
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
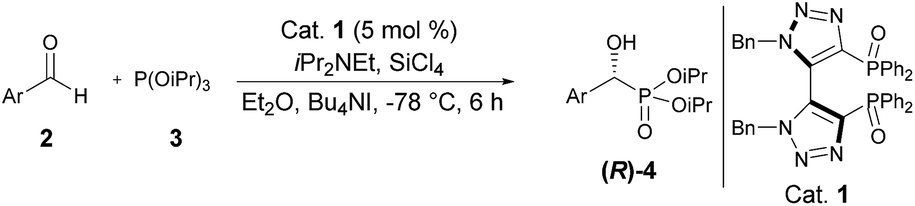
1,1′-Dibenzyl-bis-(triazolyl)diphenylphosphine dioxide: a new efficient organocatalyst for silicon tetrachloride-mediated enantioselective Abramov-type phosphonylation of aldehydes with trialkyl phosphites†
Nicolas Sevraina,
Jean-Noël Vollea,
Jean-Luc Pirat a,
Tahar Ayad*ab and
David Virieux*a
a,
Tahar Ayad*ab and
David Virieux*a
aInstitut Charles Gerhardt, CNRS UMR 5253, Ecole Nationale Supérieure de Chimie de Montpellier, 8 Rue de l'Ecole Normale, 34 296 Montpellier, France. E-mail: david.virieux@enscm.fr
bPSL Research University, Chimie Paristech-CNRS, Institut de Recherche de Chimie Paris, 75 005 Paris, France. E-mail: tahar.ayad@chimie-paristech.fr
First published on 9th November 2017
Abstract
Asymmetric phosphonylation of aldehydes with trialkyl phosphites using a combination of SiCl4 and a novel 1,1′-dibenzyl-bis-(triazolyl)diphenylphosphine dioxide organocatalyst has been developed. This protocol provides the corresponding α-hydroxyphosphonates with a broad range of functional groups and substitution patterns in excellent yields and good selectivities.
Chiral α-hydroxyphosphonates represent an important class of molecules that has been widely used in pharmaceutical and agrochemical chemistry owing to their interesting biological activities (Fig. 1).1 They are also of considerable interest because they are key precursors for the preparation of valuable α-substituted phosphonate derivatives, which encompass a wide range of biological activities, such as α-hydroxy phosphonic acids,2 and α-amino-,3 α-keto-,4 α-halo-,5 and α-acetoxy phosphonates.6 Therefore, it is not surprising that numerous methods have emerged in the past decades for their preparation in enantiomerically pure or enriched form.7 Among these methods, the asymmetric phosphonylation of aldehydes with dialkyl phosphites (Pudovik reaction),8 is without a doubt one of the most powerful synthetic tools for the stereoselective construction of P–C bonds in organic chemistry, which provides a practical access to α-hydroxyphosphonates. In contrast, an alternative approach based on the enantioselective addition of easily available and inexpensive trialkyl phosphites to aldehydes (Abramov type reaction),9 has been scarcely studied with only a very limited number of reports. In 2008, Nakajima and co-workers disclosed the first example of SiCl4-mediated asymmetric addition of triethyl phosphite to aldehydes using various C2-symmetric chiral bisphosphine dioxides as chiral Lewis base catalysts.10 Under optimized conditions, the corresponding diethyl α-hydroxyphosphonates were obtained with moderate to good yields and with low to moderate ee values, ranging from 9 to 49%. Few years later, the same group achieved similar modest selectivities up to 49% ee using newly prepared Ar-DIOP dioxide derivatives.11 In 2013, Dogan and co-workers reported the synthesis of diethyl α-hydroxyphosphonates with comparable efficiency in terms of yields and enantioselectivities by using a new phosphine oxide chiral inducer containing an aziridinyl scaffold.12 These results clearly underlined that new and more effective catalytic systems needed to be developed for enantioselective Abramov type reaction to obtain high-value chiral α-hydroxyphosphonates with enhanced enantioselectivities.
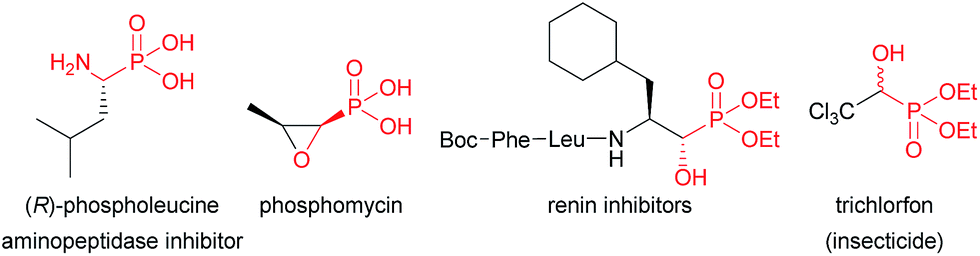 | ||
| Fig. 1 Biologically active α-hydroxy- and α-aminophosphonate derivatives. | ||
Recently, we have developed a highly convergent and atom economic synthetic route toward a new family of chiral C2-symmetric diphosphine dioxides bearing an original bis(triazolyl) backbone through a tandem Cu-mediated Huisgen reaction – oxidative coupling (Scheme 1).13 With these synthesized ligands we then envisioned that they could act as effective chiral Lewis bases for asymmetric catalysis. Indeed, achiral monotriazolyl phosphines also named clickphos showed high electron-donating behavior through N ⇨ P orbital overlap and we suspected that the bis-triazolyldiphosphines dioxides might exhibit a similar stabilization.14 Herein, we report the results of our investigation on SiCl4-mediated enantioselective Abramov-type phosphonylation of diversely substituted aldehydes with trialkyl phosphites using the novel 1,1′-dibenzyl-bis-(triazolyl)diphenyl phosphine dioxide 3 as catalyst.
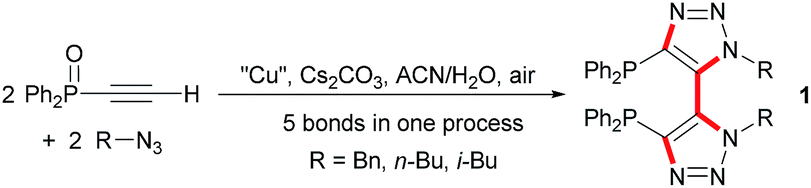 | ||
| Scheme 1 Bis-triazolylphosphine synthesis. | ||
For initial optimization of the reaction conditions, the asymmetric phosphonylation of benzaldehyde 2a with triethyl phosphite 3 as nucleophile was investigated as a model reaction system by adopting a literature procedure (Table 1).10 The first experiment was carried out in dichloromethane at −78 °C for 4 h using 10 mol% of catalyst 1 in the presence of SiCl4 and diisopropylethylamine, giving the chiral diethyl α-hydroxyphosphonate 4a in 78% yield and an encouraging enantiomeric ratio of 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :26 (entry 1). Although modest, it should be noted that this result compared favorably with the best results reported so far in the literature for the same transformation using diphosphine dioxides as Lewis bases.10–12 Interestingly, we found that the use of tetrabutylammoniun iodide as an additive, which has been reported to significantly accelerate the allylation of aldehydes with allyltrichlorosilanes,15 significantly increased the catalytic activity in terms of both yield and selectivity (entry 1 vs. 2).
:26 (entry 1). Although modest, it should be noted that this result compared favorably with the best results reported so far in the literature for the same transformation using diphosphine dioxides as Lewis bases.10–12 Interestingly, we found that the use of tetrabutylammoniun iodide as an additive, which has been reported to significantly accelerate the allylation of aldehydes with allyltrichlorosilanes,15 significantly increased the catalytic activity in terms of both yield and selectivity (entry 1 vs. 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Product | Solvent | Additive | Yieldb (%) | erc (%) |
| a Unless otherwise specified, all reactions were performed using 10 mol% of catalyst 1 with 0.5 mmol of PhCHO 2a, Bu4NI (1 equiv., 0.5 mmol), i-Pr2NEt (3 equiv., 1.5 mmol), trialkyl phosphite 3 (1.2 equiv., 0.6 mmol) and SiCl4 (1.5 equiv.) added over 10 min (0.75 mmol, 1 M in CH2Cl2) for 4 h.b Isolated yield after purification by flash chromatography.c Determined by HPLC chromatography using a Chiralcel AS-H or IC columns. Absolute configuration was attributed as R by comparison of optical rotation to literature values (see the ESI).d SiCl4 was added over 2 h with a syringe pump.e Run with 5 mol% of catalyst for 6 h.f Run with 15 mol% of catalyst for 2 h. | |||||
| 1 | R = Et, 4a | CH2Cl2 | None | 78 | 74:26 |
| 2 | R = Et, 4a | CH2Cl2 | Bu4NI | 89 | 78:22 |
| 3 | R = Et, 4a | DCE | Bu4NI | 68 | 61:39 |
| 4 | R = Et, 4a | CHCl3 | Bu4NI | 65 | 53:47 |
| 5 | R = Et, 4a | EtCN | Bu4NI | 73 | 65:35 |
| 6 | R = Et, 4a | Toluene | Bu4NI | 80 | 70:30 |
| 7 | R = Et, 4a | THF | Bu4NI | 81 | 82:18 |
| 8 | R = Et, 4a | Et2O | Bu4NI | 91 | 84.5:15.5 |
| 9d | R = Et, 4a | Et2O | Bu4NI | 89 | 85:15 |
| 10 | R = Me, 4b | Et2O | Bu4NI | 87 | 84.6:15.4 |
| 11 | R = Bu, 4c | Et2O | Bu4NI | 79 | 78.5:21.5 |
| 12 | R = iPr, 4d | Et2O | Bu4NI | 94 | 88.5:11.5 |
| 13e | R = iPr, 4d | Et2O | Bu4NI | 92 | 88:12 |
| 14f | R = iPr, 4d | Et2O | Bu4NI | 95 | 89:11 |
| 15 | R = iPr, 4d | Et2O | NaI | 70 | 85.5:14.5 |
| 16 | R = iPr, 4d | Et2O | KI | 73 | 85.5:14.5 |
| 17 | R = iPr, 4d | Et2O | I2 | 56 | 70:30 |

Encouraged by these results, the influence of various parameters such as the solvent, trialkyl phosphite reagents, catalyst loading as well as other iodide additives was evaluated. As outlined in Table 1, the stereochemical outcome of the reaction is strongly solvent-dependent. When propionitrile or halogenated solvents, such as dichloroethane or chloroform were used, compound 4a was isolated in moderate yield and low selectivity (entries 3–5), while slightly better result were obtained with a non-polar solvent such as toluene (entry 6). From this screening, it turned out that ether solvents were the most effective and that diethylether provided the best results with respect to both catalytic activity and asymmetric induction, giving 4a in excellent yield (91%) and with a good enantiomeric ratio of 84.5:15.5 (entries 7–8). An attempt to increase the selectivity of the reaction by preventing the non-selective reaction through a slow addition of SiCl4 over 2 h did not show significant improvement (entry 9).16 Next, the effect of various trialkyl phosphites on the phosphonylation reaction of 2a was studied (entries 10–12). Changing the alkyl group of the phosphite from triethyl to trimethyl led to the formation of 4a in 87% yield and with a comparable enantiomeric ratio of 84.6/15.4 (entries 8 vs. 10). A significant drop in the catalytic efficiency was observed with tributyl phosphite, while the bulky triisopropyl phosphite emerged as the best phosphonylating agent, yielding the desired diisopropyl α-hydroxyphosphonate 4d in an excellent isolated yield of 94% and a high enantiomeric ratio of 88.5/11.5 (entries 11–12). In addition, we found that increasing (15 mol%) or lowering (5 mol%) the amount of catalyst 1 resulted in almost similar catalytic activities, although a longer reaction time (6 h) was required to reach completion with 5 mol% (entries 13–14). Finally, we also examined a series of different additives including KI, NaI, I2 but tetrabutylammoniun iodide proved to be the best choice for this transformation (entries 15–17).
With the optimal conditions established, the scope of the reaction was then probed. To this end, diisopropyl phosphite 3 was reacted with a set of diversely functionalized aryl aldehydes, in diethyl ether at −78 °C for 6 h, using 5 mol% of 1,1′-dibenzyl-bis-(triazolyl)diphenylphosphine dioxide 3 as Lewis base in combination with SiCl4 in the presence of diisopropylethylamine and tetrabutylammonium iodide as an additive. As shown in Table 2, the reaction proceeded well in most cases, providing the corresponding enantioenriched α-hydroxyphosphonate products 4d–p in good to excellent yields (79 to 96%) and a good to high enantiomeric ratio up to 89:11, regardless of the electronic nature and position of the substituents present on the substrates. More specifically, reaction of benzaldehyde derivatives bearing electron donating groups at the para or meta position of the phenyl ring furnished compounds 4f–h in excellent isolated yields ranging from 93 to 95% and high enantiomeric ratios varying from 85.5:14.5 to 88.5:11.5 (entries 3–5). Comparable results were achieved with substrates bearing electron withdrawing groups such as –F or –CF3, whereas for unclear reasons, only a moderate selectivity was observed for p-Cl benzaldehyde, while maintaining a high yield of 91% (entry 6). It should be also noted that a marked drop of the catalytic efficiency in terms of both yield and selectivity was observed with hindered or ortho-substituted benzaldehyde derivatives. For instance, α-hydroxyphosphonates 4e, 4i and 4o were obtained in significantly lower yields varying from 78 to 81% and low to moderate enantiomeric ratios in the range of 67.5:32.5 to 72:28 (entries 4, 6 and 12). These results demonstrate that the reaction is highly sensitive to the steric properties of the aryl aldehyde, which could be attributed to an increased steric hindrance between the catalytic system and the ortho-substituted group of the substrate during the course of the reaction. Finally, 2-naphthyl substituted aldehyde also reacted quite well in these reaction conditions and gave the phosphonylated product 4n in 95% yield and a high enantiomeric ratio of 88.5:11.5. 2-Furaldehyde appeared, however, to be not a suitable substrate (entries 11 and 13).
|
||||
|---|---|---|---|---|
| Entry | 4, Ar | Yieldb (%) | erc (%) | |
| a Reactions run using 5 mol% of catalyst 1 with 0.5 mmol of aldehyde 2, Bu4NI (1 equiv., 0.5 mmol), i-Pr2NEt (3 equiv., 1.5 mmol), trialkyl phosphite 3 (1.2 equiv., 0.6 mmol) and SiCl4 (1.5 equiv., 0.75 mmol, 1 M in DCM) for 6 h.b Isolated yield after purification by flash chromatography.c Determined by HPLC chromatography using a Chiralcel AS-H or IC columns (see the ESI). The absolute configurations of all products were assigned to be R by analogy with 4d. | ||||
| 1 | 4d |  |
92 | 88:12 |
| 2 | 4e |  |
79 | 72:28 |
| 3 | 4f | 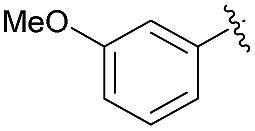 |
93 | 88.5:11.5 |
| 4 | 4g | 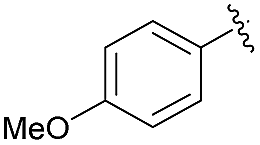 |
95 | 85.5:14.5 |
| 5 | 4h |  |
94 | 86.5:13.5 |
| 6 | 4i | 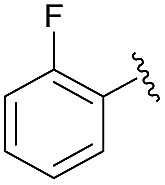 |
81 | 66.5:33.5 |
| 7 | 4j | 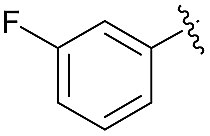 |
93 | 81:19 |
| 8 | 4k | 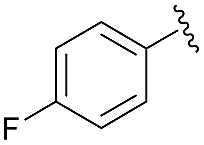 |
95 | 89:11 |
| 9 | 4l |  |
93 | 86:14 |
| 10 | 4m | 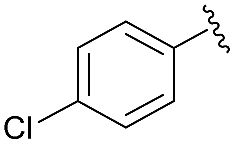 |
91 | 72:28 |
| 11 | 4n |  |
95 | 88.5:11.5 |
| 12 | 4o | 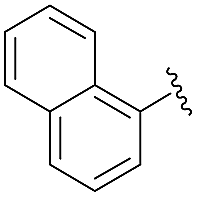 |
78 | 67.5:32.5 |
| 13 | 4p | 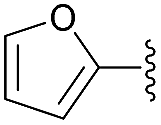 |
96 | 66:34 |
Conclusions
In summary, we have developed a new catalytic system for SiCl4 promoted enantioselective phosphonylation of aldehydes with trialkyl phosphites using a novel chiral C2-symmetric diphenylphosphine dioxide organocatalyst bearing an original bis(triazolyl) backbone. Under optimized conditions, a wide range of high-value chiral α-hydroxyphosphonates were obtained in excellent isolated yields up to 96% and very promising enantiomeric ratios up to 89:11, indicating a considerable potential for this new class of ligand in asymmetric catalysis. To our knowledge, this study represents, so far, the best reported results for the Abramov type reaction using diphosphine dioxides as chiral Lewis base catalysts.17 Further improvement of this catalytic system via ligand modification are ongoing in our laboratory and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. S. is grateful to the French Ministry of Higher Education and Scientific Research for financial support.Notes and references
- For selected examples, see: (a) D. L. Pompliano, E. Rands, M. D. Schaber, S. D. Mosser, N. J. Anthony and J. B. Gibbs, Biochemistry, 1992, 31, 3800 CrossRef CAS PubMed; (b) M. Tao, R. Bihovsky, G. J. Wells and J. P. Mallamo, J. Med. Chem., 1998, 41, 3912 CrossRef CAS PubMed; (c) O. I. Kolodiazhnyi, Russ. Chem. Rev., 2006, 75, 227 CrossRef CAS.
- R. F. Frechette, C. Ackerman, S. Beers, R. Look and J. Moore, Bioorg. Med. Chem. Lett., 1995, 5, 1801 CrossRef.
- B. Kaboudin, Tetrahedron Lett., 2003, 44, 1051 CrossRef CAS.
- T. R. Burke, M. S. Smyth, A. Otaka and P. P. Roller, Tetrahedron Lett., 1993, 34, 4125 CrossRef CAS.
- S. Kumaraswamy, R. Senthamizh Selvi and K. C. Kumaraswamy, Synthesis, 1997, 207 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor, S. Sobhani and Z. Amoozgar, Synthesis, 2004, 295 CrossRef CAS.
- For selected examples, see: (a) H. Gröger and B. Hammer, Chem.–Eur. J., 2000, 6, 943 CrossRef; (b) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2005, 16, 3295 CrossRef CAS; (c) B. Saito, H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 1978 CrossRef CAS PubMed; (d) P. Merino, E. Marqués López and R. P. Herrera, Adv. Synth. Catal., 2008, 350, 1195 CrossRef CAS; (e) N. S. Goulioukina, G. N. Bondarenko, A. V. Bogdanov, K. N. Gavrilov and I. P. Belestkaya, Eur. J. Org. Chem., 2009, 510 CrossRef CAS; (f) D. Uraguchi, T. Ito and T. Ooi, J. Am. Chem. Soc., 2009, 131, 3836 CrossRef CAS PubMed; (g) K. Suyama, Y. Sakai, K. Matsumoto, B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2010, 49, 797 CrossRef CAS PubMed; (h) D. Zhao and R. Wang, Chem. Soc. Rev., 2012, 41, 2095 RSC; (i) J. V. Alegre-Requena, E. Marqués-López, P. J. Sanz Miguel and R. P. Herrera, Org. Biomol. Chem., 2014, 12, 1258 RSC; (j) R. P. Herrera, Chem. Rec., 2017, 17, 1 CrossRef PubMed.
- (a) A. N. Pudovik, Dokl. Akad. Nauk SSSR, 1950, 73, 499 CAS; (b) A. N. Pudovik and I. V. Konovalova, Synthesis, 1979, 96 Search PubMed.
- V. S. Abramov, Dokl. Akad. Nauk SSSR, 1950, 73, 487 CAS.
- K. Nakanishi, S. Kotani, M. Sugiura and M. Nakajima, Tetrahedron, 2008, 64, 6415 CrossRef CAS.
- Y. Ohmaru, N. Sato, M. Mizutani, S. Kotani, M. Sugiura and M. Nakajima, Org. Biomol. Chem., 2012, 10, 4562 CAS.
- O. Dogan, M. Isci and M. Aygun, Tetrahedron: Asymmetry, 2013, 24, 562 CrossRef CAS.
- C. Laborde, M.-M. Wei, A. Van der Lee, E. Deydier, J.-C. Daran, J.-N. Volle, R. Poli, J.-L. Pirat, E. Manoury and D. Virieux, Dalton Trans., 2015, 44, 12539 RSC.
- (a) S. Spinella, Z.-H. Guan, J. Chen and X. Zhang, Synthesis, 2009, 3094 CAS; (b) Q. Dai, W. Gao, D. Liu, L. M. Kapes and X. Zhang, J. Org. Chem., 2006, 71, 3928 CrossRef CAS PubMed.
- (a) J. D. Short, S. Attenoux and D. J. Berrisford, Tetrahedron Lett., 1997, 38, 2351 CrossRef CAS; (b) S. Kotani, S. Hashimoto and M. Nakajima, Tetrahedron, 2007, 63, 3122 CrossRef CAS.
- Nakajima reported that adding SiCl4 over 2 h improved the selectivity in the Abramov type reaction by preventing the non-selective reaction promoted by SiCl4 itself (ref. 10).
- First disulfonimide catalyzed enantioselective Abramov reaction using silylphosphites with 10 to 99% ee values has been reported, see: J. Guin, Q. Wang, M. Van Gemmeren and B. List, Angew. Chem., Int. Ed., 2015, 54, 355 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10919a |
| This journal is © The Royal Society of Chemistry 2017 |