since the changes of dissipation (ΔD) significantly exceeded 10% of the corresponding values for Δf,43 the adsorbed layers were considered to have viscoelastic properties and the mass adsorption was analyzed by the Voigt-based model implemented in Qtools (Biolin Scientific, Sweden),44 using the combined Δf and ΔD data at three harmonic overtones (n = 5, 7 and 9).
Results and discussion
Characterization of Au@MNPs
The core–shell nanostructure of Au@MNPs was characterized by TEM, DLS and XRD. TEM images showed that MNPs with a narrow size distribution and average size of ∼13 nm (Fig. 2A) were synthesized by the well-established protocol with modifications. DLS measurements were conducted for further characterization of MNPs, providing an average particle size distribution of 13.4 ± 3.4 nm, which correlated with TEM results. Unlike the previously reported multistep synthesis of 10 nm MNPs,33,37 we obtained 13 nm MNPs in a modified single-step synthesis. Mediation of the incubation time at 200 °C and reaction time at 300 °C is the key to increasing the diameter of MNPs beyond 10 nm (data not shown). Narrowly distributed Au@MNPs with average diameter of ∼27 nm (Fig. 2B) were fabricated by reduction of Au(OOCCH3)3 onto the 13 nm MNPs, indicating an Au shell of ∼7 nm. This addition of Au shell was also characterized by DLS measurements, showing an increased average particle size distribution of 27.5 ± 7.3 nm in comparison to 13 nm MNPs. In contrast to Au@MNPs with an Au shell of 5 nm synthesized by three iterative reductions of Au(OOCCH3)3 onto the core MNPs,33 this current procedure is proved to be able to increase the thickness of the Au shell to 7 nm in a single reduction by controlling the reaction time and the concentration of the Au precursor. Moreover, this synthesis method exhibits good repeatability in average size control over Au@MNPs with relative standard deviation (RSD) of 10.2% obtained by DLS measurements of 3 Au@MNPs samples synthesized from different batches.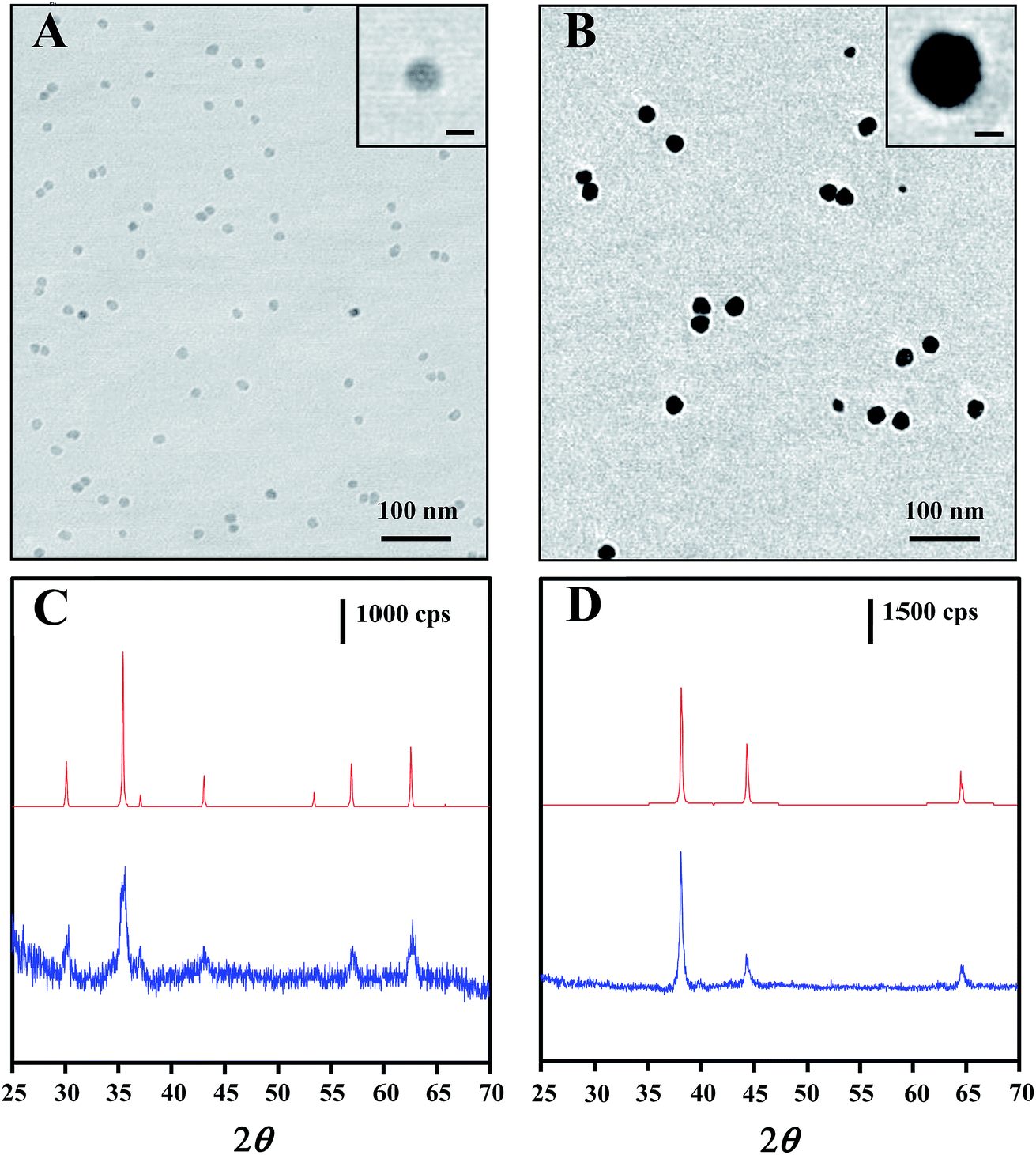 | ||
| Fig. 2 TEM images of (A) 13 nm core MNPs and (B) 27 nm Au@MNPs, (C) XRD spectra for magnetite powder standard (top), 13 nm core MNPs (bottom), (D) Au powder standards (top) and 27 nm Au@MNPs (bottom). Scale bars in the inset windows of each TEM image correspond to 10 nm. cps = counts per s. | ||
The core–shell composition of Au@MNPs was further confirmed by XRD spectra of 13 nm core MNPs and 27 nm Au@MNPs. For core MNPs (Fig. 2C bottom), five diffraction peaks at 30.0°, 35.5°, 43.1°, 57.0° and 62.6° are evident in spectrum, which correlate with the characteristic diffraction peaks of standard Fe3O4 powder (Fig. 2C top) and indicate an MNP crystal structure consistent with the cubic spinel type structure of magnetite.37 By contrast, the XRD spectrum of Au@MNPs (Fig. 2D bottom) matched the spectrum peaks for standard Au powder (Fig. 2D top) and exhibited dominant peaks at 38.2°, 44.4°, and 64.6°, which are indexed to (111), (200), and (220) planes of gold in the cubic phase.45 Due to the heavy-atom effect of Au,46 we observed no characteristic peak of Fe3O4 in the spectrum of Au@MNPs, suggesting a full Au-shell encapsulation of the MNPs.
Optical and magnetic properties of Au@MNPs
The optical properties of Au@MNPs were evaluated via UV/vis absorption spectroscopy and SERS measurements. It has been well documented that colloidal dispersions of metals exhibit absorption bands in the ultraviolet and visible ranges, due to surface plasma resonances.47 Particularly, Au NPs stabilized by citrates exhibit a surface plasmon peak at 520 nm.48 As shown in Fig. 3A, the UV-vis absorbance spectra for commercial GNPs (30 nm; spectrum a) and Au@MNPs (spectrum b) show an absorbance peak around 520 nm, which is absent for Fe3O4 core particles (spectrum c). This result provides additional evidence that the MNPs are coated with Au and demonstrates that the optical character of Au@MNPs is near to that of pure GNPs.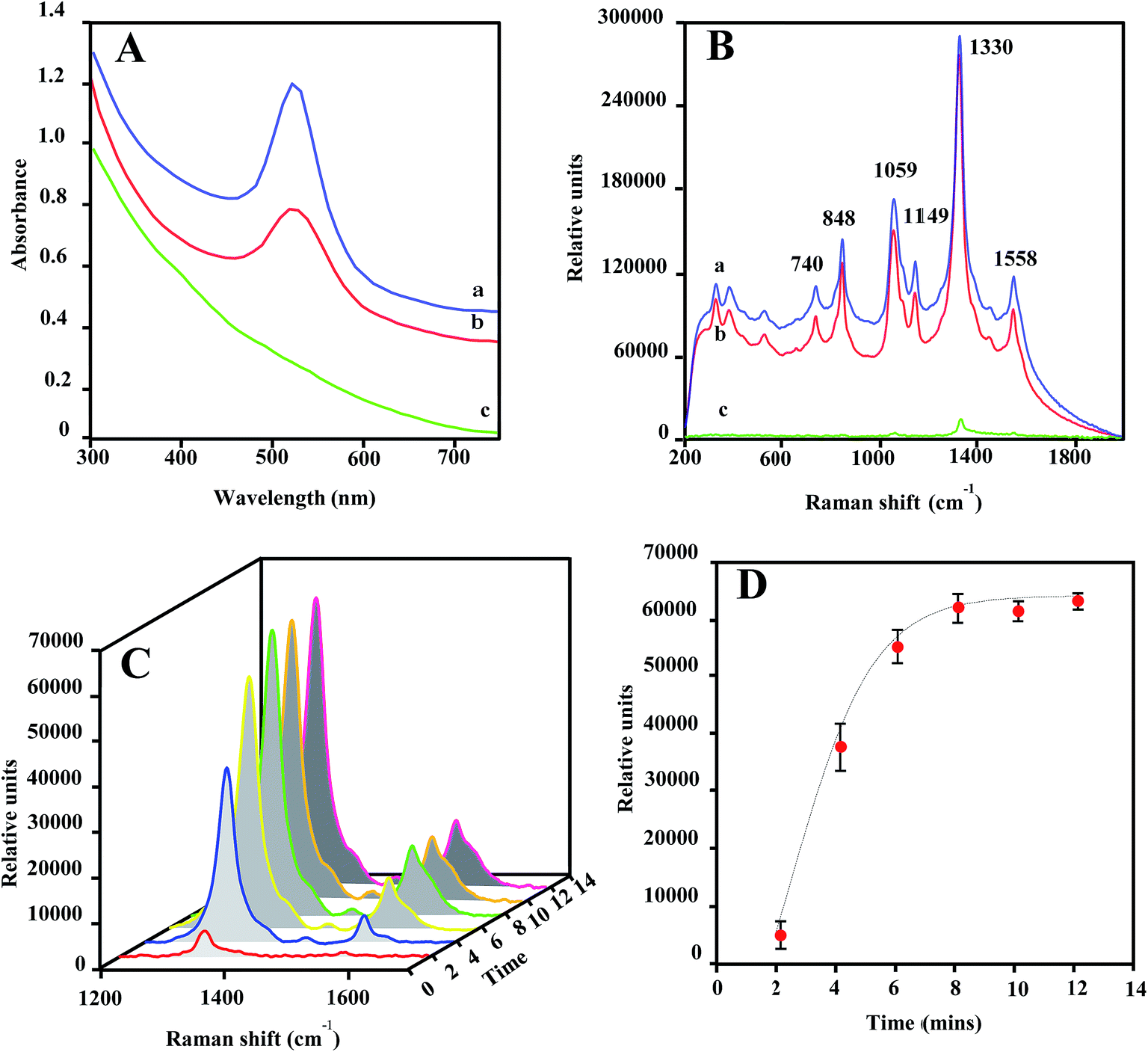 | ||
| Fig. 3 (A) UV-vis spectra for a colloidal suspension of commercially supplied 30 nm GNPs (a), 27 nm Au@MNPs (b), and 13 nm MNPs (c) in H2O. (B) SERS spectra for 20 μL air-dried NP samples spiked with DSNB (0.25 mM) 30 nm GNPs (a), 27 nm Au@MNPs (b) and 13 nm MNPs (c). Measurements were conducted using equivalent NP concentrations (2.5 × 1011 particles per mL). SERS spectra are taken at an integration time of 15 s. (C) SERS spectra in 1200–1700 cm−1 for an aqueous solution of DSNB-Au@MNPs in the microfluidic channel as a function of flowing time when magnetically concentrated at the focal spot by an external magnet. (D) Kinetic plot of peak height at 1330 cm−1 (solid circle) and fitting curve (dash curve). SERS spectra are taken at an integration time of 5 s. (0.25 mg mL−1 of DSNB-Au@MNPs). | ||
SERS enhancement is dependent on the plasmon absorption wavelength of plasmonic nanostructures and laser wavelength used in measurements.49–51 As shown in Fig. 3B, laser excitation (785 nm) of dried DSNB-labeled GNPs (spectrum a) and Au@MNPs (spectrum b) samples exhibited characteristic vibrational peaks at 740, 848, 1059, 1149, 1330, and 1558 cm−1, which are consistent with signature spectra for DSNB as previously reported,39,52 and weakly intensified in dried bare MNP samples spiked with the same amount of DSNB (spectrum c). Moreover, the signal-peak heights of 1059 and 1330 cm−1 are comparable in spectra of DSNB-conjugated GNPs and Au@MNPs, indicating that the Au character on both NPs showed similar SERS activity, which is absent in MNP samples due to the nonplasmonic iron oxide surfaces. On the basis of these results, we expect that the surface plasmon resonance of Au@MNPs is sufficiently tuned to the excitation frequency of the laser, and using Au@MNPs to substitute the bare MNPs in magnetic-capture-based SERS assays could substantially improve SERS signaling.
The magnetic recovery of Au@MNPs was demonstrated in a straight microfluidic channel using a Raman system equipped with a 100× magnification digital camera (Fig. S1†). Au@MNPs labeled with DSNB (DSNB-Au@MNPs) was injected into the microfluidic channel at a flow rate of 10 μL min−1 and concentrated at a specific spot by a small magnet below. The integrated digital camera was able to precisely aim the laser beam on the concentrated pellets with a 25 μm spot size for laser interrogation. During the magnetic focusing, the intensities of SERS peaks at 1330 and 1558 cm−1 gradually increased as a function of focusing time (Fig. 3C) and reached stabilization after 8 min, indicating the magnetic response of Au@MNPs (Fig. 3D). Moreover, the plateaued SERS signal intensity after 8 min revealed that the saturated surface coverage of concentrated pellets plays a key role in supporting stable SERS signaling. To ensure signal reproducibility, all SERS measurements of magnetically concentrated sample pellets in this paper are taken after 12 min of magnetic focusing time at a constant 10 μL min−1 flow rate.
Characterization of NP assemblies
XPS compositional data were compiled for the confirmation of GNP and Au@MNP assemblies. The wide-scan XPS survey spectra for DSNB-GNPs, Reporter-GNPs and Capture-Au@MNPs, provided binding energy peaks that demonstrate the presence of Au, S, C and N (Table 1). The S (2p3/2) and S (2p1/2) doublet peaks (161.9 eV and 163.2 eV) in all three NP samples are clear evidence of sulfur bound to gold as a thiolate.53 The single peak of P (2p) at 134.3 eV is diagnostic for the phosphoester bonds of the DNA backbone.54 In addition, the conjugation of amine-modified reporter probes to the DSNB-GNPs was confirmed by the spectra for the N (1s) region. Two peaks were evident in the DSNB-GNP spectrum: a higher energy peak at 405.3 eV corresponding to the nitro functional group,55 and a lower energy peak at 401.3 eV indicating the succinimidyl N.56 In contrast to the DSNB-GNPs, an additional peak at 399.5 eV appeared in the reporter-DSNB spectrum (Fig. 4), which is attributed to heterocyclic N atoms.57 Meanwhile a decrease in the intensity of the succinimidyl N peak (401.1 ev) was detected. These results reflect the immobilization of reporter probes via amide linkage and the removal of succinimidyl N. Two weak N (1s) peaks located at 398.7 and 400.2 eV were obtained for the Capture-Au@MNPs, which we ascribe to intracyclic and exocyclic nitrogen atoms.58 C (1s) peaks are in accordance with the binding energies of carbon species including hydrocarbons, carbon bound to nitrogen and oxygen, amide carbon and urea carbon with characteristic binding energies (BEs) of approximately 285, 286–287.8, 288, and 289 eV, respectively.59| Atomic composition | DSNB-GNPs | Reporter-GNPs | Capture-Au@PMPs |
|---|---|---|---|
| Au (4f7/2) | 84 | 84 | 84 |
| S (2p3/2) | 161.8 | 161.9 | 161.8 |
| S (2p1/2) | 163.2 | 163.2 | 163.1 |
| P (2p) | nd | 134.4 | 134.3 |
| N (1s) | 401.3, 405.3 | 399.5, 401.3, 405.3 | 400.6, 402.4 |
| C (1s) | 285.1, 286.3, 288.1, 289.1 | 285.1, 286.4, 288.2, 289.0 | 284.9, 286.5, 288.2, 289.0 |
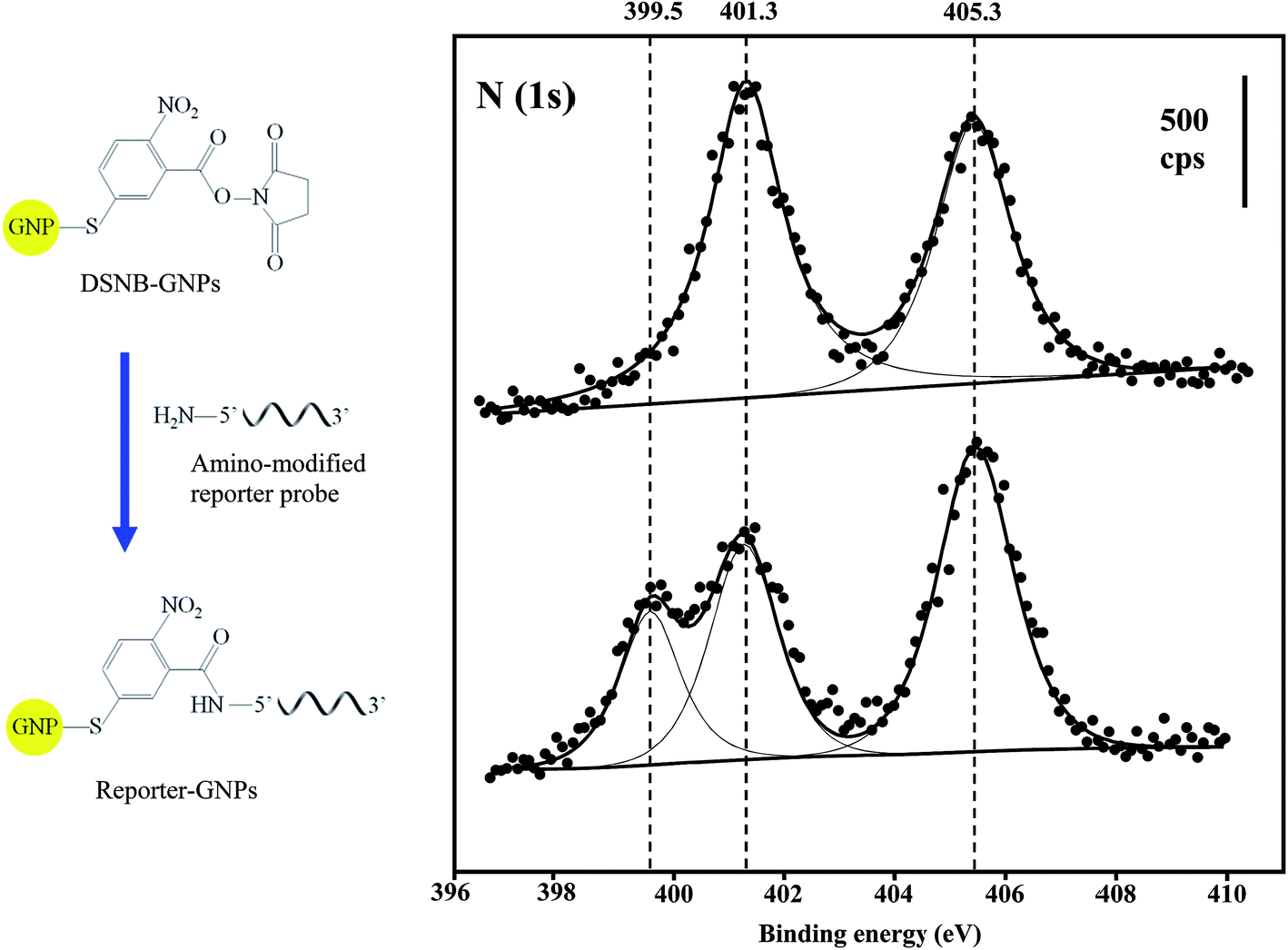 | ||
| Fig. 4 XPS N (1s) spectra for DSNB-GNPs (top spectrum) and Reporter-GNPs (bottom spectrum). The solid circles represent raw data measurements, and the solid lines are fitted peaks from deconvoluted raw data. cps = counts per s. (Left is the schematic showing the conjugation of the reporter oligonucleotide probe to GNPs via DSNB.) | ||
SERS detection of magnetically captured miR-141 target sequence
A SERS detection assay of the target miRNA sequence was conducted first by incubating Reporter-GNPs and Capture-Au@MNPs with miR-141 target oligonucleotide in capped glass vials for 1 h and then injecting them into the microfluidic channel and concentrating them at a specific spot in microchannel by a small external magnet. Upon laser excitation, an intense spectral peak profile specific for DSNB was recorded in samples containing 1 nM target sequence (Fig. 5A spectrum a). The detection specificity was demonstrated by a series of control reactions, consisted of the absence of the target sequence (spectrum d) and the nonspecific target miR-429 (spectrum c), reflecting the nonspecific binding of probes. An additional control assay, employing DSNB-GNPs unconjugated with reporter probes to substitute Reporter-GNPs, provide background spectra (spectrum b) that are similar to (spectrum c) and (spectrum d).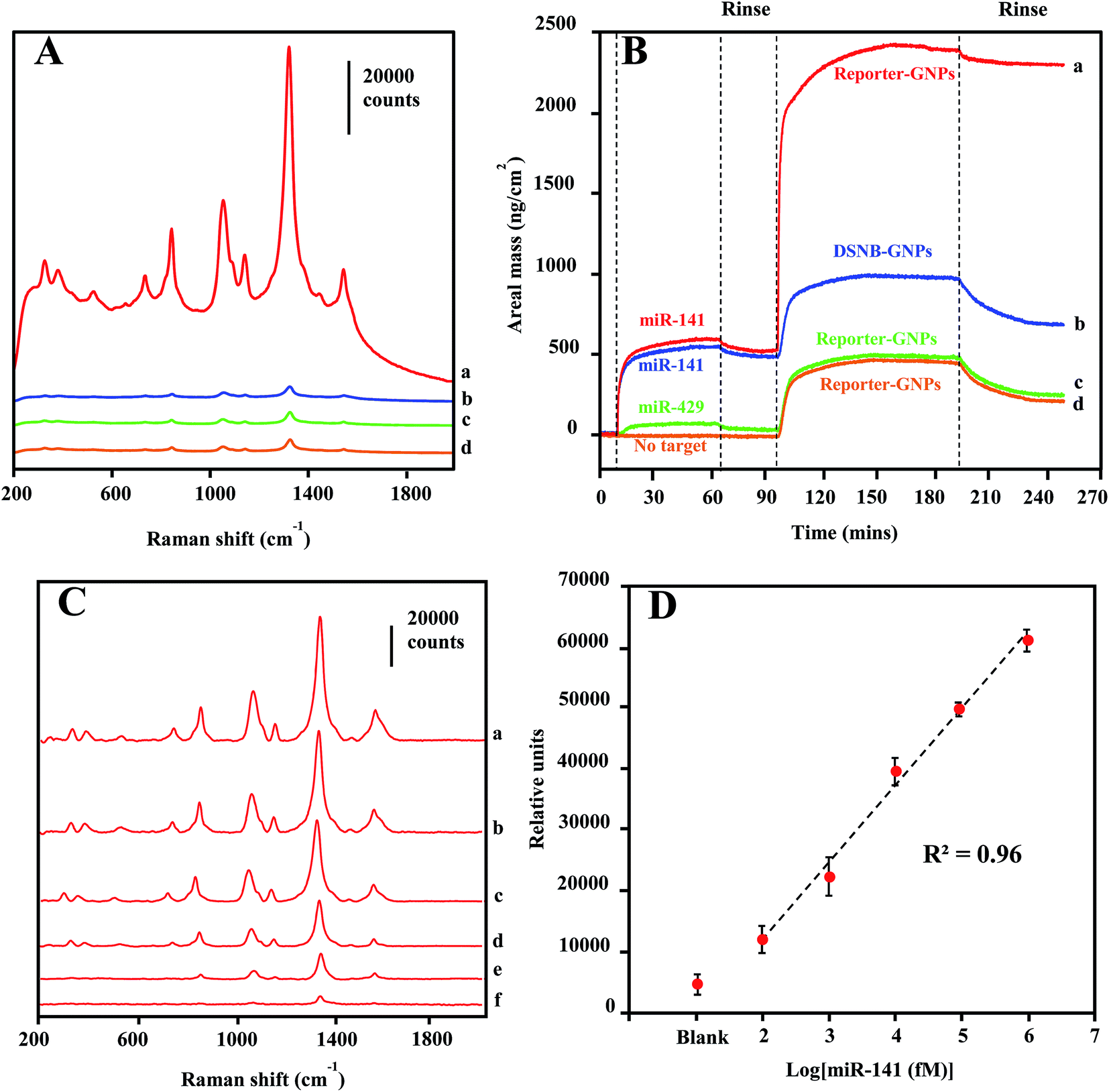 | ||
| Fig. 5 (A) SERS spectra for magnetic pull-down hybridization reactions (a) containing Reporter-GNPs, Capture-Au@MNPs, and target miR-141, (b) containing DSNB-GNPs, Capture-Au@MNPs, and target miR-141, (c) containing Reporter-GNPs, Capture-Au@MNPs, and nonspecific target miR-429, (d) containing only Reporter-GNPs and Capture-Au@MNPs. (B) QCM-D monitoring the response of capture-probe functionalized gold surfaces to (a) target miR-141 sequence (476 ng cm−2) and sequentially to Reporter-GNPs (1750 ng cm−2), (b) target miR-141 sequence (460 ng cm−2) and sequentially to nonspecific DSNB-GNPs (201 ng cm−2), (c) nonspecific miR-429 (27 ng cm−2) and sequentially to Reporter-GNPs (229 ng cm−2), and (d) the absence of target sequence (5 ng cm−2) sequentially to Reporter-GNPs (212 ng cm−2). (C) Stacked spectra for magnetic concentrates of hybridization reactions containing dilutions of target miR-141 in buffer solution: 1 nM (a), 100 pM (b), 10 pM (c), 1 pM (d), 100 fM (e) and the absence of miR-141 (f). (D) Plot of peak height at 1330 cm−1 vs. logarithm concentration of target miR-141. Error bars denote the standard deviation for three replicate measurements at each dilution. | ||
Oligonucleotide interactions involved in the specificity of miR-141 detection were confirmed by QCM-D analysis. As shown in Fig. 5B, the real monitoring of the sequential depositions of target miR-141 sequence and Reporter-GNPs onto the capture oligonucleotide-coated Au surfaces are consistent with the recognition events observed in the SERS detections. The high-level areal mass depositions for both target (476 ng cm−2) and Reporter-GNPs (1750 ng cm−2) revealed the complementary oligonucleotide interactions and the formation of hybridization complexes on the recognition Au surface (curve a). By contrast, control measurements in which the target sequence was absent (curve d) or substituted by a nonspecific oligonucleotide miR-429 (curve c) only yielded minimal areal mass change and low-level subsequent deposition of Reporter-GNPs (∼210–230 ng cm−2). Additionally, we examined the specificity of Reporter-GNPs in a negative control hybridization reaction using the substitution of DSNB-GNP (curve b), in which a low-level adsorption of DSNB-GNPs (201 ng cm−2) was observed after target sequence binding to the recognition Au surface. The low-level adsorptions of assembled GNPs are routinely observed in all negative controls, which are likely attributed to incomplete washings of nonspecifically deposited GNPs.
The LOD sensitivity for miRNA target sequence was determined by acquiring spectra for hybridization reactions containing dilution of the target oligonucleotide, and by identifying the lowest concentration, at which the spectra intensity of the 1330 cm−1 peak is distinguishable from that in absence of target miR-141. As shown in Fig. 5C, the spectra were background-corrected, and there is a progressive reduction in overall peak intensification as the concentration of target sequence decreased from 1 nM to 100 fM. The peak height of the 1330 cm−1 peaks were plotted as a function of target miR-141 concentration (fM) in Fig. 5D. The peak height is linearly regressed versus the logarithm concentration of target miR-141 with R2 of 0.96, indicating a conservative LOD for miRNA at 100 fM. This current LOD sensitivity is a hundredth of the LOD sensitivity (10 pM) previously reported in SERS detection of small DNA sequences,35 using the same DSNB-conjugated GNPs as Raman reporters, but using silica-coated MNPs as the magnetic-capture substrates. Since short oligonucleotides exhibit similar hybridization efficiencies,60 these results have implications that the plasmonic Au shell of Au@MNPs could enable a hot-spot enrichment after magnetic concentration, which would substantially improve signal intensity of SERS analytes and therefore increase the sensitivity of magnetically capturing target microRNAs, and potentially for other biomarkers. The wide-range quantification of target sequences could meet the challenge in miRNA diagnosis, which is that the concentrations of miRNAs may span several orders of magnitude.61 Moreover, using a microfluidic device in this assay significantly reduced the sample volume to 120 μL, corresponding to an attomole sensitivity for target miR-141 sequence detection.
SERS detection of miRNA in serum
There has been increasing interest in using circulating miRNAs in serum as cancer biomarkers, due to their high-level stability62 and noninvasive diagnosis.4 To test the practical application of the current SERS-based magnetic-capture assay in serum miRNA detection, we investigated the LOD for miR-141 in diluted serum (20%). The concentration-dependent SERS spectra of miR-141 from 200 pM to 200 nM are shown in Fig. 6A. In Fig. 6B, by plotting the 1330 cm−1 peak height vs. logarithm of miRNA concentration, a linear relationship was observed with R2 of 0.94, indicating a LOD of 200 pM for serum miR-141 detection which is a 2000-fold higher than that of miR-141 in buffer solution. This dramatic sensitivity reduction is probably due to either serum-protein interference with the target-sequence-mediated tethering of Capture-Au@MNPs and Reporter-GNPs, or the protein dissociation of Raman reporter molecules on the GNPs surface63 or a combination of both effects.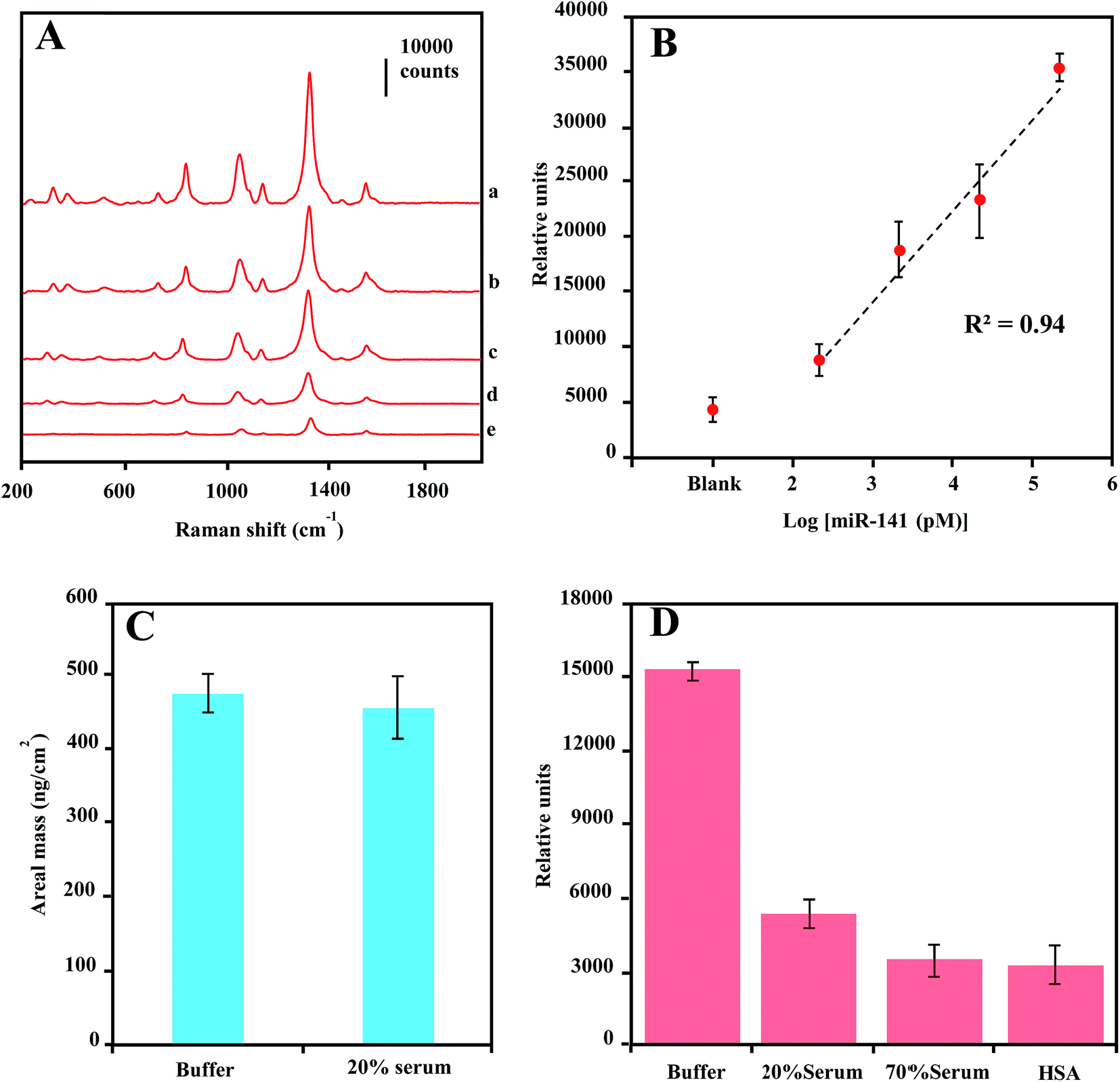 | ||
| Fig. 6 (A) Stacked spectra for magnetic concentrates of hybridization reactions containing dilutions of target miR-141 in buffer solution: 200 nM (a), 20 nM (b), 2 nM (c), 200 pM (d) and the absence of miR-141 (e), spectra integration time is 5 s. (B) Plot of 1330 cm−1 peak height from spectra of magnetic concentrates vs. logarithm of target miR-141 concentration in 20% serum solution. (C) QCM-D measurement of target miR-141-sequence adsorption on capture-probe-functionalized gold surface in buffer and 20% serum solution. (D) Peak heights at 1330 cm−1 from spectra of Reporter-GNPs dispersed in buffer, 20% serum, 70% serum and 40 mg mL−1 HSA buffer solution. Error bars denote the standard deviation for three replicate measurements at each dilution. Spectral integration time is 15 s. | ||
To verify the cause for the sensitivity reduction, we first conducted QCM measurement of areal mass depositions of miR-141 on the capture oligonucleotide-coated Au surfaces in 20% serum. Compared with the measurement conducted in buffer solution, there is no significant difference in areal mass depositions of miR-141 from buffer to 20% serum (Fig. 6C), indicating that serum proteins offered little interference in the hybridization of small oligonucleotide sequences.64 We further measured the SERS enhancement of Reporter-GNPs spikes in PBS buffer solution, 20% serum, 70% serum and 40 mg mL−1 human serum albumin (HSA) solution. As shown in Fig. 6D, the 1330 cm−1 peak height from spectra of Reporter-GNPs reduced nearly three folds as the serum concentration increased, from buffer solution to 20% serum. Additionally, the high-concentration 70% serum and HSA solution also showed a strong impact on SERS intensity of Reporter-GNPs and reduced the signal intensity over four fold, compared to that in buffer solution. These results indicate that the probable mechanism by which the reduction of LOD sensitivity occurs is the serum-protein-induced dissociation of Raman reporter molecules from the GNP surface, as proteins tend to bind to and denature on metal surfaces.65,66
Although the SERS signal reduction of free Reporter-GNPs from buffer to 20% serum is not comparable to that of captured Reporter-GNPs in magnetic concentrates, we are of the opinion that the protein displacement of Raman reporter molecules, DSNB, would reduce the number of reporter oligonucleotide probes conjugated to GNPs via DSNB linkers, which would have a great impact on the formation of hybridization complexes and therefore dramatically decrease the corresponding sensitivity. Moreover, the disassociated reporter oligonucleotide probes would bind to target sequences and block the tethering of Reporter-GNPs to Capture-Au@MNPs, resulting in less intensification of the SERS signal for target detection.
Conclusions
In the work presented here, we have demonstrated a magnetic-capture-based SERS assay for detection of microRNAs, in which miR-141 was selected as target sequence and captured by hybridization reactions with complementary probes conjugated with Raman-active GNPs and Au@MNPs. SERS response to the magnetically concentrated hybridization complexes, GNPs/miR-141/Au@MNPs, featured high specificity to the target miR-141 sequence. QCM-D monitoring of the formation of hybridization complexes on Au surfaces revealed the RNA/DNA interactions involved in the specificity of miR-141 detection, and positively correlated with recognition events interrogated by SERS measurements. The recorded LOD of current SERS assay is estimated to be 100 fM, a 100-fold improvement over the LOD (10 pM) reported in a similar magnetic pull-down assay for small DNA detection, but using nonplasmonic Silica@MNPs as magnetic substrates. The enhanced detection sensitivity is mainly due to the plasmonic Au shell of Au@MNPs that has added benefits of decreasing nonplasmonic iron oxide surfaces in hybridization complexes and increasing the probability of forming SERS hot spots with nearby nanostructured Au contents.Although the LOD was not as low as those in earlier reports using enzymes or oligonucleotides to trigger the target amplification, we believe further optimization of the plasmonic activity of NPs should improve the detection limit to meet the requirement of enzyme-free diagnostic settings. It will be of interest in our future studies to investigate the dependence on particle size and shape associated with surface plasmon resonance and SERS enhancement in magnetic pull-down assays. Additionally, as suggested by hybridization reactions conducted in serum solutions, a reliable and reproducible SERS signaling of clinical sample solutions will require SERS tags with protection shells; that will prevent the serum-protein-induced dissociation of Raman reporter molecules on the GNP surface. Part of our ongoing work is to develop a SERS tag with protection shells, such as silica coating. Nevertheless, the simple design of the current assay and the adaption of a microfluidic format provided opportunities to enable a rapid and affordable point-of-care setting for detections of miRNAs, and potentially of other biomolecular targets by constructing a series of recognition probes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC-21404090), Zhejiang Provincial Natural Science Foundation of China (ZJNSF-LQ14B050001).References
- E. A. Miska, Curr. Opin. Genet. Dev., 2005, 15, 563–568 CrossRef CAS PubMed
.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS PubMed
- M. Schubert, K. Junker and J. Heinzelmann, J. Cancer Res. Clin. Oncol., 2016, 142, 1673–1695 CrossRef CAS PubMed
- G. A. Calin and C. M. Croce, Nat. Rev. Cancer, 2006, 6, 857–866 CrossRef CAS PubMed
- P. S. Mitchell, R. K. Parkin, E. M. Kroh, B. R. Fritz, S. K. Wyman, E. L. Pogosova-Agadjanyan, A. Peterson, J. Noteboom, K. C. O'Briant, A. Allen, D. W. Lin, N. Urban, C. W. Drescher, B. S. Knudsen, D. L. Stirewalt, R. Gentleman, R. L. Vessella, P. S. Nelson, D. B. Martin and M. Tewari, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10513–10518 CrossRef CAS PubMed
- Y. Gao, B. Feng, S. Han, K. Zhang, J. Chen, C. Li, R. Wang and L. Chen, Cell. Physiol. Biochem., 2016, 38, 427–448 CrossRef CAS PubMed
- M. H. Mo, L. Chen, Y. Fu, W. Wang and S. W. Fu, J. Cancer, 2012, 3, 432–448 CrossRef CAS PubMed
- A. Valoczi, C. Hornyik, N. Varga, J. Burgyan, S. Kauppinen and Z. Havelda, Nucleic Acids Res., 2004, 32, e175 CrossRef PubMed
- N. Redshaw, T. Wilkes, A. Whale, S. Cowen, J. Huggett and C. A. Foy, BioTechniques, 2013, 54, 155–164 CrossRef CAS PubMed
- J. M. Thomson, J. Parker, C. M. Perou and S. M. Hammond, Nat. Methods, 2004, 1, 47–53 CrossRef CAS PubMed
- B. N. Johnson and R. Mutharasan, Analyst, 2014, 139, 1576–1588 RSC
- A. D. Mehta, M. Rief and J. A. Spudich, J. Biol. Chem., 1999, 274, 14517–14520 CrossRef CAS PubMed
- J. Su, D. Wang, L. Norbel, J. Shen, Z. Zhao, Y. Dou, T. Peng, J. Shi, S. Mathur, C. Fan and S. Song, Anal. Chem., 2017, 89, 2531–2538 CrossRef CAS PubMed
- K. Yang, Y. J. Hu and N. Dong, Biosens. Bioelectron., 2016, 80, 373–377 CrossRef CAS PubMed
- A. Pallaoro, M. R. Hoonejani, G. B. Braun, C. D. Meinhart and M. Moskovits, ACS Nano, 2015, 9, 4328–4336 CrossRef CAS PubMed
- L. M. Tong, T. Zhu and Z. F. Liu, Chem. Soc. Rev., 2011, 40, 1296–1304 RSC
- S. M. Nie and S. R. Emery, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed
- P. G. Etchegoin, P. D. Lacharmoise and E. C. Le Ru, Anal. Chem., 2009, 81, 682–688 CrossRef CAS PubMed
- Y. Q. Wang, B. Yan and L. X. Chen, Chem. Rev., 2013, 113, 1391–1428 CrossRef CAS PubMed
- K. Hering, D. Cialla, K. Ackermann, T. Dorfer, R. Moller, H. Schneidewind, R. Mattheis, W. Fritzsche, P. Rosch and J. Popp, Anal. Bioanal. Chem., 2008, 390, 113–124 CrossRef CAS PubMed
- J. D. Driskell, A. G. Seto, L. P. Jones, S. Jokela, R. A. Dluhy, Y. P. Zhao and R. A. Tripp, Biosens. Bioelectron., 2008, 24, 917–922 CrossRef CAS PubMed
- J. D. Driskell, O. M. Primera-Pedrozo, R. A. Dluhy, Y. Zhao and R. A. Tripp, Appl. Spectrosc., 2009, 63, 1107–1114 CrossRef CAS PubMed
- H.-N. Wang, B. M. Crawford, A. M. Fales, M. L. Bowie, V. L. Seewaldt and T. Vo-Dinh, J. Phys. Chem. C, 2016, 120, 21047–21055 CAS
- J. D. Driskell and R. A. Tripp, Chem. Commun., 2010, 46, 3298–3300 RSC
- H. Zhang, Y. Liu, J. Gao and J. Zhen, Chem. Commun., 2015, 51, 16836–16839 RSC
- J. J. Li, Q. Xi, W. F. Du, R. Q. Yu and J. H. Jiang, Analyst, 2016, 141, 2384–2387 RSC
- Y. Pang, C. Wang, J. Wang, Z. Sun, R. Xiao and S. Wang, Biosens. Bioelectron., 2016, 79, 574–580 CrossRef CAS PubMed
- J. Zheng, D. Ma, M. Shi, J. Bai, Y. Li, J. Yang and R. Yang, Chem. Commun., 2015, 51, 16271–16274 RSC
- X. Li, S. Ye and X. Luo, Chem. Commun., 2016, 52, 10269–10272 RSC
- A. M. Michaels, J. Jiang and L. Brus, J. Phys. Chem. B, 2000, 104, 11965–11971 CrossRef CAS
- T. Kang, H. Kim, J. M. Lee, H. Lee, Y. S. Choi, G. Kang, M. K. Seo, B. H. Chung, Y. Jung and B. Kim, Small, 2014, 10, 4200–4206 CrossRef CAS PubMed
- W. Zhou, Y. F. Tian, B. C. Yin and B. C. Ye, Anal. Chem., 2017, 89, 6120–6128 CrossRef CAS PubMed
- H. Zhang, M. H. Harpster, W. C. Wilson and P. A. Johnson, Langmuir, 2012, 28, 4030–4037 CrossRef CAS PubMed
- J. Neng, M. H. Harpster, W. C. Wilson and P. A. Johnson, Biosens. Bioelectron., 2013, 41, 316–321 CrossRef CAS PubMed
- H. Zhang, M. H. Harpster, H. J. Park, P. A. Johnson and W. C. Wilson, Anal. Chem., 2011, 83, 254–260 CrossRef CAS PubMed
- B. H. Jun, M. S. Noh, J. Kim, G. Kim, H. Kang, M. S. Kim, Y. T. Seo, J. Baek, J. H. Kim, J. Park, S. Kim, Y. K. Kim, T. Hyeon, M. H. Cho, D. H. Jeong and Y. S. Lee, Small, 2010, 6, 119–125 CrossRef CAS PubMed
- S. H. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. X. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed
- L. Wang, J. Luo, Q. Fan, M. Suzuki, I. S. Suzuki, M. H. Engelhard, Y. Lin, N. Kim, J. Q. Wang and C. J. Zhong, J. Phys. Chem. B, 2005, 109, 21593–21601 CrossRef CAS PubMed
- D. S. Grubisha, R. J. Lipert, H. Y. Park, J. Driskell and M. D. Porter, Anal. Chem., 2003, 75, 5936–5943 CrossRef CAS PubMed
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed
- M. H. Harpster, H. Zhang, A. K. Sankara-Warrier, B. H. Ray, T. R. Ward, J. P. Kollmar, K. T. Carron, J. O. Mecham, R. C. Corcoran, W. C. Wilson and P. A. Johnson, Biosens. Bioelectron., 2009, 25, 674–681 CrossRef CAS PubMed
- J. Neng, M. H. Harpster, H. Zhang, J. O. Mecham, W. C. Wilson and P. A. Johnson, Biosens. Bioelectron., 2010, 26, 1009–1015 CrossRef CAS PubMed
- C. C. White and J. L. Schrag, J. Chem. Phys., 1999, 111, 11192–11206 CrossRef CAS
- M. V. Voinova, M. Jonson and B. Kasemo, Biosens. Bioelectron., 2002, 17, 835–841 CrossRef CAS PubMed
- L. Y. Wang, J. Luo, Q. Fan, M. Suzuki, I. S. Suzuki, M. H. Engelhard, Y. H. Lin, N. Kim, J. Q. Wang and C. J. Zhong, J. Phys. Chem. B, 2005, 109, 21593–21601 CrossRef CAS PubMed
- T. Mielke, C. Villa, P. C. Edwards, G. F. X. Schertler and M. P. Heyn, J. Mol. Biol., 2002, 316, 693–709 CrossRef CAS PubMed
- J. A. Creighton and D. G. Eadon, J. Chem. Soc., Faraday Trans., 1991, 87, 3881–3891 RSC
- K. R. Brown, D. G. Walter and M. J. Natan, Chem. Mater., 2000, 12, 306–313 CrossRef CAS
- M. Kahraman, P. Daggumati, O. Kurtulus, E. Seker and S. Wachsmann-Hogiu, Sci. Rep., 2013, 3, 3396 CrossRef PubMed
- C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2003, 107, 7426–7433 CrossRef CAS
- N. G. Greeneltch, M. G. Blaber, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. C, 2013, 117, 2554–2558 CAS
- J. D. Driskell, K. M. Kwarta, R. J. Lipert, M. D. Porter, J. D. Neill and J. F. Ridpath, Anal. Chem., 2005, 77, 6147–6154 CrossRef CAS PubMed
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS
- S. K. Kulkarni, A. S. Ethiraj, S. Kharrazi, D. N. Deobagkar and D. D. Deobagkar, Biosens. Bioelectron., 2005, 21, 95–102 CrossRef CAS PubMed
- O. Bjorneholm, A. Nilsson, E. O. F. Zdansky, A. Sandell, B. Hernnas, H. Tillborg, J. N. Andersen and N. Martensson, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 10353–10365 CrossRef
- J. Wei, P. Hing and Z. Q. Mo, Surf. Interface Anal., 1999, 28, 208–211 CrossRef CAS
- P. A. Johnson and R. Levicky, Langmuir, 2003, 19, 10288–10294 CrossRef CAS
- B. A. Cavic, M. E. McGovern, R. Nisman and M. Thompson, Analyst, 2001, 126, 485–490 RSC
- C. Y. Lee, P. Gong, G. M. Harbers, D. W. Grainger, D. G. Castner and L. J. Gamble, Anal. Chem., 2006, 78, 3316–3325 CrossRef CAS PubMed
- W. Grange, M. Duckely, S. Husale, S. Jacob, A. Engel and M. Hegner, PLoS Biol., 2008, 6, 343–351 CAS
- W. T. Liu, H. L. Guo and J. H. Wu, Appl. Environ. Microbiol., 2007, 73, 73–82 CrossRef CAS PubMed
- R. Fiammengo, Biomarkers Med., 2017, 11, 69–86 CrossRef CAS PubMed
- X. Chen, Y. Ba, L. Ma, X. Cai, Y. Yin, K. Wang, J. Guo, Y. Zhang, J. Chen, X. Guo, Q. Li, X. Li, W. Wang, Y. Zhang, J. Wang, X. Jiang, Y. Xiang, C. Xu, P. Zheng, J. Zhang, R. Li, H. Zhang, X. Shang, T. Gong, G. Ning, J. Wang, K. Zen, J. Zhang and C. Y. Zhang, Cell Res., 2008, 18, 997–1006 CrossRef CAS PubMed
- K. Rijal and R. Mutharasan, Anal. Chem., 2007, 79, 7392–7400 CrossRef CAS PubMed
- M. Lin, L. He, J. Awika, L. Yang, D. R. Ledoux, H. Li and A. Mustapha, J. Food Sci., 2008, 73, T129–T134 CrossRef CAS PubMed
- R. S. Golightly, W. E. Doering and M. J. Natan, ACS Nano, 2009, 3, 2859–2869 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10918k |
| This journal is © The Royal Society of Chemistry 2017 |