
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystallization of ultrathin poly(3-hydroxybutyrate) films in blends with small amounts of poly(L-lactic acid): correlation between film thickness and molecular weight of poly(L-lactic acid)†
Khasanaha,
Isao Takahashi *b,
Kummetha Raghunatha Reddy‡
a and
Yukihiro Ozaki*a
*b,
Kummetha Raghunatha Reddy‡
a and
Yukihiro Ozaki*a
aDepartment of Chemistry, School of Science and Technology, Kwansei Gakuin University, Gakuen 2-1, Sanda, Hyogo 669-1337, Japan. E-mail: ozaki@kwansei.ac.jp
bDepartment of Physics, School of Science and Technology, Kwansei Gakuin University, Gakuen 2-1, Sanda, Hyogo 669-1337, Japan. E-mail: suikyo@kwansei.ac.jp
First published on 14th November 2017
Abstract
The crystallization behavior of poly(3-hydroxybutyrate) (PHB) ultrathin films in blends with small amounts of poly(L-lactic acids) (PLLAs) was investigated using grazing incidence X-ray diffraction (GIXD) and infrared-reflection absorption spectroscopy (IRRAS). Ultrathin films of PHB/PLLA with the same blend ratio of 80/20 (w/w) and two different film thicknesses (30 and 13 nm) were prepared using PHB (Mw = 650![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and PLLA with Mw's ranging from 300000 to 710 g mol−1 to explore the effects of molecular weight on the crystallization of PHB in different confined environments. In the 30 nm thick films, the intensity of PHB crystalline Bragg reflections was strikingly affected by the blended PLLA molecules. Middling molecular weight PLLAs (Mw = 23000–13100 g mol−1) significantly inhibited the crystallization of PHB compared with higher molecular weight PLLAs (Mw ≥ 50000 g mol−1). In the 13 nm thick films, thickness confinement was revealed to play an important role in crystallization inhibition as both higher and middling molecular weight PLLAs inhibited the crystallization of PHB. Furthermore, IRRAS showed that, for the 30 nm thick films, the addition of a small amount of PLLA (Mw ≥ 13100 g mol−1) only altered the crystalline structure of PHB in the highly ordered state. In contrast, such PLLAs greatly affected the PHB crystals in both intermediate and highly ordered states in the 13 nm thick films. Unlike the behavior of PLLA in bulk PHB, the lower molecular weight PLLAs (Mw ≤ 3600 g mol−1) showed limited effects on the crystallinity and crystalline structure of PHB in both thicknesses of thin films. Several factors, such as phase separation caused by free surface and interface effects, entanglement of PLLA chains, and molecular size of PLLA, are very likely to be responsible for the particular crystallization behavior of PHB in the studied blends.
000 g mol−1) and PLLA with Mw's ranging from 300000 to 710 g mol−1 to explore the effects of molecular weight on the crystallization of PHB in different confined environments. In the 30 nm thick films, the intensity of PHB crystalline Bragg reflections was strikingly affected by the blended PLLA molecules. Middling molecular weight PLLAs (Mw = 23000–13100 g mol−1) significantly inhibited the crystallization of PHB compared with higher molecular weight PLLAs (Mw ≥ 50000 g mol−1). In the 13 nm thick films, thickness confinement was revealed to play an important role in crystallization inhibition as both higher and middling molecular weight PLLAs inhibited the crystallization of PHB. Furthermore, IRRAS showed that, for the 30 nm thick films, the addition of a small amount of PLLA (Mw ≥ 13100 g mol−1) only altered the crystalline structure of PHB in the highly ordered state. In contrast, such PLLAs greatly affected the PHB crystals in both intermediate and highly ordered states in the 13 nm thick films. Unlike the behavior of PLLA in bulk PHB, the lower molecular weight PLLAs (Mw ≤ 3600 g mol−1) showed limited effects on the crystallinity and crystalline structure of PHB in both thicknesses of thin films. Several factors, such as phase separation caused by free surface and interface effects, entanglement of PLLA chains, and molecular size of PLLA, are very likely to be responsible for the particular crystallization behavior of PHB in the studied blends.
1. Introduction
Polymer blending is an effective way to create new and desired physical properties.1–3 Poly(3-hydroxybutyrate) (PHB), a thermoplastic polyester produced by bacteria, is a promising alternative material for substituting conventional plastics derived from petroleum. The physical and mechanical properties of PHB are similar to those of commercial isotactic polypropylene; moreover, it exhibits many desirable features, including biodegradability and biocompatibility.4–6 PHB has often been blended with other polymers to reduce its high crystallinity and to modify unfavorable properties, such as brittleness, low thermal stability, and poor processability.7–12 Poly(L-lactic acid) (PLLA) is also a biodegradable thermoplastic polyester; it can be produced from renewable sources and is already used in many commercial applications.13–17 The blending of biodegradable polymers should be very beneficial for environmental sustainability and is expected to be used in many applications. Thus, PHB/PLLA blends have become a popular research topic in polymer blend studies, in which the structure, morphology, miscibility, and crystallization behaviors have been extensively investigated for further applications.11,18–29PHB shows biphasic separation in blends with high molecular weight PLLA (Mw > 18000 g mol−1), but it is miscible over the whole composition range of low molecular weight PLLA (Mw < 18000 g mol−1) in the melt.26–28 Similarly, high molecular weight PLLA (Mw = 680000 g mol−1) was reported to be miscible in the melt with low molecular weight atactic PHB (Mw = 9400 g mol−1).24,25 Moreover, the miscibility of PHB and PLLA in their blends also greatly depends on the concentrations and crystallization dynamics of PLLA and PHB.11–23 Most of the PHB/PLLA blend studies have been conducted using bulk samples, whereas investigations of PHB/PLLA ultrathin blend films are still limited.11,18–29,46
In general, the properties of polymers in thin and ultrathin films are considerably different from those in the bulk, such as glass transition temperature,30–32 morphology,33,34 mobility,35,36 and crystallization,37–40 due to confinement effects as well as surface and interface effects. The preferred orientation of molecular chains is another phenomenon peculiar to crystalline polymer thin films, which often brings about useful anisotropic features.41,44 Therefore, investigations of thin and ultrathin polymer films can be very important for exploring specific crystallization phenomena at the surface and interface region.
Previously, we found that PHB ultrathin films (thickness ∼52 nm) formed two kinds of crystalline structures, that is, a highly ordered structure and a less ordered structure (the latter has been recognized as the intermediate state), which yield infrared (IR) bands at 1723 and 1731 cm−1, respectively.45 The band at 1731 cm−1, due to the less ordered structure, appears during the melt-crystallization process, which is usually hard to detect in bulk PHB. Similarly, the corresponding band shows up in miscible PHB/PLLA (Mw of PLLA = 13100 g mol−1) ultrathin films with similar thicknesses.46 In addition, the crystallization of PHB is extremely reduced with the addition of ≥22 wt% PLLA. This result indicates that the addition of another component, even by a relatively small amount, can be quite effective at controlling the crystallization of polymer thin films. Since the miscibility of PHB and PLLA depends on the molecular weight, varying the molecular weight of a small amount of PLLA in PHB/PLLA blend ultrathin films should reveal a crystallization process of PHB that may be different from the bulk.
In the present study, we investigated the molecular weight dependence of a small amount of PLLA on the crystallization of ultrathin PHB films and its correlation with film thickness, using a wide range of PLLAs with different molecular weights (300000–710 g mol−1). Based on a previous study,46 the present study deals with only a small amount of PLLA (20 wt%) with the aim of studying the effect of PLLA molecular weight on the crystallization of PHB in ultrathin films. Ultrathin films generally have thickness up to 100 nm; however, films where the thickness is close to the polymer coil size (∼10 nm) show more specific confinement effects than conventional ultrathin films.62 For distinguishing that geometrical confinement effect and molecular weight dependence, we prepared blend films with two different thicknesses: 30 nm and 13 nm. Two surface-sensitive techniques were employed to observe the crystallization behavior of PHB in the ultrathin blend films: grazing incidence X-ray diffraction (GIXD) and infrared-reflection absorption spectroscopy (IRRAS). We found that the inhibition of PHB crystallization in the blends strongly depended on the molecular weight of the PLLA and the film thickness. Moreover, several important factors that influenced the ability of a small amount of PLLAs to inhibit the crystallization of PHB are also discussed. Controlling the crystallinity of PHB films using a small addition of another polymer, as presented in this study, would make a great advance for thin film technology and will greatly expand the range of PHB applications.
2. Experimental
2.1. Materials and sample preparation
Bacterially synthesized PHB with a weight-averaged molecular weight (Mw) of 650000 g mol−1, purchased from Aldrich Co., Ltd., was dissolved in hot chloroform, precipitated in methanol, and vacuum-dried at 60 °C for several days to remove impurities. Eight kinds of PLLAs with different molecular weights were obtained from Polysciences Inc. (Mw = 300000; 100000; and 50000 g mol−1) and Polymer Source Inc. (Mw = 23000; 13100; 6900; 3600; and 710 g mol−1). Blends of PHB/PLLAs were prepared by dissolving the prescribed amount of PHB and PLLA in hot chloroform.
Ultrathin films were deposited on flat Au and Si (100) substrates for IRRAS and GIXD measurements, respectively, by spin coating for 45 s. The films were dried under low vacuum at 60 °C for 12 h. To advance the crystallization, annealing was carried out by keeping the films in a vacuum oven at 120 °C which is close to the melting point of PHB ultrathin films45 for 5 h. The temperature was then slowly decreased to room temperature before the measurement of GIXD. Two types of film thicknesses, 30 ± 3 nm and 13 ± 2 nm, were formed by changing the concentration of solutions and rotating speeds.
2.2. Infrared spectroscopy
All of the IRRAS spectra were measured using a Thermo Nicolet 6700 FTIR spectrometer equipped with an MCT detector at 2 cm−1 resolution over 256 scans to ensure a high signal-to-noise ratio. A reflection attachment (Spectra-Tech. FT80 RAS) with an incident angle of 80° was attached to the spectrometer together with a rotatable wire-grid KRS5-substrate polarizer (ST Japan).2.3. X-ray diffraction
GIXD profiles were collected using a Rigaku Ultima IV X-ray diffractometer (Rigaku Co., Japan) where CuKα (λ = 1.542 Å) radiation was generated at 40 kV and 40 mA. The system was equipped with a scintillation detector and cross beam optics for automatic alignment coupled with a thin film attachment. In order to investigate the entire crystalline area in the films, the angle of incidence was set to 0.19° (corresponding to 1.14θc) for the incident X-ray to fully penetrate into the films. GIXD profiles were collected at room temperature with a scanning speed of 0.3° min−1 and interval of 0.1°. Film thicknesses were determined from the X-ray reflectivity profiles after analysis with in-house developed nonlinear curve-fitting software.3. Results and discussion
3.1. Crystallinity and crystallization behavior of PHB as studied by GIXD
Fig. 1(a) shows out-of-plane GIXD profiles collected at room temperature from the 30 nm thick films of neat PHB and PHB/PLLA blends (80/20), in which PHB was blended with eight molecular weights of PLLAs. An obvious Bragg reflection, centered at 2θ = 13.6°, originated from the crystallites of PHB in the films and was indexed to be the (020) reflection.42–47 Two other weak reflections also appear at around 2θ = 16.6° and 20.3° in Fig. 1(a)(1)–(5), which are the (200/110) and (203) reflections of crystalline PLLA, respectively.46 However, several diffraction profiles in Fig. 1(a)(6)–(8) did not exhibit any reflections from PLLA, indicating that the PLLAs did not crystallize in those films. The critical molecular weight for crystallizable PLLA may be exists. Moreover, the results displayed in Fig. 1(a) reveal that PHB and several kinds of PLLA (Mw ≥ 13100 g mol−1) crystallized separately in those blends.
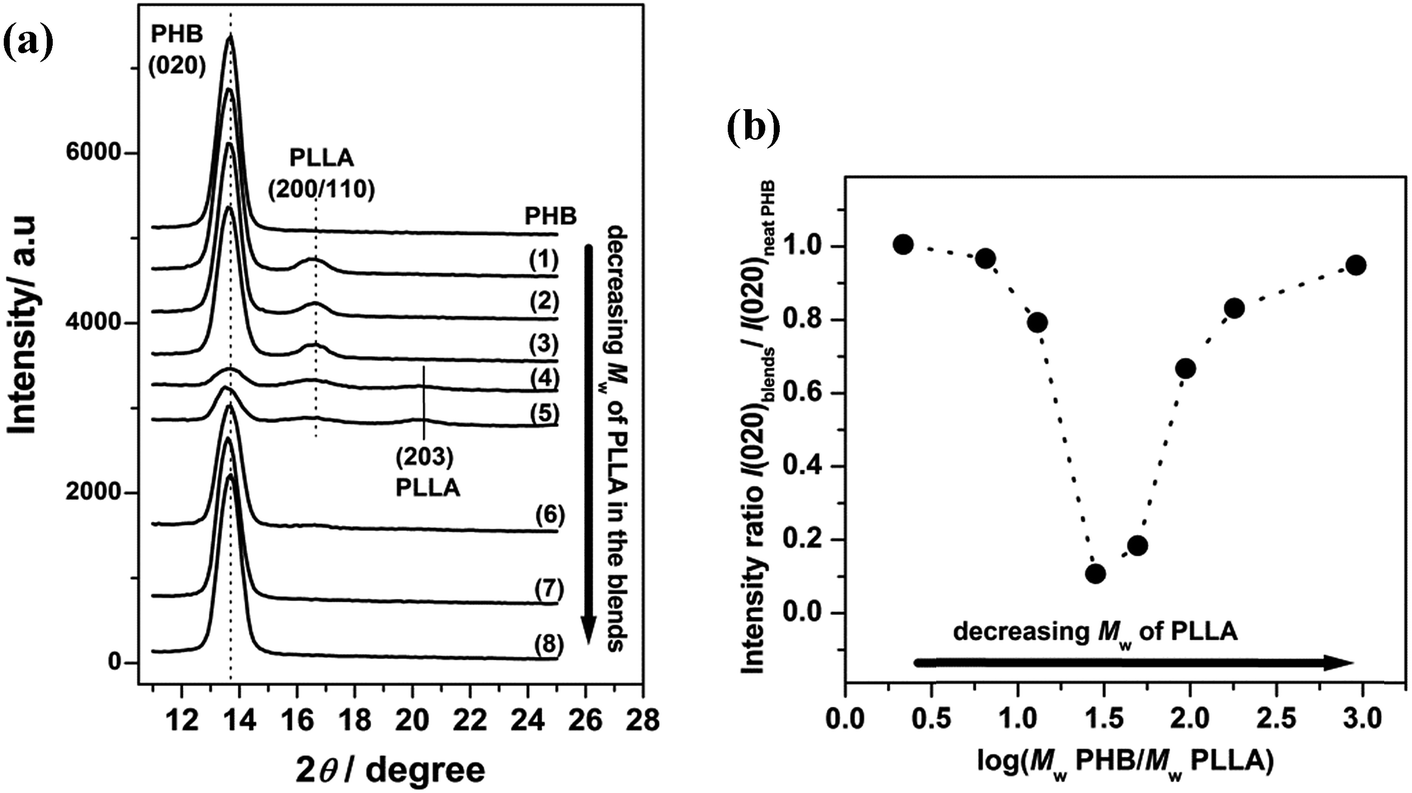 | ||
| Fig. 1 (a) Out-of-plane GIXD profiles of 30 nm thick films of neat PHB and PHB/PLLA blends (80/20) with different Mw of PLLA: (1) 300000; (2) 100000 (3) 50000; (4) 23000; (5) 13100; (6) 6900; (7) 3600; and (8) 710 g mol−1. (b) Plot of (020) intensity ratio of PHB as a function of log(Mw PHB/Mw PLLA). | ||
The appearance of only the (020) reflection of PHB indicates that the b-axis of the PHB crystallites was almost aligned normal to the film surface (the ac planes are parallel to the film surface).42,45,47 This alignment also fixed the type of PHB lamellae to be the edge-on lamellae. The fact that the (020) reflection of PHB appears in all of the PHB/PLLA blends indicates that the preferred orientation of PHB crystallites in the blends is the same as that in neat PHB. Even though PLLA does not disturb the alignment and lamellae type of PHB in the blends, the intensity of the (020) reflection of PHB showed a distinct variation as the molecular weight of PLLA varied. The (020) peak, which is sharp and strong and does not overlap with any other peaks, has often been used to characterize the degree of PHB crystallinity. Since the blend ratio and illuminated area of the X-ray beam were fixed in this study, the intensity variation of the (020) reflection would simply indicate a change in the crystalline volume of PHB in the blends.
Fig. 1(b) shows a plot of (020) intensity ratio of PHB as a function of the log ratio of PHB to PLLA Mw. The (020) intensity ratio is calculated from the integrated intensity of (020) reflection in the blends to that of neat PHB. As can be seen from Fig. 1(b), the intensity ratio slightly decreased with decreasing PLLA molecular weight (Mw = 300000–50000 g mol−1, hereinafter called the higher molecular weight PLLAs) in the blends. The intensity ratio dramatically decreased in the blends with PLLA Mw = 23000–13100 g mol−1 (hereinafter called the middling molecular weight PLLAs). Furthermore, the intensity ratio increased again in the blends where Mw of PLLA was ≤6900 g mol−1 (hereinafter called the lower molecular weight PLLAs). These results clearly indicate that the inhibition of PHB crystallization by the addition of a small amount of PLLA strongly depends on the PLLA molecular weight. The drastic decrease of intensity ratio for the middling molecular weight PLLAs blended PHB films should be noticed. In the case of blends with the higher molecular weight PLLAs, it is quite probable that those PLLAs were also immiscible with PHB, even in the thin films, and that the origin of the moderate decrease in the crystalline volume of PHB in those films might be common in bulk PHB/PLLA blends. The lower molecular weight PLLAs also showed a slight effect on the PHB crystallization, demonstrating a sharp contrast to the behavior of those in bulk samples, in which a noticeable change is usually observed.26–28 Several factors would affect such a PHB crystallization behavior in the PHB/PLLA ultrathin blend films. Before we discuss those possible factors, we should investigate the effect of thickness confinement using the thinner (13 nm) PHB/PLLA blend films.
Fig. 2 displays the out-of-plane GIXD profiles of the 13 nm thick films of neat PHB and PHB/PLLA blends with various molecular weights of PLLA. PHB is still able to crystallize in the film as the (020) reflection was clearly observed for the neat PHB, although the intensity of this (020) peak was much lower than that of the 30 nm thick film; that is, the (020) intensity divided by 13 nm (thickness) (=26 nm−1) was significantly smaller than that of the 30 nm thick film (=68 nm−1). It has been widely accepted that the degree of crystallinity of a polymer often decreases with reducing thickness.40,48–50 As shown in the GIXD profiles of (1)–(5) in Fig. 2, no (020) reflection of PHB was observed in the blends with higher and middling molecular weight PLLAs (Mw ≥ 13100 g mol−1), indicating that the crystallization of PHB ultrathin films is fully inhibited by the addition of higher and middling molecular weight PLLAs. Thus, reducing the film thickness seems to enhance the ability of higher and middling molecular weight PLLAs to inhibit PHB crystallization. The (020) reflection also appears in the lower molecular weight PLLA blends, as depicted in (7) and (8), which may be similar to the results observed in the corresponding films depicted in Fig. 1(a). These results indicate that the lower molecular weight PLLAs do not significantly affect the crystallization of PHB ultrathin films. However, it is noted that the (020) reflection profile of (6) in Fig. 2 was found to be very weak if we compare it to that of (6) in Fig. 1(a). In this case, the effect of thickness confinement in the 13 nm thick film also seems to enhance the ability of PLLA with Mw 6900 g mol−1 to inhibit the crystallization of PHB as occurred by the higher and middling molecular weights of PLLAs.
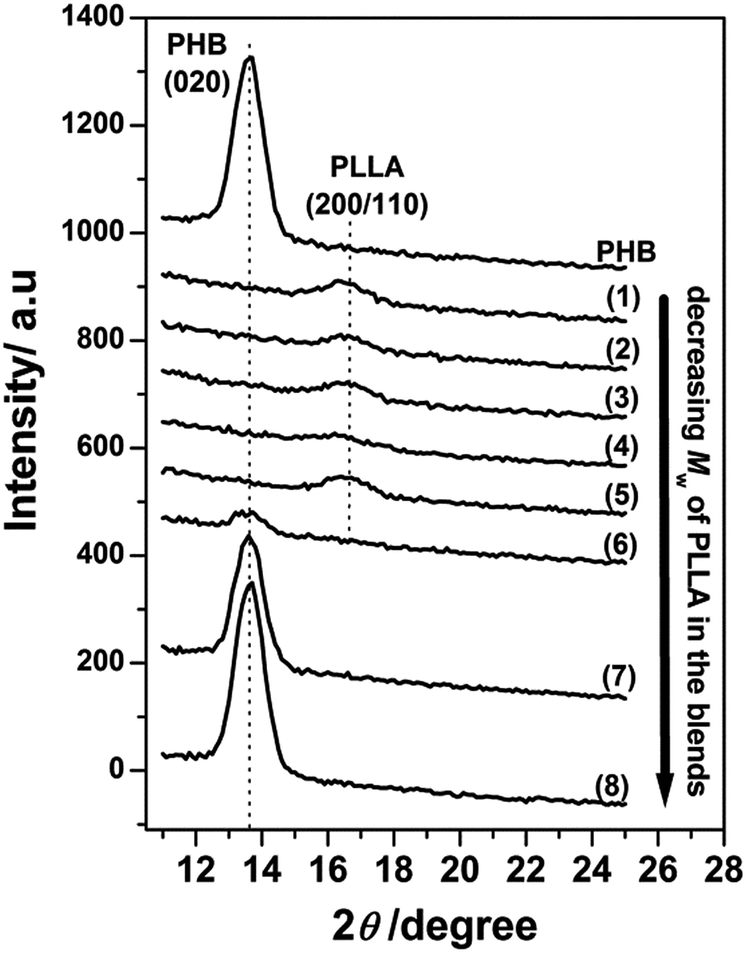 | ||
| Fig. 2 Out-of-plane GIXD profiles of 13 nm thick films of neat PHB and PHB/PLLA blends (80/20) with different Mw of PLLA: (1) 300000; (2) 100000 (3) 50000; (4) 23000; (5) 13100; (6) 6900; (7) 3600; and (8) 710 g mol−1. | ||
Furthermore, the higher molecular weight PLLAs still crystallized as the (200/110) reflection appears in Fig. 2(1)–(5). The very weak (200/110) reflections indicate that only a tiny fraction of the PLLA chains were able to crystallize in those blends. Similar to the results in Fig. 1(a)(6)–(8), the lower molecular weight PLLAs in Fig. 2(6)–(8) did not exhibit any PLLA reflection peaks, suggesting that the lower molecular weight PLLAs did not crystallize, irrespective of thickness confinement. Additionally, thermal annealing could not induce the crystallization of PHB in those blends in the 13 nm thick films, as shown in Fig. 3, indicating strong inhibition of PHB crystallization by the small amount of PLLA in these blend films. In addition, the crystallization of PLLAs was promoted to a certain extent by the thermal annealing as shown in Fig. 3(2)–(6) where increase in intensity from crystalline PLLA is noticed.
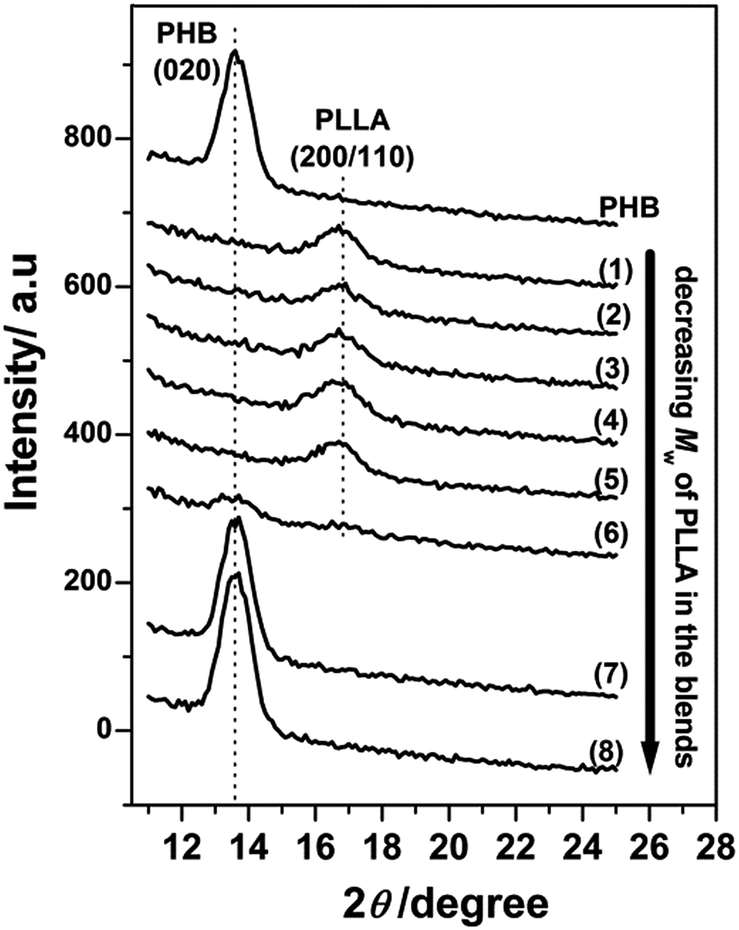 | ||
| Fig. 3 Out-of-plane GIXD profiles of annealed 13 nm thick films of neat PHB and PHB/PLLA blends (80/20) with different Mw of PLLA: (1) 300000; (2) 100000 (3) 50000; (4) 23000; (5) 13100; (6) 6900; (7) 3600; and (8) 710 g mol−1. | ||
The crystallization behavior of PHB in the PHB/PLLA ultrathin films shown in Fig. 1 and 2 demonstrates a striking contrast with the behavior observed in bulk PHB/PLLA blends. In bulk PHB/PLLA blends, it has been claimed that low molecular weight PLLAs are miscible with PHB and easily affect the crystallization of PHB. In contrast, the high molecular weight PLLAs were immiscible with PHB because of the phase separation between PHB and PLLA.26–28 However, in the present study on ultrathin films, a small amount of the lower molecular weight PLLAs did not show significant effects on the crystallization of PHB, indicating that those two components are very likely to be immiscible. The higher molecular weight PLLAs also showed phase separation with PHB in the 30 nm thick films, which was similar to the behavior in the bulk. Reducing the film thickness could be considered to make the confinement effect stronger, such that it can overcome the phase separation between high molecular weight PLLA and PHB. The present results are discussed and analyzed more completely in the following sections. The next several factors would be worth considering to understand the complex behavior of ultrathin PHB/PLLA film crystallization.
000 g mol−1.52,53 Long PLLA chains, which can be entangled, would effectively trap PHB molecules and disturb the crystallization of PHB chains. The PLLA chains with Mw ≥ 13100 g mol−1 may be adequate for the entanglement with PHB molecules and inhibit crystallization. On the other hand, the chain length of lower molecular weight PLLAs (Mw ≤ 3600 g mol−1) should be too short for entanglement.3.2. Size of PLLA molecules
Since the crystallization of PHB was revealed to have a complex thickness dependence, the relationship with the molecular size of PLLA should be considered; the molecular size of each PLLA is listed in Table 1 in the ESI.† Some might consider that small PLLA molecules (lower molecular weight PLLAs) would hinder the crystallization of PHB through interlamellar segregation because such shorter PLLA molecules are expected to easily diffuse into the PHB matrix and act as an effective obstacle for lamellae folding of PHB. However, our GIXD results have proved that such small molecules are ineffective at hindering lamellae folding. Small molecules of PLLAs most probably aggregate during the crystallization process. As a consequence, being unable to affect the crystallization of PHB. Similarly, big molecules of high molecular weight PLLAs would also be ineffective at reducing the crystallinity of PHB if the phase separation (as mentioned in point 1) between PHB and PLLA occurs. However, the molecules, whose sizes are comparable or exceed half of the film's thickness, might effectively inhibit the crystallization of PHB because phase separation would not be expected if part of the PLLA molecules that can infiltrate through the films are pinned by the interaction with the substrate.The crystallization behavior of PHB in the PHB/PLLA ultrathin blend films of 30 and 13 nm thick films observed in the present study is summarized in Fig. 4 and compared with bulk blends. As shown in Fig. 4, the inhibition effect by a small amount of PLLA on the crystallization of PHB is quite different from bulk blends. The long chains of high molecular weight PLLAs do not hinder the crystallization of PHB in bulk but effectively inhibit that in 13 nm thick films. Similarly, the shorter chains of low molecular weight PLLAs effectively inhibit the crystallization of PHB in the bulk but do not have much effect in ultrathin films.
 | ||
| Fig. 4 Scheme of PHB crystallization behavior in ultrathin PHB/PLLA (80/20 w/w) films (30 and 13 nm thick) in comparison with that of bulk blends (summarized from ref. 26–28). | ||
3.3. Conformation and crystalline structure of PHB as studied by IRRAS
In order to explore the chain conformation and crystalline structure of PHB in the PHB/PLLA ultrathin films on the molecular scale, IRRAS measurements were conducted. Fig. 5 shows the IRRAS spectra (bottom panel) and their second derivatives (upper panel) in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching region of 30 nm thick films of PHB and those of PHB/PLLA (80/20) blends with various molecular weights of PLLA. PHB shows two bands at 1726 and 1748 cm−1, which can be assigned to the crystalline and amorphous CO stretching modes, respectively.45 With the addition of PLLAs, the intensity of the CO crystalline band at 1726 cm−1 is significantly reduced in the spectra of the blends where the molecular weight of PLLA ≥ 6900 g mol−1 (see spectra (1)–(6) in the bottom panel of Fig. 5). These results reveal that the presence of PLLA appreciably decreases the crystallization of PHB. According to the IRRAS selection rule,60,61 the weak absorbance of the CO crystalline band of PHB in the IRRAS spectra of the blends indicates that the CO group is aligned more parallel to the substrate's surface. In the previous studies, the crystalline CO groups of PHB in the ultrathin films were also reported to be nearly parallel to the substrate surface (along the a-axis).45,54
O stretching region of 30 nm thick films of PHB and those of PHB/PLLA (80/20) blends with various molecular weights of PLLA. PHB shows two bands at 1726 and 1748 cm−1, which can be assigned to the crystalline and amorphous CO stretching modes, respectively.45 With the addition of PLLAs, the intensity of the CO crystalline band at 1726 cm−1 is significantly reduced in the spectra of the blends where the molecular weight of PLLA ≥ 6900 g mol−1 (see spectra (1)–(6) in the bottom panel of Fig. 5). These results reveal that the presence of PLLA appreciably decreases the crystallization of PHB. According to the IRRAS selection rule,60,61 the weak absorbance of the CO crystalline band of PHB in the IRRAS spectra of the blends indicates that the CO group is aligned more parallel to the substrate's surface. In the previous studies, the crystalline CO groups of PHB in the ultrathin films were also reported to be nearly parallel to the substrate surface (along the a-axis).45,54
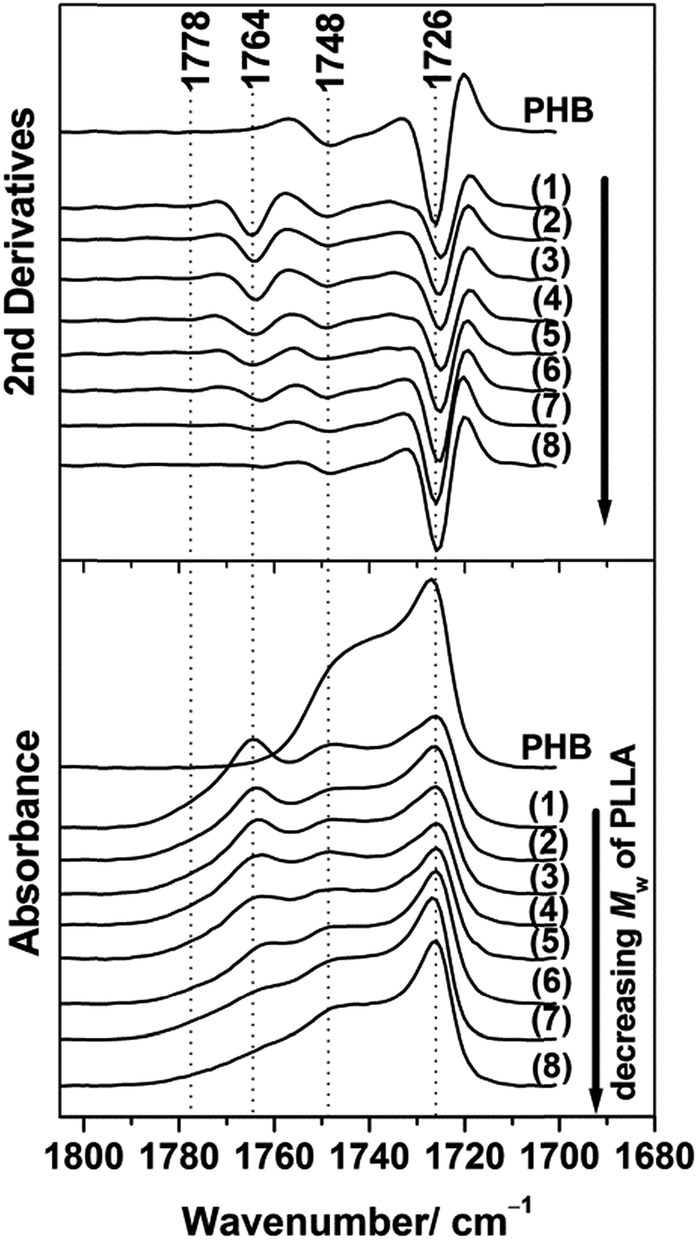 | ||
| Fig. 5 IRRAS spectra (bottom) and their second derivatives (upper) in the CO stretching region of 30 nm thick films of PHB and PHB/PLLA (80/20) blends where Mw of PLLA are (1) 300000; (2) 100000 (3) 50000; (4) 23000; (5) 13100; (6) 6900; (7) 3600; and (8) 710 g mol−1. | ||
Further reduction of the molecular weight of PLLA (Mw ≤ 3600 g mol−1) in the blends yielded spectra that were more similar to that of neat PHB, as seen from (7)–(8) in Fig. 5. This indicates that these low molecular weight PLLAs neither stopped the crystallization nor changed the preferred alignment of the PHB crystallites. This result was consistent with the results obtained from the GIXD measurements (profiles (7) and (8) in Fig. 1(a) and 2).
The two bands at 1764 and 1778 cm−1 in all of the blends were ascribed to the CO stretching mode of amorphous PLLA and that of fairly structural defect of PLLA, respectively.23,54,55 The intensity of the amorphous band at 1764 cm−1 of PLLA also decreased in the blends with decreasing the molecular weight of PLLA and eventually became very weak, as observed for the blend with the lowest molecular weight PLLA (see (8) in Fig. 5). The lowest molecular weight PLLA eventually aggregated in the films as described above, and aggregated short PLLA chains are irrelevant to crystallization.
In the previous study, we revealed the behavior of two crystalline structures in PHB ultrathin films with different structural orderings, that is, highly ordered and less ordered (intermediate) structures that can be characterized by bands at 1724 and 1731 cm−1, respectively. They can be recognized after deconvolution of the IRRAS spectra of PHB ultrathin films where several overlapped bands coexist in the CO stretching region of PHB due to the coexistence of the two crystalline structures.45 In this study, the CO stretching band region of the IRRAS spectra shown in Fig. 5 can be decomposed into several components using the GRAMS software. Fig. 6(a) displays the result of decomposition, showing the bands of the highly ordered structure (1724 cm−1), the less ordered (intermediate) state (1731 cm−1), two amorphous states arising from different conformations of PHB (1739 and 1748 cm−1), one amorphous band of PLLA (1764 cm−1), and another band arising from structural defects of PLLA (1778 cm−1).46
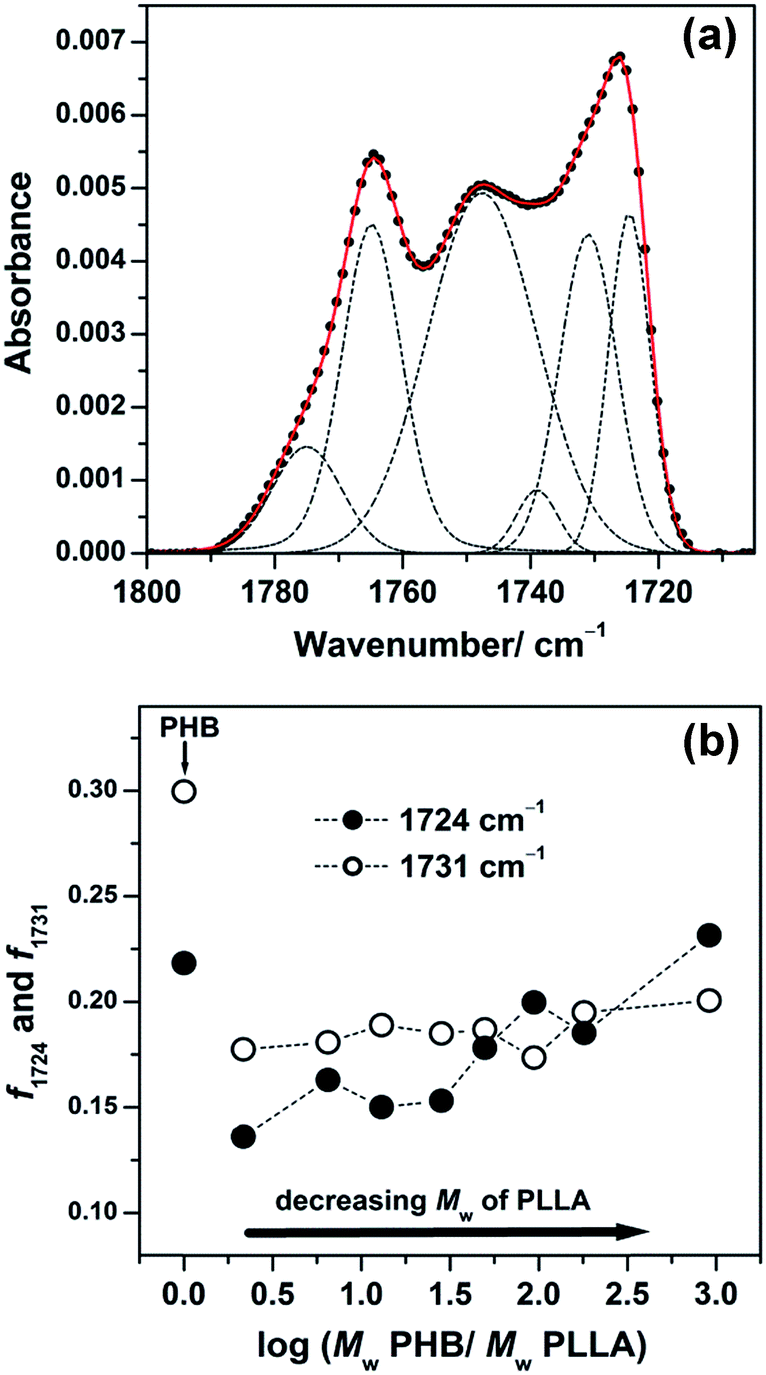 | ||
| Fig. 6 (a) Decomposition of an observed IRRAS spectrum in the CO stretching region of the 30 nm thick film shown in Fig. 5(1) of PHB/PLLA blends with Mw of PLLA = 300000 g mol−1. (b) Plots of integrated intensity of the 1724 cm−1 (highly ordered) and 1731 cm−1 (intermediate) bands normalized by the sum of integrated intensity of all fractions in each blend. | ||
The integrated intensity of each crystalline structure at 1724 and 1731 cm−1 is plotted in Fig. 6(b) after normalization by the sum of all fractions in each blend. In comparison with the intensity of the corresponding bands of neat PHB, the intensity of both the intermediate (1731 cm−1) and the highly ordered crystalline (1724 cm−1) bands became weaker in the blends, suggesting that the presence of PLLAs suppresses the formation of PHB crystals in the blends. While the integrated intensity of the intermediate state looked almost unchanged among the blend samples, the plot of the highly ordered crystalline band showed a gradual increase in the blends with decreasing molecular weight of PLLA. This indicated that the variation in the molecular weight of PLLA does not affect the crystal formation of the intermediate state, but the higher molecular weight PLLA disturbs the crystal formation of the highly ordered state.
Fig. 7 shows the IRRAS spectra (bottom) and their second derivatives (upper) in the CO stretching region of the 13 nm thick film of PHB and PHB/PLLA (80/20) blends with various molecular weights of PLLA. PHB still shows two bands due to the crystalline and amorphous CO bands at 1726 and 1749 cm−1, respectively. However, the intensity of the 1726 cm−1 band was lower than that of the 30 nm thick films, presumably because the volume of crystallites decreased. Since a small amount of PLLAs (Mw ≥ 13100 g mol−1) greatly suppressed the intensity and altered the band shape of the CO crystalline band of PHB, as noticeably observable in (1)–(5) of Fig. 7, we conclude that those PLLAs disturb both the crystallinity and the crystalline structure of PHB.
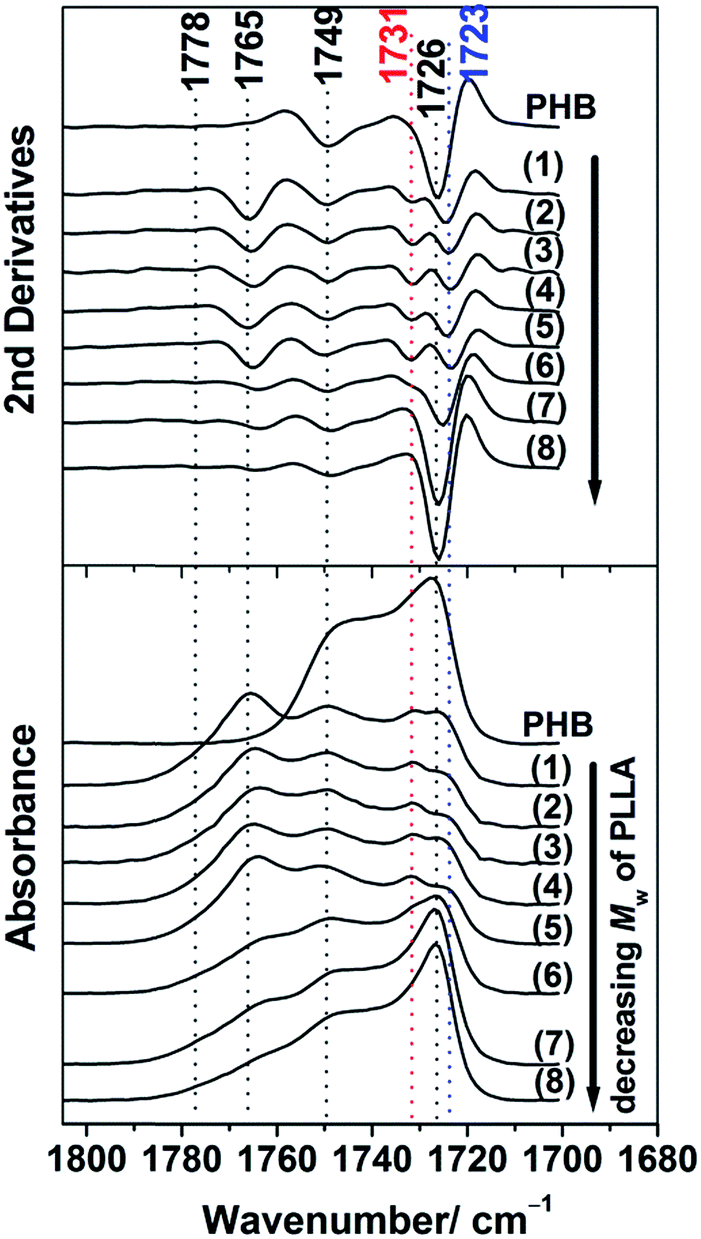 | ||
| Fig. 7 IRRAS spectra (bottom) and their second derivatives (upper) in the CO stretching region of 13 nm thick films of PHB and PHB/PLLA (80/20) blends where Mw of PLLA are (1) 300000; (2) 100000 (3) 50000; (4) 23000; (5) 13100; (6) 6900; (7) 3600; and (8) 710 g mol−1. | ||
The high and middling molecular weight PLLAs caused the CO crystalline band of PHB at 1726 cm−1 to split into two bands at 1723 and 1731 cm−1, which presumably correspond to the highly ordered and intermediate crystalline structures of PHB, respectively. However, the IRRAS results would be inconsistent with the GIXD profiles shown as (1)–(5) in Fig. 2, in which no reflection from PHB crystals was recognized in the corresponding blend samples. The inconsistency between GIXD and IRRAS may arise from the different sensitivity of these techniques in detecting very tiny crystallites.46 In the GIXD samples, the PHB crystals were too small and the amount of crystallites was also too small to be detected by the X-ray diffractometer equipped with a conventional X-ray tube. The intensity of CO crystalline bands observed in the blends with low molecular weight PLLAs was found to be rather similar to that of neat PHB at 1726 cm−1 ((7) and (8) in Fig. 7). This clearly indicates that the low molecular weight PLLAs are unable to alter the crystallization of PHB, which is independent of the film thickness. Furthermore, the intensity of the PLLA amorphous band at 1765 cm−1, which can be used as an indicator of PLLA chain entanglement, decreased as the molecular weight of PLLAs decreased, which agrees with the scheme discussed above.
To explore the crystallization behavior in the intermediate and highly ordered states, the spectra shown in Fig. 7 were also decomposed. A typical result is shown in Fig. 8(a) and displays the deconvoluted IRRAS spectra in the CO stretching region of the 13 nm thick films of PHB/PLLA (Mw = 300000 g mol−1). Plots of the integrated intensity of the 1724 cm−1 (highly ordered) and 1731 cm−1 (intermediate) bands, normalized by the sum of the integrated intensity in each blend, are depicted in Fig. 8(b). Similar to Fig. 6(b), the intensity of both intermediate and highly ordered crystalline structures decreased markedly after the addition of PLLAs. However, the intensity of the intermediate state (1731 cm−1) showed a small increase with decreasing PLLA molecular weight. Furthermore, the intensity of the highly ordered crystalline state (1724 cm−1) greatly reduced at first and subsequently increased with decreasing PLLA molecular weight. These results suggest that the molecular weight dependence of PLLA on the crystallization of PHB is greater for thinner films.
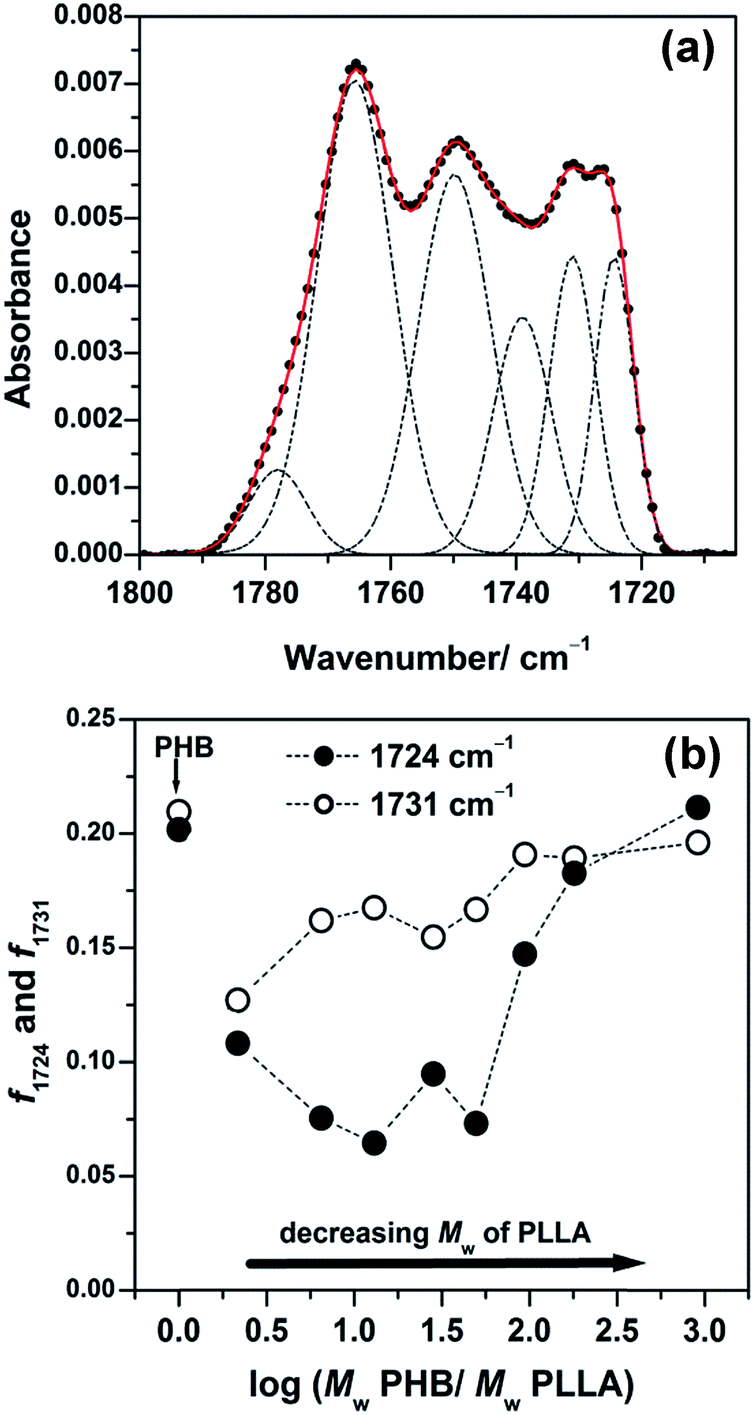 | ||
| Fig. 8 (a) Decomposition of an observed IRRAS spectrum in the CO stretching region of the 13 nm thick film shown in Fig. 7(1) of PHB/PLLA blends with Mw of PLLA = 300000 g mol−1. (b) Plots of integrated intensity of 1724 cm−1 (highly ordered) and 1731 cm−1 (intermediate) bands normalized by the sum of integrated intensity of all fractions in each blend. | ||
In the C–H stretching region, weak intermolecular hydrogen bonding between carbonyl and methyl groups (CH3⋯OC) of PHB was found at an anomalously high frequency region.56–59 The weak hydrogen bonding bands of PHB were still recognizable in the 30 nm and 13 nm thick films, as shown in Fig. 9. In the 30 nm thick film, the weak hydrogen bonding band appeared at 3011 cm−1 as a weak shoulder band, while in the 13 nm thick film, it shifted to 3009 cm−1 and the peak became more apparent. These results may indicate that the hydrogen bonds in the 13 nm thick film were weaker than the hydrogen bonds formed in the 30 nm thick film. However, the slightly higher intensity of the 3009 cm−1 bands (indicated in Fig. 9(b)) would reflect that, in the 13 nm thick film, the number of bonds oriented nearly perpendicular to the film surface seemed to be larger than that in the 30 nm thick films.
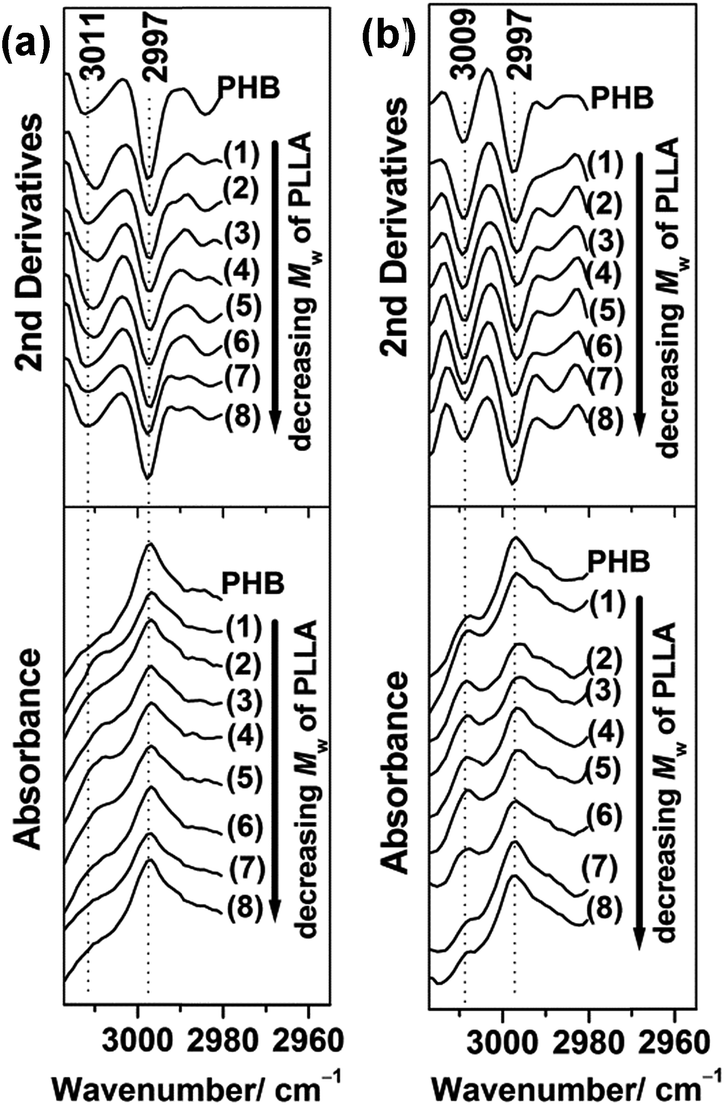 | ||
| Fig. 9 IRRAS spectra (bottom) and their second derivatives (upper) in the 3018–2980 cm−1 region of (a) 30 nm and (b) 13 nm thick films of PHB and PHB/PLLA (80/20) blends where Mw of PLLA are (1) 300000; (2) 100000 (3) 50000; (4) 23000; (5) 13100; (6) 6900; (7) 3600; and (8) 710 g mol−1. | ||
4. Conclusions
We investigated the crystallization behavior of PHB in PHB/PLLA ultrathin films with thicknesses of 30 and 13 nm as a function of PLLA molecular weight using GIXD and IRRAS measurements. Eight PLLAs with different molecular weights (ranging from 300000 to 710 g mol−1) were added to PHB (Mw = 650000 g mol−1) in order to explore the effect of a small amount of PLLA on the crystallization behavior of PHB in ultrathin film blends. The PHB/PLLA ratio was fixed at 80/20 (w/w) for all of the blends studied. The crystallization of PHB showed a strong dependency on the molecular weight of PLLA and film thickness. In the 30 nm thick films, phase separation occurred between PHB and higher molecular weight PLLAs (Mw ≥ 50000 g mol−1) in the blends; therefore, those PLLAs were less affected the crystallization of PHB. The miscible PHB and PLLA blends were obtained when the middling molecular weight PLLAs (Mw = 23000–13100 g mol−1) were used, yielding dramatic decreases in the crystallinity of PHB. It was quite noteworthy that the crystallization of PHB was fully inhibited in the Mw ≥ 6900 g mol−1 PLLA blends when the film thickness was reduced to 13 nm.
IRRAS demonstrated that, for the 30 nm thick films, the addition of a small amount of PLLA significantly suppressed the intensity of the PHB crystalline band at 1726 cm−1. However, the PLLAs did not affect the crystalline structures of PHB significantly. Furthermore, for the 13 nm thick films, both the intensity and the crystalline structures of PHB were remarkably altered by a small amount of PLLA (Mw ≥ 6900 g mol−1). Both GIXD and IRRAS results consistently showed that the lower molecular weight PLLAs (Mw ≤ 3600 g mol−1) did not affect the crystallinity and crystalline structures of PHB. Furthermore, several factors, such as the free surface and interface effects, entanglement of PLLA chains, and molecular size of PLLA, must be taken into account to comprehend the complex crystallization behavior of PHB in PHB/PLLA ultrathin films.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported in part by the Japan Society for the Promotion of Science (Grant-in-Aid for Scientific Research(C) 24560033).References
- L. A. Utracki, P. Mukhopadhyay and R. K. Gupta, Polymer Blends: Introduction, in Polymer Blends Handbook Second Edition, ed. L. A. Utracki and C. A. Wilkie, Springer, New York, 2014, pp. 19–36 Search PubMed.
- L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576–602 CrossRef CAS.
- L. M. Robeson, Polym. Eng. Sci., 1984, 24, 587–597 CAS.
- Q. Zhao and G. Cheng, Biodegradable PHB/PEG Derivatives and their Degradation Behavior, in New Frontiers in Polymer Research, ed. R. K. Bregg, Nova Science Publishers Inc., New York, 2006, pp. 99–124 Search PubMed.
- K. Sudesh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef CAS.
- M. M. Satkowski, D. H. Melik, J.-P. Autran, P. R. Green, I. Noda and L. A. Schechtman, Physical and Processing properties of polyhydroxyalkanoate copolymers, in Biopolymers, Polyesters II-Properties and chemical synthesis, ed. Y. Doi and A. Steinbüchel, Wiley-VCH, Weinheim, 2001, vol. 3b, pp. 231–263 Search PubMed.
- M. Avella and E. Martuscelli, Polymer, 1988, 29, 1731–1737 CrossRef CAS.
- M. Pizzoli, M. Scandola and G. Ceccorulli, Macromolecules, 1994, 27, 4755–4761 CrossRef CAS.
- P. Xing, L. Dong, Y. An, Z. Feng, M. Avella and E. Martuscelli, Macromolecules, 1997, 30, 2726–2773 CrossRef CAS.
- Y. An, L. Dong, L. Li, Z. Mo and Z. Feng, Polymer, 1999, 35, 365–369 CAS.
- T. Furukawa, H. Sato, R. Murakami, J. Zhang, Y. Duan, I. Noda, S. Ochiai and Y. Ozaki, Macromolecules, 2005, 38, 6445–6454 CrossRef CAS.
- T. Kabe, T. Tsuge, K. Kasuya, A. Takemura, T. Hikima, M. Takata and T. Iwata, Macromolecules, 2012, 45, 1858–1865 CrossRef CAS.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- H. Tsuji, Poly(Lactic Acid), in Bio-Based Plastics: Materials and Applications, ed. S. Kabasci, John Wiley & Sons, Ltd., United Kingdom, 2014, pp. 171–239 Search PubMed.
- (a) S. Suzuki and Y. Ikada, Medical Applications, in Poly(Lactic acid) Synthesis, Structures, Properties, Processing and Applications, ed. R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, pp. 445–454 Search PubMed; (b) S. Obuchi and S. Ogawa, Packaging and Other Commercial Applications, in Poly(Lactic acid) Synthesis, Structures, Properties, Processing and Applications, ed. R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, pp. 457–467 Search PubMed; (c) M. Mochizuki, Textile Applications, in Poly(Lactic acid) Synthesis, Structures, Properties, Processing and Applications, ed. R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, pp. 469–475 Search PubMed; (d) A. Hiraishi, Environmental Applications, in Poly(Lactic acid) Synthesis, Structures, Properties, Processing and Applications, ed. R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, pp. 445–486 Search PubMed.
- J. R. Dorgan, H. J. Lehermeier, L.-I. Palade and J. Cicero, Macromol. Symp., 2001, 175, 55–66 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- C. Sheng, T. Zhang, Y. Yuan, L. Zhou, Y. Duan and J. Zhang, Polym. Int., 2014, 63, 1270–1277 CrossRef CAS.
- C. Vogel, E. Wessel and H. W. Siesler, Macromolecules, 2008, 41, 2975–2977 CrossRef CAS.
- Y. Hu, H. Sato, J. Zhang, I. Noda and Y. Ozaki, Polymer, 2008, 49, 4204–4210 CrossRef CAS.
- T. Furukawa, H. Sato, R. Murakami, J. Zhang, I. Noda, S. Ochiai and Y. Ozaki, Polymer, 2007, 48, 1749–1755 CrossRef CAS.
- T. Furukawa, H. Sato, R. Murakami, J. Zhang, I. Noda, S. Ochiai and Y. Ozaki, Polymer, 2006, 47, 3132–3140 CrossRef CAS.
- J. Zhang, H. Sato, T. Furukawa, H. Tsuji, I. Noda and Y. Ozaki, J. Phys. Chem. B, 2006, 110, 24463–24471 CrossRef CAS PubMed.
- J. W. Park, Y. Doi and T. Iwata, Biomacromolecules, 2004, 5, 1557–1566 CrossRef CAS PubMed.
- I. Ohkoshi, H. Abe and Y. Doi, Polymer, 2000, 41, 5985–5992 CrossRef CAS.
- J.-S. Yoon, W.-S. Lee, K.-S. Kim, I.-J. Chin, M.-N. Kim and C. Kim, Eur. Polym. J., 2000, 36, 435–442 CrossRef CAS.
- N. Koyama and Y. Doi, Polymer, 1997, 38, 1589–1593 CrossRef CAS.
- E. Blümm and A. J. Owen, Polymer, 1995, 36, 4077–4081 CrossRef.
- L. Zhang, C. Xiong and X. Deng, Polymer, 1996, 37, 235–241 CrossRef CAS.
- C. Yang, R. Onitsuka and I. Takahashi, Eur. Phys. J. E: Soft Matter Biol. Phys., 2013, 36, 66 CrossRef PubMed.
- D. S. Fryer, P. F. Nealey and J. J. de Pablo, Macromolecules, 2000, 33, 6439–6447 CrossRef CAS.
- J. A. Forrest and J. Mattsson, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 61, R53–R56 CrossRef CAS.
- K. Taguchi, H. Miyaji, K. Izumi, A. Hoshino, Y. Miyamoto and R. Kokawa, Polymer, 2001, 42, 7443–7447 CrossRef CAS.
- H. Abe, Y. Kikkawa, T. Iwata, H. Aoki, T. Akehata and Y. Doi, Polymer, 2000, 41, 867–874 CrossRef CAS.
- E. K. Lin, R. Kolb, S. K. Satija and W. Wu, Macromolecules, 1999, 32, 3753–3757 CrossRef CAS.
- B. Frank, A. P. Gast, T. P. Russell, H. R. Brown and C. Hawker, Macromolecules, 1996, 29, 6531–6534 CrossRef CAS.
- Y. Wang, S. Ge, M. Rafailovich, J. Sokolov, Y. Zou, H. Ade, J. Lüning, A. Lustiger and G. Maron, Macromolecules, 2004, 37, 3319–3327 CrossRef CAS.
- H. Schönherr and C. W. Frank, Macromolecules, 2003, 36, 1188–1198 CrossRef.
- C. W. Frank, V. Rao, M. M. Despotopoulou, R. F. W. Pease, W. D. Hinsberg, R. D. Miller and J. F. Rabolt, Science, 1996, 273, 912–915 CAS.
- M. M. Despotopoulou, C. W. Frank, R. D. Miller and J. F. Rabolt, Macromolecules, 1996, 29, 5797–5804 CrossRef CAS.
- Y. Ma, W. Hu and G. Reiter, Macromolecules, 2006, 39, 5159–5164 CrossRef CAS.
- X. Sun, L. Guo, H. Sato, Y. Ozaki, S. Yan and I. Takahashi, Polymer, 2011, 52, 3865–3870 CrossRef CAS.
- S. Yan, Macromolecules, 2003, 36, 339–345 CrossRef CAS.
- H. Chang, Q. Guo, D. Shen, L. Li, Z. Qiu, F. Wang and S. Yan, J. Phys. Chem. B, 2010, 114, 13104–13109 CrossRef CAS PubMed.
- Khasanah, K. R. Reddy, S. Ogawa, H. Sato, I. Takahashi and Y. Ozaki, Macromolecules, 2016, 49, 4202–4210 CrossRef CAS.
- X. Sun, A. Tokuda, Y. Oji, T. Nakatani, H. Tsuji, Y. Ozaki, S. Yan and I. Takahashi, Macromolecules, 2012, 45, 2485–2493 CrossRef CAS.
- K. Mori, S. Mukoyama, Y. Zhang, H. Sato, Y. Ozaki, H. Terauchi, I. Noda and I. Takahashi, Macromolecules, 2008, 41, 1713–1719 CrossRef CAS.
- M. M. Despotopoulou, R. D. Miller, J. F. Rabolt and C. W. Frank, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2335–2349 CrossRef CAS.
- K. Urayama, M. Tsuji and D. Neher, Macromolecules, 2000, 33, 8269–8279 CrossRef CAS.
- Q. M. Zhang, H. Xu, F. Fang, Z.-Y. Cheng, F. Xia and H. You, J. Appl. Phys., 2001, 89, 2613–2616 CrossRef CAS.
- O. E. Farrance, R. A. L. Jones and J. K. Hobbs, Polymer, 2009, 50, 3730–3738 CrossRef CAS.
- J. J. Cooper-White and M. E. Mackay, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1803–1814 CrossRef CAS.
- J.-F. Zhang and X. Sun, Poly(lactic acid)-Based Bioplastics, in Biodegradable Polymers for Industrial Applications, ed. R. Smith, Woodhead Publishing Limited, England, 2005, p. 255 Search PubMed.
- H. Sato, R. Murakami, K. Mori, Y. Ando, I. Takahashi, I. Noda and Y. Ozaki, Vib. Spectrosc., 2009, 51, 132–135 CrossRef CAS.
- J. Zhang, H. Tsuji, I. Noda and Y. Ozaki, J. Phys. Chem. B, 2004, 108, 11514–11520 CrossRef CAS.
- H. Wang and K. Tashiro, Macromolecules, 2015, 49, 581–594 CrossRef.
- H. Sato, Y. Ando, J. Dybal, T. Iwata, I. Noda and Y. Ozaki, Macromolecules, 2008, 41, 4305–4312 CrossRef CAS.
- H. Sato, K. Mori, R. Murakami, Y. Ando, I. Takahashi, J. Zhang, H. Terauchi, F. Hirose, K. Senda, K. Tashiro, I. Noda and Y. Ozaki, Macromolecules, 2006, 29, 1525–1531 CrossRef.
- H. Sato, R. Murakami, A. Padermshoke, F. Hirose, K. Senda, I. Noda and Y. Ozaki, Macromolecules, 2004, 37, 7203–7213 CrossRef CAS.
- S. A. Francis and A. H. Ellison, J. Opt. Soc. Am., 1959, 49, 131–138 CrossRef CAS.
- J. Umemura, Reflection-Absorption Spectroscopy of Thin Film on Metallic Substrates, in Handbook of Vibrational Spectroscopy, ed. J. M. Chalmers and P. R. Griffiths, Wiley, Chichester, UK, 2002, vol. 2, pp. 982−998 Search PubMed.
- Y.-X. Liu and E.-Q. Chen, Coord. Chem. Rev., 2010, 254, 1011–1037 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10996b |
| ‡ Present address: SABIC Research and Technology Pvt. Ltd., Chikkadunnasandra Village, Anekal Taluk, Off Sarjapur-Attibele State Highway, Bengaluru Area 562125, India. |
| This journal is © The Royal Society of Chemistry 2017 |