
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg(I)/persulfate-catalyzed decarboxylative coupling of α-oxocarboxylates with organotrifluoroborates in water under room temperature†
Sheng Chang *ab,
Jian Feng Wangc,
Lin Lin Donga,
Dan Wanga,
Bo Feng*a and
Yuan Tai Shia
*ab,
Jian Feng Wangc,
Lin Lin Donga,
Dan Wanga,
Bo Feng*a and
Yuan Tai Shia
aCollege of Pharmacy, Jilin Medical University, Jilin, Jilin 132013, China. E-mail: jmu_changsheng@126.com; furfolgbec84@163.com; Tel: +86 432 6456 0532
bState Key Laboratory of Medicinal Chemical Biology, NanKai University, Tianjin, 300071, China
cDepartment of Radiotherapy, China-Japan Union Hospital of Jilin University, Changcun, Jilin 130033, China
First published on 8th November 2017
Abstract
The decarboxylative coupling reaction of α-oxocarboxylates and organotrifluoroborates was carried out smoothly in the presence of catalytic AgNO3 using K2S2O8 as oxidant to generate diarylketone products in high yields. The method is novel, simple, safe and efficient. Both aryl substituted potassium α-oxocarboxylates and organotrifluoroborates proceeded smoothly in water under room temperature. The utilization of α-oxocarboxylates as acylating agent presents some elements of interest.
Introduction
Arylketone is a common functionality in many types of organic compounds, for example, pharmaceuticals and natural products.1 For organic chemists, it is also an extremely useful functional group for further manipulations. Consequently, it continues to inspire the development of methods for the construction of this structural element. Classical routes to synthesize aryl ketones mainly rely on the Friedel–Crafts acylation of aromatic compounds2 and the oxidation of the corresponding secondary alcohols.3 Transition-metal-catalyzed reactions provide many opportunities for the synthesis of aryl ketones. The coupling reaction of acylating agents with aryl halides (or aryl metallic compounds and their equivalents) has been developed under the catalysis of transition metals.4Transition metal catalyzed decarboxylative coupling reactions have received much attention for their applications in the construction of C–C and C-hetero bonds in recent years.5 Among various types of carboxylic acids (carboxylate), α-oxocarboxylates have emerged as a novel class of acyl surrogates after releasing molecular CO2.6 Theoretically, there are three types of acyl reagents from the decarboxylation of α-oxocarboxylates: acyl anions, acyl cations and acyl radicals.7 The direct decarboxylative acylation of potassium α-oxocarboxylates with aryl bromides/chlorides have been demonstrated by Gooßen's group, with the Pd(II)/Cu(I) catalytic system being particularly efficient with the assistance of P-based and N-based ligands.8a–8c Then they demonstrated a one-pot three-component decarboxylative coupling with α-oxocarboxylates, amines and aryl bromides for the synthesis of azomethines.8d In 2014, Ji et al. reported a palladium/copper-catalyzed decarboxylative coupling of aryl iodides with α-oxocarboxylates without the need of ligands.8e α-Oxocarboxylates providing acyl anions with Pd/Cu catalysis by this decarboxylation type, to afford diaryl ketones under harsh conditions (or derivatives) (Scheme 1, I). Recently, You and co-workers reported a transition-metal-free decarboxylation of α-oxocarboxylates with α-bromo-acetophenone.9a In 2017, the room-temperature coupling/decarboxylation reaction between in situ generated α-oxocarboxylates with α-bromoketones was described by their group. The transformation shows excellent regioselectivity for 1,2- and 1,3-diketones by solvent-controlled.9b Acyl cations were generated in this decarboxylation manner, with the coupling of α-oxocarboxylates with sp3 carbon center (Scheme 1, II). Acyl radicals were formed via Ag(I)/persulfate-catalysis of potassium α-oxocarboxylates by the third decarboxylation type. Lei's group described the first example for realizing isocyanide insertion by using an acyl radical via the oxidative radical decarboxylation, to construct 6-acyl phenanthridine structure (Scheme 1, III).10a Synthesis of oxazoles by Ag(I)/persulfate-catalyzed oxidative decarboxylation–cyclization of α-oxocarboxylates and isocyanides was reported by them for the next year.10b Therefore, it is still necessary to develop more Ag(I)/persulfate-catalyzed oxidative decarboxylation of potassium α-oxocarboxylates.
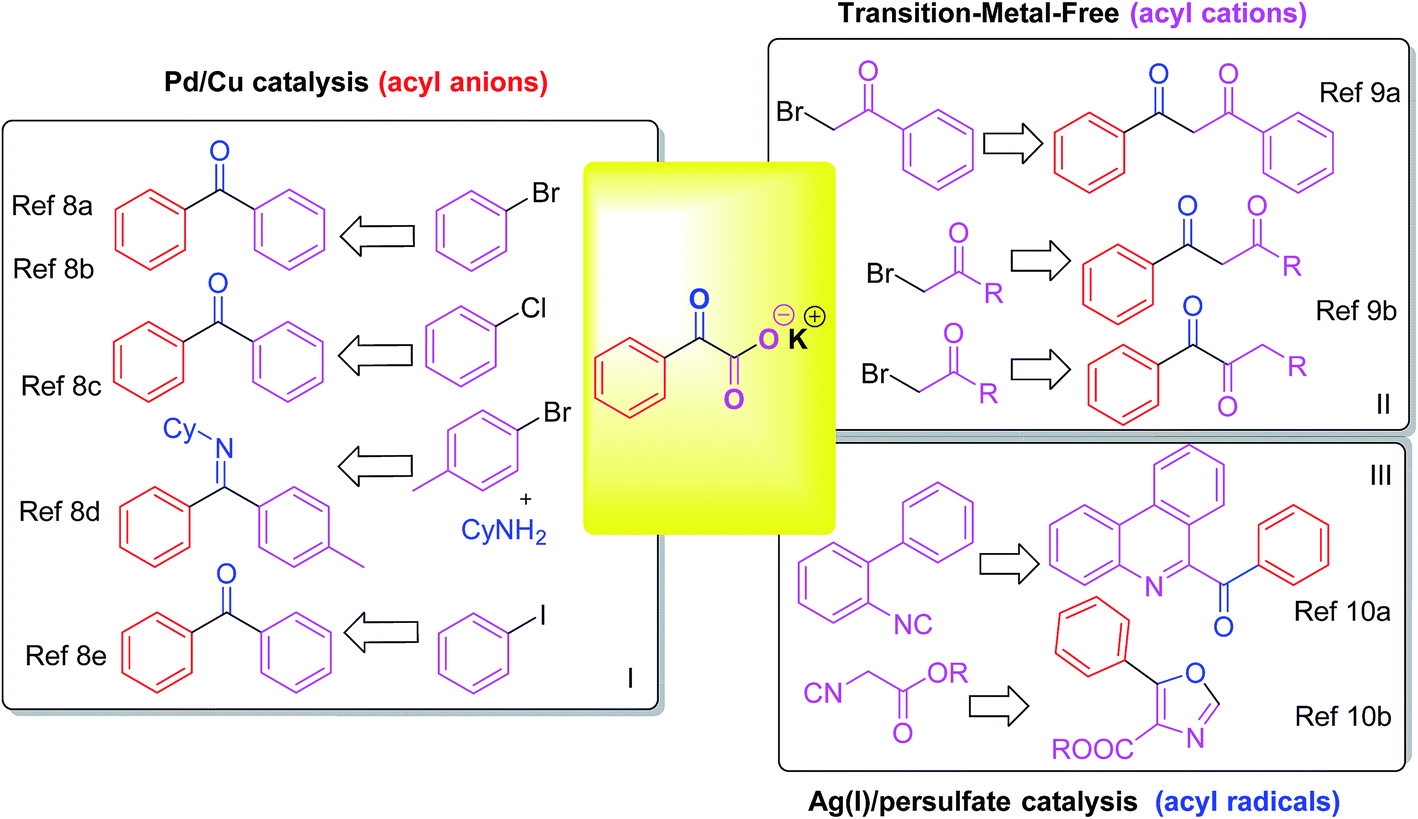 | ||
| Scheme 1 Decarboxylation coupling of α-oxocarboxylates. | ||
Interestingly, there is a novel recognition that α-oxocarboxylic acids can serve as aryl radical precursors via oxidative silver-promoted carbon–carbon bond cleavage in recent years.11 Ag(I)/persulfate catalytic systems have been shown to be effective for this transformation, including direct C–H acylation of arenes12 and oxidative radical decarboxylation–cyclization.13 In the presence of silver(I) salts, a persulfate anion disproportionates into a sulfate dianion and a sulfate radical anion, which could react with α-oxocarboxylates through an radical process, providing an acyl radical.
As continued effort on coupling of organoboronic reagents and carbonylative coupling,14 we report herein a novel method for the silver-catalyzed, decarboxylative acylation of aryltrifluoroborates to form the corresponding diarylketones. It has been successfully developed using α-oxocarboxylates as acylating reagents, as well as a catalytic amount of a silver salt and a cheap inorganic oxidant. The reaction demonstrates a broad substrate scope and excellent functional-group tolerance. This catalytic method should be valuable in the synthesis of aryl-carbonyl motifs.
Results and discussion
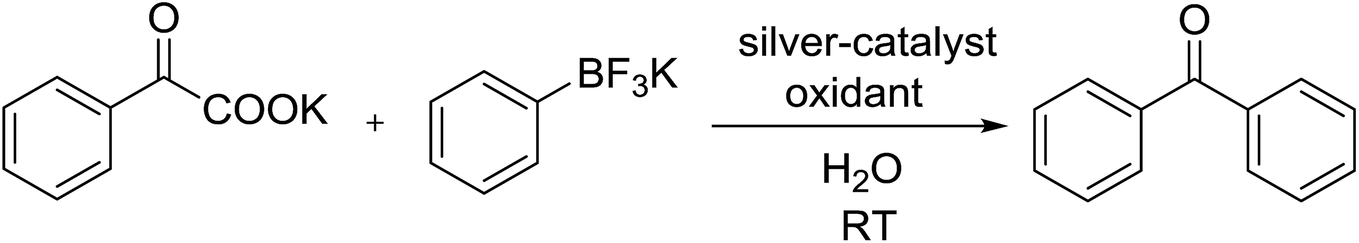
Initially, we optimized the reaction conditions using potassium oxophenylacetate and phenyltrifluoroborate as model substrate in acetone with 5 mol% AgNO3 and 1.5 equiv. K2S2O8 at room temperature for 1 h under air. Only trace amount of cross-coupling biphenyl ketone products was observed (Table 1, entry 1). Then we explored the silver-catalyzed reactions with a variety of reaction medium (Table 1), and 22% of yield were obtained when CH3CN was used as a solvent (Table 1, entry 2). No better results were obtained with polar aprotic solvents such as DMF, DMSO, or NMP(N-methyl-2-pyrrolidone) (Table 1, entries 3–5). The solvents (DCE, DCM, CHCl3 and CCl4) were tested, and DCE gave the best result (Table 1, entries 6–9). Three additional ethereal solvents [1,4-dioxane, DME and THF] were surveyed without better results (Table 1, entries 10–12). However, the reaction did not proceed in toluene, EA or t-BuOH (Table 1, entries 13–15). Considered that the infusibility of both potassium α-oxocarboxylates and aryltrifluoroborate in organic solvent, water could be much better medium candidate for this transformation. Water is an easily available, nontoxic, noninflammable, and renewable solvent. The unusual properties of water can lead to unusual reactivity that is not seen in traditional organic solvents. Consequently, the use of water as a solvent in synthetic organic chemistry and materials science has spread throughout the chemical community. The combination of water and metal catalysis has led in recent years to the development of a huge number of new and greener synthetic methodologies. It also provides opportunities for alternative reactivity and simplified product isolation compared to organic solvents. To our delight, sharply increase was observed with the application of water (Table 1, entry 16).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Yieldb (%) | Entry | Solvent | Yieldb (%) |
| a Reaction conditions: potassium oxophenylacetate (1.0 mmol), phenyltrifluoroborate (1.05 mmol), AgNO3 (0.05 mmol), K2S2O8 (1.5 mmol), solvent (2 mL), 25 °C, 1 h.b Isolated yields. | |||||
| 1 | Acetone | Trace | 9 | CCl4 | 31 |
| 2 | CH3CN | 22 | 10 | 1,4-Dioxane | 26 |
| 3 | DMF | 15 | 11 | DME | 38 |
| 4 | DMSO | 20 | 12 | THF | 41 |
| 5 | NMP | 12 | 13 | Toluene | Trace |
| 6 | DCE | 58 | 14 | EA | Trace |
| 7 | DCM | 46 | 15 | t-BuOH | Trace |
| 8 | CHCl3 | 53 | 16 | H2O | 86 |
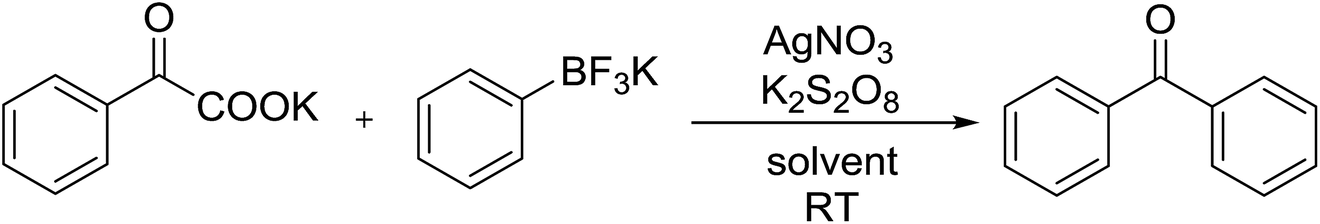
We began to screen many oxidants for the assistance of the regeneration of Ag(I). Many oxidants have been tested and showed different influence on the reaction (Table 2, entries 1–10). The cross-coupling could not proceed with Cu(OAc)2, oxygen and DDQ (Table 2, entries 2–4). PhI(OAc)2 and BPO (dibenzoyl peroxide) can enhance the efficiency to about 26–34% (Table 2, entries 5–6). In addition, inorganic peroxide are efficient to the cross-coupling. Oxone and (NH4)2S2O8 can afford diphenylketone with the yield of 70% and 74% (Table 2, entries 7–8), then Na2S2O8 performed better and K2S2O8 performed best (86%) (Table 2, entries 9–10). Then the effect of silver salts was investigated, and many silver(I) compounds such as AgOAc, Ag2CO3, and AgOTf were also applicable for this reaction, albeit in the slightly decreased yield (Table 2, entries 11–13). AgBF4, AgOTf and Ag2O were inferior to AgOTf (Table 2, entries 14–16). Meanwhile, the use of Ag2SO4, AgSbF6 and AgF as catalysts gave no desired products (Table 2, entries 17–19). And 86% of yield were obtained when AgNO3 was used as a catalyst (Table 2, entry 20). Therefore, AgNO3/K2S2O8 in water at room temperature under air for 1 hour was chosen as the optimal conditions for the synthesis of diarylketone from α-oxocarboxylates and aryltrifluoroborate.
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant | Yieldb (%) | Entry | Silver salts | Yieldc (%) |
| a Reaction conditions: potassium oxophenylacetate (1.0 mmol), phenyltrifluoroborate (1.05 mmol), silver salts (0.05 mmol), oxidant (1.5 mmol), H2O (2 mL), 25 °C, 1 h.b Isolated yields using AgNO3 as the silver salt.c Isolated yields using K2S2O8 as the oxidant. | |||||
| 1 | — | — | 11 | AgOAc | 84 |
| 2 | Cu(OAc)2 | Trace | 12 | Ag2CO3 | 81 |
| 3 | O2 (1 atm) | Trace | 13 | AgOTf | 79 |
| 4 | DDQ | Trace | 14 | AgBF4 | 43 |
| 5 | PhI(OAc)2 | 26 | 15 | AgOTf | 57 |
| 6 | BPO | 34 | 16 | Ag2O | 59 |
| 7 | Oxone | 77 | 17 | Ag2SO4 | — |
| 8 | (NH4)2S2O8 | 74 | 18 | AgSbF6 | — |
| 9 | Na2S2O8 | 82 | 19 | AgF | — |
| 10 | K2S2O8 | 86 | 20 | AgNO3 | 86 |
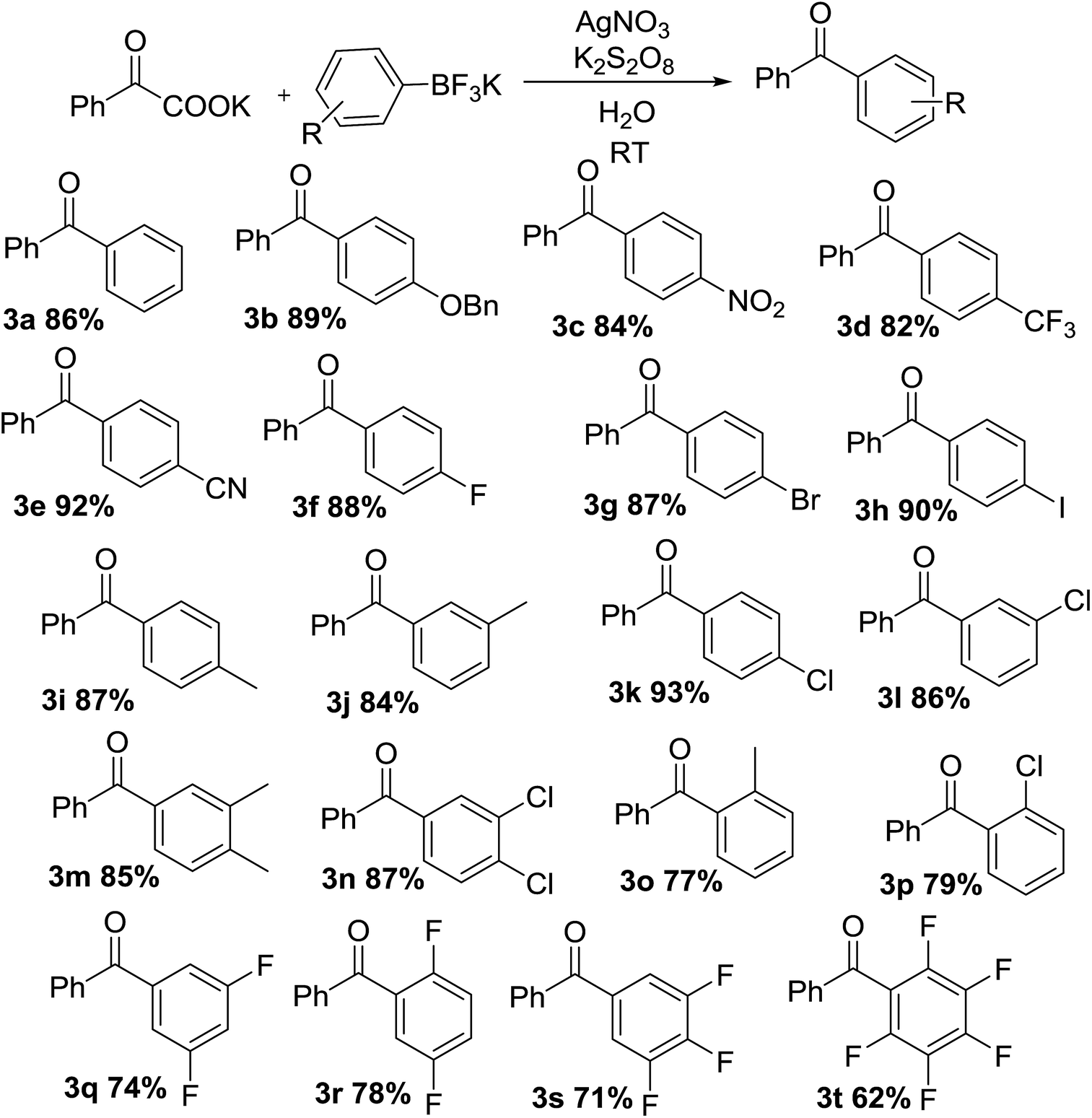
With optimal conditions in hand, the scope of this decarboxylative coupling reaction method was evaluated next (Tables 3 and 4). As shown, the reaction exhibits a broad substrate scope, with respect to both of the α-oxocarboxylates and the organotrifluoroborates coupling partners. Importantly, the reaction is compatible with electron-neutral (3a), electron-rich (3b), and electron-deficient organotrifluoroborate precursors (3c–f), furnishing the products in good yields. The mild reaction conditions are compatible with a range of sensitive functional groups, such as nitro-(3c), trifluoromethyl-(3d), cyan-(3e), and even halides (fluoro-, bromo-, iodo-) (3f–h), affording diaryl ketone products with a bouquet of functional handles poised for further functionalization. Although, the organotrifluoroborate component bearing functional groups at different positions, such as methyl and chloro at 3- or 4-position of the phenyl ring were obtained in good yields (3i–l). Moreover, meta, para-di-methyl phenyltrifluoroborate and meta, para-di-chloro phenyltrifluoroborate (3m and 3n) underwent the desired cross-coupling in high yields. Substrates having ortho methyl (3o) or ortho-chloro (3p) substituents at phenyl moiety gave slightly lower reactivity, presumably due to steric reason. Finally, we were pleased to find that the reaction conditions could be readily extended to the arylation of a range of polyfluorinated substrates (3q–t). Importantly, polyfluorinated biaryl building blocks are of great interest in materials chemistry, because of favorable physicochemical properties.
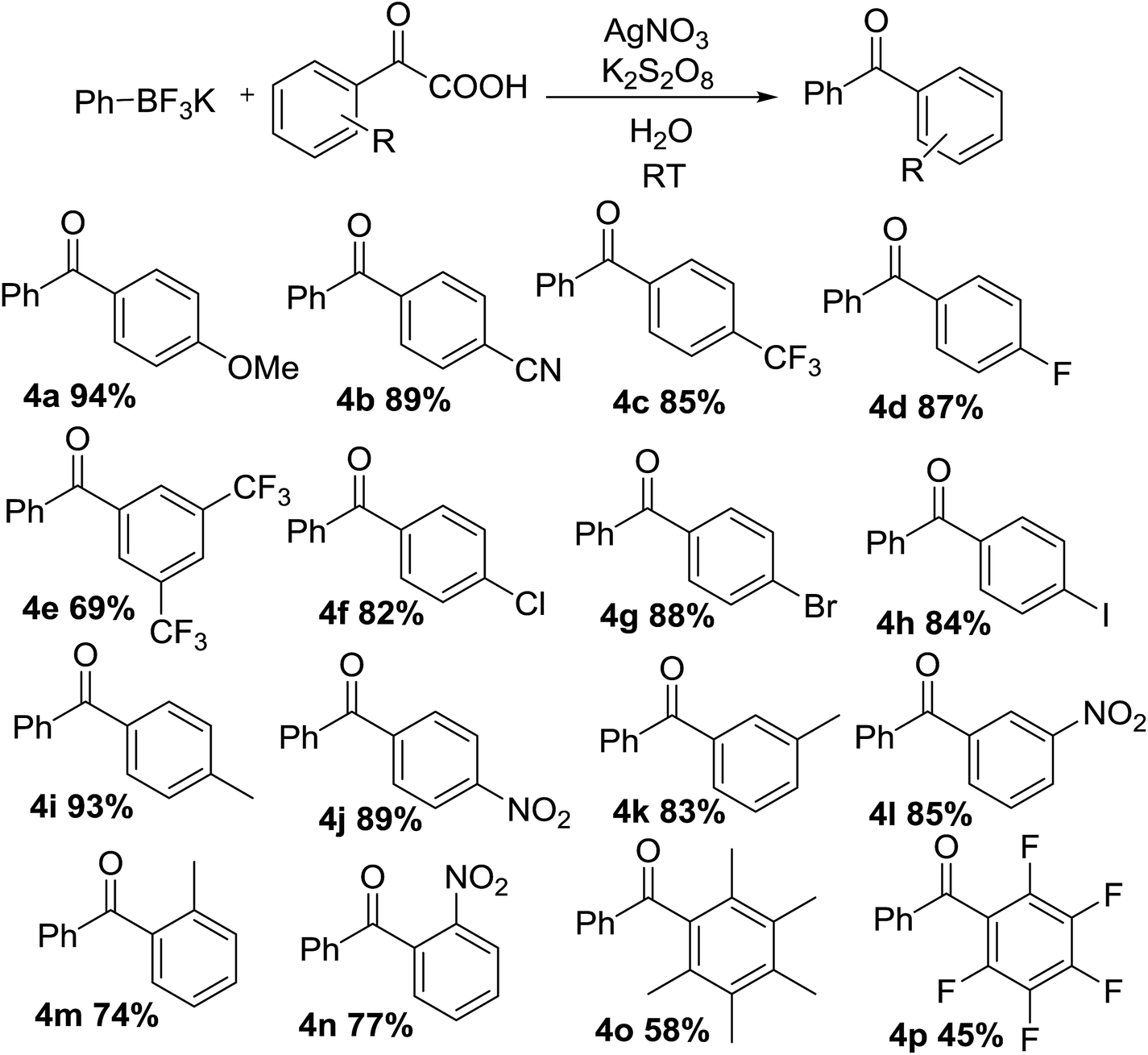
The scope, with respect to the α-oxocarboxylates component, is also very broad (Table 4), including electron-rich (4a), and electron-deficient (4b–d). The reaction was shown to tolerate a wide variety of functionality on the aryl ring including methoxy-, cyan-, fluoro- and trifluoromethyl- (4a–d). Yields were reduced when using highly electron deficient arenes with substituents such as two CF3 (4e). The reaction could accommodate substrates containing a chloro, bromo or iodo substituent, which can later be applied to various cross-coupling reactions with metal catalysts (4f–h). Good yield was achieved using para-methyl and para-nitro substituents, to afford corresponding diarylmethanone (4i–j). Meta-substituted examples such as 3-methyl and 3-nitro (4k–l) also gave desired products in excellent yield. The ortho-substituted example (4m–n) were also tolerated however in reduced yield for sterically hindered site-selectivity. Furthermore, the formation of sterically demanding per-methylated and per-fluorinated product was smoothly in this process (4o–4p). The high selectivity of Ag(I)/persulfate-catalyzed system across a range of sterically and electronically diverse α-oxocarboxylates and organotrifluoroborate precursors is a particularly noteworthy feature of this reaction manifold.
To further explore the potential of this decarboxylative coupling method, several (E)-styryl potassium trifluoroborate derivatives were examined (Scheme 2). (E)-styryl potassium trifluoroborate underwent decarboxylative coupling with potassium 4-methoxy-oxophenylacetate in moderate yield. Decarboxylative coupling of (E)-styryl potassium trifluoroborate with potassium 4-chloro-oxophenylacetate also provided chalcone in good to excellent yields. It is worth noting that when the hydroxyl group was introduced to ortho-position of potassium oxophenylacetate, the decarboxylative coupling with vinyl potassium trifluoroborate gave acceptable conversion to afford chromone, showing the robustness of our protocol and demonstrating potential for practical applications. This synthetic method could further be extended to a large-scale decarboxylative acylation. The desired products (3a, 3i, 3k, 4f, 4i) were obtained with slightly decreased yields on 10 mmol scale (78% for 3a, 72% for 3i, 86% for 3k, 77% for 4f, and 81% for 4i).
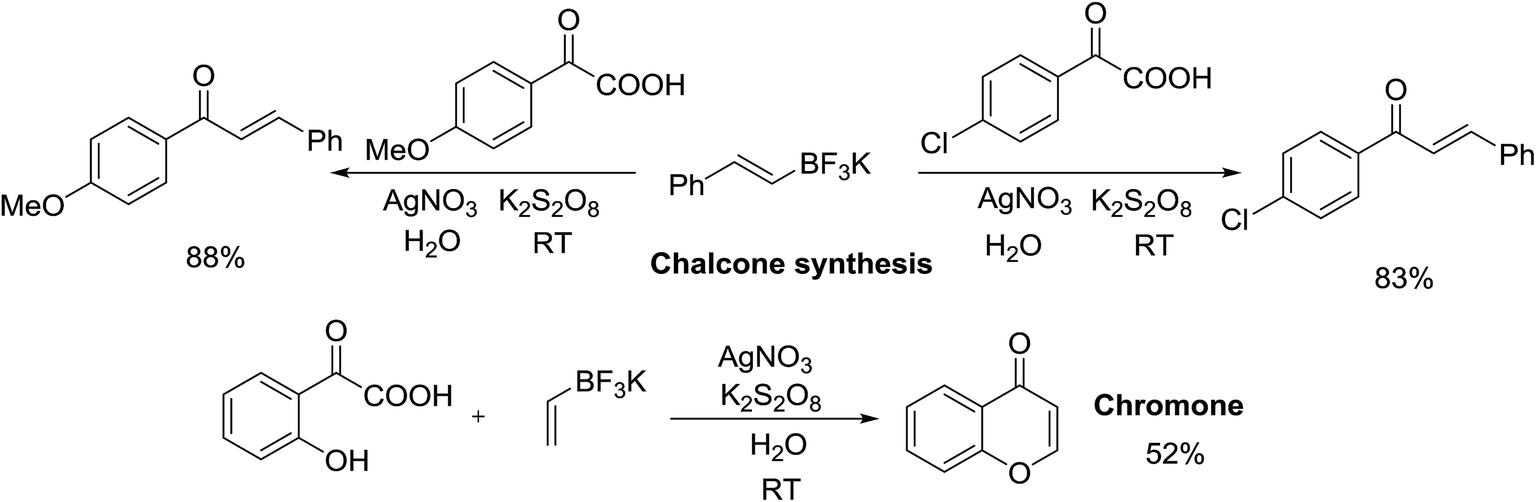 | ||
| Scheme 2 Applications of Ag(I)/persulfate-catalyzed decarboxylative coupling. | ||
With regard to our results and literature reports,10 we propose the following possible reaction mechanism (Scheme 3). Monovalent silver catalyst was oxidized by K2S2O8 to persulfate anion radical and divalent silver ion (S3-1†) (the persulfate anion radical may also oxidize Ag(I) to Ag(II) (S3-2†)). Then the Ag(II) generated in situ reacted with the α-oxocarboxylates formed acyl radical complexes and CO2 (S3-3†). Cross-coupling product obtained after the attack of acyl radical to aryltrifluoroborate (S3-4†), which could be terminated by addition of radical scavengers such as hydroquinone or 2,6-di-tert-butylphenol, suggesting that the cross-coupling reactions proceed via a radical mechanism. Potassium trifluoroborate radical was finally quenched by oxidation of persulfate anion (S3-5†). Finally, 1.5 equiv. of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) was added to the reaction, and the acyl radical was trapped successfully by TEMPO, affording the corresponding adduct in 67% yields (Scheme 3, S3-6†).
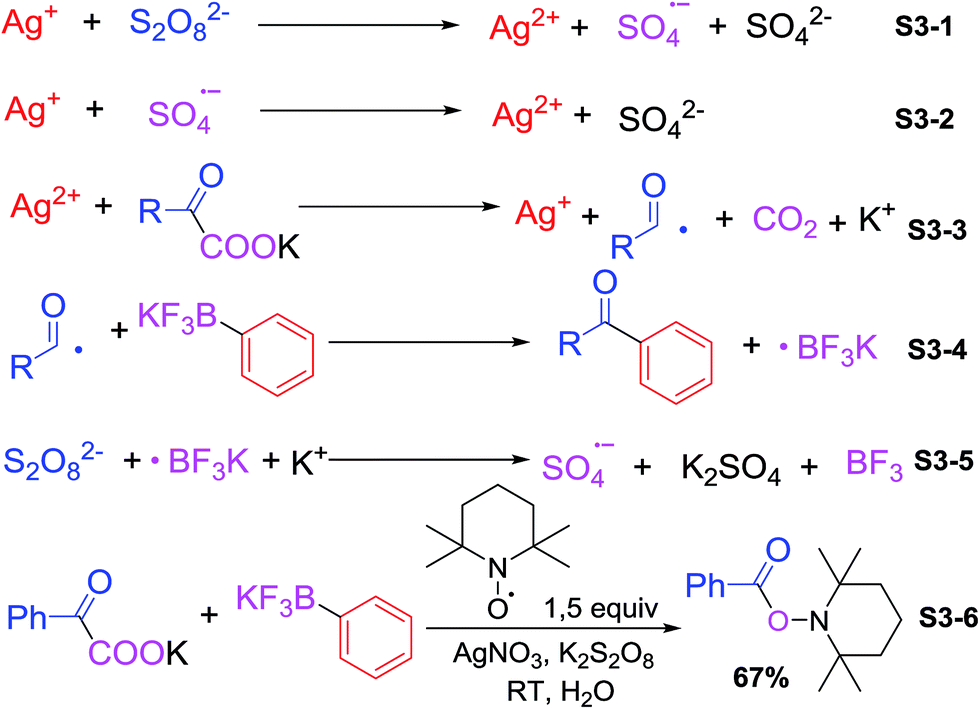 | ||
| Scheme 3 Possible mechanism. | ||
Conclusions
In conclusion, we have demonstrated a general method for highly selective decarboxylative coupling of α-oxocarboxylates with aryltrifluoroborates enabled by versatile Ag(I)/persulfate-catalyzed system. The development of this coupling further highlights the beneficial use of inexpensive Ag(I) catalysts for decarboxylation, while exploiting K2S2O8 promoter. We anticipate that this strategy will find widespread use as a practical alternative to the classic ketone construction in organic synthesis. Most notably, this new protocol features the synthesis of chalcone and chromone derivatives. Further mechanistic studies and extension to a variety of coupling partners are currently underway in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the State Key Laboratory of Medicinal Chemical Biology (2017001), National innovative undertaking plan of college students (201713743012), Young talent training plan Of Jilin Science and Technology Bureau (20156429), and NSFC (81773692).References
- (a) H. Surburg and J. Panten, Common Fragrance and Flavor Materials, Wiley, Weinheim, Germany, 2006 Search PubMed; (b) Y. Deng, Y.-W. Chin, H. Chai, W. J. Keller and A. D. Kinghorn, J. Nat. Prod., 2007, 70, 2049 CrossRef CAS PubMed; (c) K. R. Romins, G. A. Freeman, L. T. Schaller, J. R. Cowan, S. S. Gonzales, J. H. Tidwell, C. W. Andrews, D. K. Stammers, R. J. Hazen, R. G. Ferris, S. A. Short, J. H. Chan and L. R. Boone, J. Med. Chem., 2006, 49, 727 CrossRef PubMed; (d) B. M. O'Keefe, N. Simmons and S. F. Martin, Org. Lett., 2008, 10, 5301 CrossRef PubMed; (e) S. Rahimipour, C. Palivan, F. Barbosa, I. Bilkis, Y. Koch, L. Weiner, M. Fridkin, Y. Mazur and G. Gescheidt, J. Am. Chem. Soc., 2003, 125, 1376 CrossRef CAS PubMed; (f) D. Ma, Y. Jiang, F. Chen, L.-K. Gong, K. Ding, Y. Xu, R. Wang, A. Ge, J. Ren, J. Li, J. Li and Q. Ye, J. Med. Chem., 2006, 49, 456 CrossRef CAS PubMed; (g) Y. Nishizuka, Nature, 1988, 334, 661 CrossRef CAS PubMed; (h) M. R. Hansen and L. H. Hurley, Acc. Chem. Res., 1996, 29, 249 CrossRef CAS.
- (a) G. Sartori and R. Maggi, Chem. Rev., 2006, 106, 1077 CrossRef CAS PubMed; (b) G. A. Olah, Friedel–Crafts Chemistry., Wiley, New York, 1973 Search PubMed.
- (a) J. E. Bäckvall, Modern Oxidation Methods., Wiley-VCH, Weinheim, 2010 Search PubMed; (b) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed.
- For reviews see: (a) C. Pan, X. Jia and J. Cheng, Synthesis, 2012, 44, 677 CrossRef CAS; (b) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC; (c) J. Ruan and J. Xiao, Acc. Chem. Res., 2011, 44, 614 CrossRef CAS PubMed; (d) L. Kollár, Modern Carbonylation Methods., Wiley, Weinheim, 2008 Search PubMed; (e) A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed; (f) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC.
- (a) P. Hu, M. Zhang, X. Jie and W. Su, Angew. Chem., Int. Ed., 2012, 51, 227 CrossRef CAS PubMed; (b) P. Hu, Y. Shang and W. Su, Angew. Chem., Int. Ed., 2012, 51, 5945 CrossRef CAS PubMed; (c) J. Cornella and I. Larrosa, Synthesis, 2012, 653 CAS; (d) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed; (e) O. Baudoin, Angew. Chem., Int. Ed., 2007, 46, 1373 CrossRef CAS PubMed; (f) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (g) T. Satoh and M. Miura, Synthesis, 2010, 3395 CrossRef CAS; (h) L. J. Goosen, N. Rodríguez and K. Goosen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed; (i) D.-X. Liu, F.-L. Li, H.-X. Li, J. Gao and J.-P. Lang, Tetrahedron, 2014, 70, 2416 CrossRef CAS; (j) W.-J. Gong, D.-X. Liu, F.-L. Li, J. Gao, H.-X. Li and J.-P. Lang, Tetrahedron, 2015, 71, 1269 CrossRef CAS; (k) D.-X. Liu, F.-L. Li, H.-X. Li, W.-J. Gong, J. Gao and J.-P. Lang, Eur. J. Org. Chem., 2014, 4817 CrossRef CAS; (l) R.-T. He, J.-F. Wang, H.-F. Wang, Z.-G. Ren and J.-P. Lang, Dalton Trans., 2014, 43, 9786 RSC.
- (a) Q. Jiang, J. Jia, B. Xu, A. Zhao and C.-C. Guo, J. Org. Chem., 2015, 80, 3586 CrossRef CAS PubMed; (b) N. Zhang, D. Yang, W. Wei, L. Yuan, F. Nie, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 3258 CrossRef CAS PubMed; (c) W.-P. Mai, G.-C. Sun, J.-T. Wang, G. Song, P. Mao, L.-R. Yang, J.-W. Yuan, Y.-M. Xiao and L.-B. Qu, J. Org. Chem., 2014, 79, 8094 CrossRef CAS PubMed; (d) K. Yan, D. Yang, W. Wei, F. Wang, Y. Shuai, Q. Li and H. Wang, J. Org. Chem., 2015, 80, 1550 CrossRef CAS PubMed.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed.
- (a) L. J. Gooßen, F. Rudolphi, C. Oppel and N. Rodríguez, Angew. Chem., Int. Ed., 2008, 47, 3043 CrossRef PubMed; (b) L. J. Gooßen, B. Zimmermann, C. Linder, N. Rodríguez, P. P. Lange and J. Hartung, Adv. Synth. Catal., 2009, 351, 2667 CrossRef; (c) L. J. Gooßen, B. Zimmermann and T. Knauber, Angew. Chem., Int. Ed., 2008, 47, 7103 CrossRef PubMed; (d) F. Rudolphi, B. Song and L. J. Gooßen, Adv. Synth. Catal., 2011, 353, 337 CrossRef CAS; (e) Y. Ji, X. Yang and W. Mao, Appl. Organomet. Chem., 2014, 28, 678 CrossRef CAS.
- (a) Z. He, X. Qi, S. Li, Y. Zhao, G. Gao, Y. Lan, Y. Wu, J. Lan and J. You, Angew. Chem., Int. Ed., 2015, 54, 855 CrossRef CAS PubMed; (b) Z. He, X. Qi, Z. She, Y. Zhao, S. Li, J. Tang, G. Gao, Y. Lan and J. You, J. Org. Chem., 2017, 82, 1403 CrossRef CAS PubMed.
- (a) J. Liu, C. Fan, H. Yin, C. Qin, G. Zhang, X. Zhang, H. Yi and A. Lei, Chem. Commun., 2014, 50, 2145 RSC; (b) Y. Ma, Z. Yan, C. Bian, K. Li, X. Zhang, M. Wang, X. Gao, H. Zhang and A. Lei, Chem. Commun., 2015, 51, 10524 RSC.
- (a) K. Cheng, B. Zhao and C. Qi, RSC Adv., 2014, 4, 48698 RSC; (b) N. Zhang, D. Yang, W. Wei, L. Yuan, F. Nie, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 3258 CrossRef CAS PubMed; (c) L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380 RSC; (d) S. Zhang, L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2013, 11, 4308 RSC; (e) H. Wang, L.-N. Guo and X.-H. Duan, Org. Biomol. Chem., 2013, 11, 4573 RSC; (f) H. Wang, H. Yang, Y. Li and X.-H. Duan, RSC Adv., 2014, 4, 8720 RSC; (g) H. Wang, L.-N. Guo and X.-H. Duan, Org. Lett., 2012, 14, 4358 CrossRef CAS PubMed.
- (a) W. P. Mai, B. Sun, L. Q. You, L. R. Yang, P. Mao, J. W. Yuan, Y. M. Xiao and L. B. Qu, Org. Biomol. Chem., 2015, 13, 2750 RSC; (b) R. Suresh, R. S. Kumaran, V. Senthilkumar and S. Muthusubramanian, RSC Adv., 2014, 4, 31685 RSC; (c) H. Wang, S. L. Zhou, L. N. Guo and X. H. Duan, Tetrahedron, 2015, 71, 630 CrossRef CAS.
- (a) Z. Y. Lin, Y. L. Chen, C. S. Lee and C.-P. Chuang, Eur. J. Org. Chem., 2010, 3876 CrossRef CAS; (b) T. Liu, Q. Ding, Q. Zong and G. Qiu, Org. Chem. Front., 2015, 2, 670 RSC; (c) H. Wang, L. N. Guo and X. H. Duan, Adv. Synth. Catal., 2013, 355, 2222 CrossRef CAS; (d) H. Yang, L. N. Guo and X. H. Duan, RSC Adv., 2014, 4, 52986 RSC; (e) W. P. Mai, G. C. Sun, J. T. Wang, G. Song, P. Mao, L. R. Yang, J. W. Yuan, Y. M. Xiao and L. B. Qu, J. Org. Chem., 2014, 79, 8094 CrossRef CAS PubMed.
- (a) C. Y. Hao, D. Wang, Y. W. Li, L. L. Dong, Y. Jin, X. R. Zhang, H. Y. Zhu and S. Chang, RSC Adv., 2016, 6, 86502 RSC; (b) S. Chang, Y. Jin, X. R. Zhang and Y. B. Sun, Tetrahedron Lett., 2016, 57, 2017 CrossRef CAS; (c) S. Chang, Y. B. Sun, X. R. Zhang, L. L. Dong, H. Y. Zhu, H. W Lai and D. Wang, Appl. Organomet. Chem., 2017 DOI:10.1002/aoc.3970.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10924e |
| This journal is © The Royal Society of Chemistry 2017 |