
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed aerobic oxidative C–O bond formation for the synthesis of 3,5-disubstituted isoxazoles from enone oximes†
Yadong Sun *,
Ablimit Abdukader,
Haiyan Zhang,
Wanle Yang and
Chenjiang Liu
*,
Ablimit Abdukader,
Haiyan Zhang,
Wanle Yang and
Chenjiang Liu
The Key Laboratory of Oil and Gas Fine Chemicals, Ministry of Education & Xinjiang Uygur Autonomous Region, Urumqi Key Laboratory of Green Catalysis and Synthesis Technology, School of Chemistry and Chemical Engineering, Physics and Chemistry Detecting Center, Xinjiang University, Urumqi 830046, China. E-mail: syd19791016@163.com
First published on 8th December 2017
Abstract
A direct access to 3,5-disubstituted isoxazoles has been accomplished through an intramolecular oxidative coupling reaction of enone oximes using a catalytic quantity of Cu(OAc)2. This method features an inexpensive metal catalyst, molecular oxygen as a green oxidant, good functional group tolerance and readily available starting materials. This attractive method for the synthesis of isoxazole derivatives is of great significance due to the product's versatile reactivity for further transformations.
Transition-metal-catalyzed direct C–H functionalization toward C–C and C–Het (Het = heteroatom) bonds formation is one of the most challenging classes of oxidation reactions, and has attracted extensive interest in the past few decades.1,2 However, most efforts have been focused on using noble metal catalysts, such as Rh, Ru, Pd, etc.3–8 Compared with the methods based on noble-metals, copper-catalyzed methods for organic synthesis are obviously economically attractive.9,10 In view of the increasing demand for environmentally benign organic synthesis and green chemistry, molecular oxygen is undoubtedly the ideal terminal oxidant, because of its remarkable advantages, such as being inexpensive, high atom efficiency, and in most cases with nontoxic water as the byproduct.11 To date, the copper-catalyzed aerobic cross-coupling between two C–H bonds or C–H and Het–H bonds have been a green and atom-economic approach for construction of heterocycles.12–20 Recently, Buchwald and Nagasawa described the copper-catalyzed aerobic oxidative cyclization of amidines and benzanilides to give benzimidazoles and benzoxazoles, respectively.21,22
Isoxazole derivatives represent a privileged class of five-membered nitrogen heterocycles and are extensively found in numerous biologically active molecules with anticancer, antidepressant, antibiotic, and analgesic activities.23–26 Moreover, they are used as key building blocks in materials science and as various complex synthetic intermediate in organic synthesis.27,28 Owing to their good reactivity and synthetic utility, the development of more efficient synthetic methods for isoxazoles is of great importance to organic chemists.29–40 Among them, there are two classical strategies for construction of 3,5-disubstituted isoxazoles: 1,3-dipolar cycloaddition and oxidative cyclization (Scheme 1). Nonetheless, the latter methods commonly require the use of stoichiometric amounts of oxidants, such as TEMPO, hypervalent iodine, DDQ, metal salt and so on, which caused heavy burden to the environment.41–46 It is no doubt that the catalyzed oxidation with pure oxygen or air as the oxidant is a green solution. Herein, we disclose a new copper(II)-catalyzed synthesis of 3,5-disubstituted isoxazoles from enone oximes through a C–H functionalization/C–O bond-forming process that uses oxygen as the terminal oxidant and water as the only direct waste product.
 | ||
| Scheme 1 Two classical strategies for the construction of 3,5-disubstituted isoxazoles. | ||
Inspired by the recent development of copper-catalyzed aerobic oxidative C–O bond formation for the construction of heterocycles47 and our continuing interest in developing C–Het bond formation reactions,48 we envisioned a direct synthesis of 3,5-disubstituted isoxazoles via copper-catalyzed dehydrogenative coupling reaction of enone oximes involving intramolecular C–O bond formation. Therefore, we first studied the reaction of chalcone oxime 1aa in DMSO using Cu(OAc)2 as the catalyst under 1 atm of O2 without adding any additives. To our delight, the desired isoxazole 2aa was obtained in 30% yield (Table 1, entry 1). This result prompted us to screen suitable reaction conditions (Table 1). Further investigation revealed that the additive played a critical role for this transformation (entries 2–8). Using Brønsted acid such as HOAc as the reaction additive provided poor result of desired product (entry 2). Detailed examination of base as the additive suggested that Cs2CO3, NaHCO3, Na2CO3, Et3N, and pyridine gave moderate yields and DABCO was the best choice (entries 3–8). We speculated that DABCO possibly act as both ligand and base to promote the product formations. The effects of different copper salts were also studied, such as CuBr and CuCl were employed as the catalyst and exhibited lower efficiencies than Cu(OAc)2 (entries 9 and 10). Solvent evaluation revealed that DMSO was the best solvent, while other solvents just led to moderate yields (entries 11 and 12). Lowering the temperature was not suitable for this transformation (entry 13). The reaction was less effective when proceeding under air atmosphere (entry 14). In the absence of the copper source, no desired product was obtained (entry 15). Thus, the optimal reaction conditions consist of Cu(OAc)2 (10 mol%) and DABCO (30 mol%) in DMSO at 100 °C under an oxygen atmosphere.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Yieldb (%) |
| a Reaction conditions: 1aa (0.20 mmol), catalyst (0.02 mmol), additive (0.06 mmol) in solvent (2 mL), 100 °C, 12 h.b Isolated yield.c Reaction was performed at 80 °C.d Reaction was performed under air. | ||||
| 1 | Cu(OAc)2 | — | DMSO | 30 |
| 2 | Cu(OAc)2 | HOAc | DMSO | 15 |
| 3 | Cu(OAc)2 | Cs2CO3 | DMSO | 53 |
| 4 | Cu(OAc)2 | NaHCO3 | DMSO | 45 |
| 5 | Cu(OAc)2 | Na2CO3 | DMSO | 58 |
| 6 | Cu(OAc)2 | Et3N | DMSO | 40 |
| 7 | Cu(OAc)2 | Pyridine | DMSO | 34 |
| 8 | Cu(OAc)2 | DABCO | DMSO | 85 |
| 9 | CuBr | DABCO | DMSO | 46 |
| 10 | CuCl | DABCO | DMSO | 59 |
| 11 | Cu(OAc)2 | DABCO | DMF | 66 |
| 12 | Cu(OAc)2 | DABCO | o-Xylene | 62 |
| 13c | Cu(OAc)2 | DABCO | DMSO | 73 |
| 14d | Cu(OAc)2 | DABCO | DMSO | 70 |
| 15 | — | DABCO | DMSO | 0 |
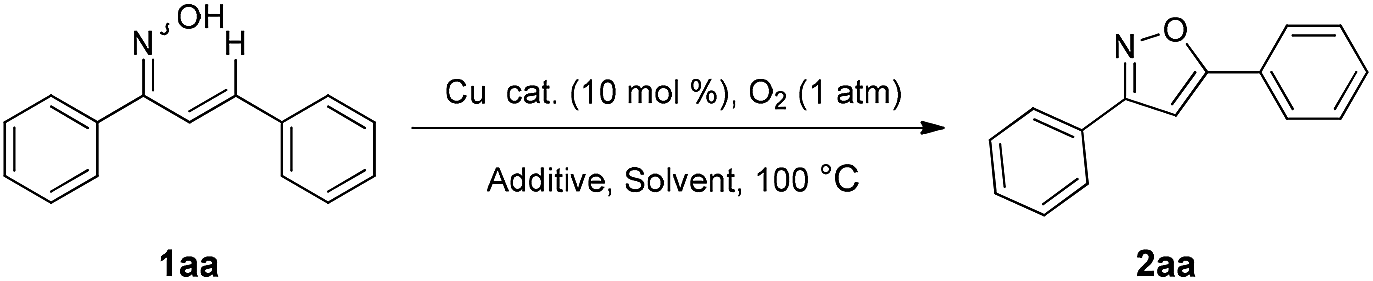
Under the optimized conditions, we sought to explore the scope and generality of the process. As the representative results shown in Table 2 attest, this methodology is compatible with a variety of electron-donating and electron-withdrawing groups. Substituted enone oximes derived from mono-substituted arylaldehydes possessing electron-donating groups such as –CH3 and –OCH3, all gave 3,5-disubstituted isoxazoles 2ab–2ad in good yields (Table 2). Similarly substrates possessing electron-withdrawing groups such as –Cl, –Br, –NO2 underwent C–H functionalization to give 3,5-disubstituted isoxazoles 2ae–2aj in excellent yields. As can be seen from Table 2, compared to substrates bearing electron-donating groups, electron-withdrawing substrates gave better yields. Nevertheless, same substituents at the para, meta, and ortho positions were also effective substituents in this transformation furnishing the corresponding products (2ae, 2af, and 2ag) in good yields. Substituted enone oximes derived from multi-substituted arylaldehydes reacted well in this transformation (2ak, 2al, and 2am). In a similar manner, 5-(naphthalen-2-yl)-3-phenylisoxazole 2an was obtained in 85% yield. Very interestingly, enone oxime 1ao with two potential reaction centers selectively underwent the desired transformation to deliver the corresponding product 2ao in 83% yield.
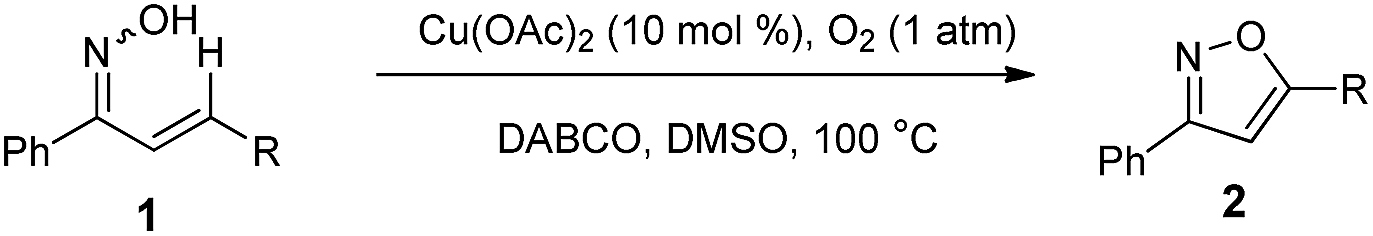
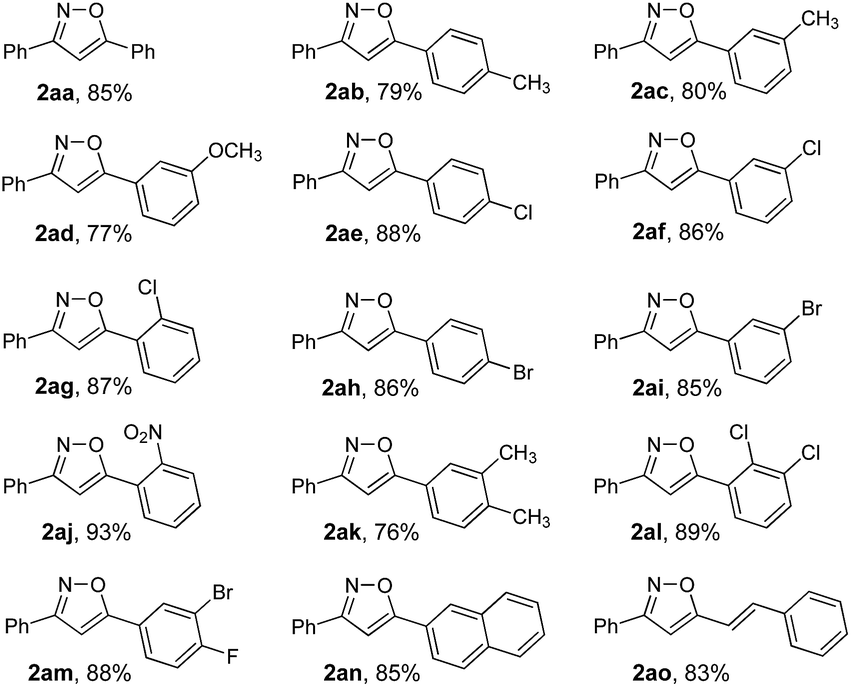
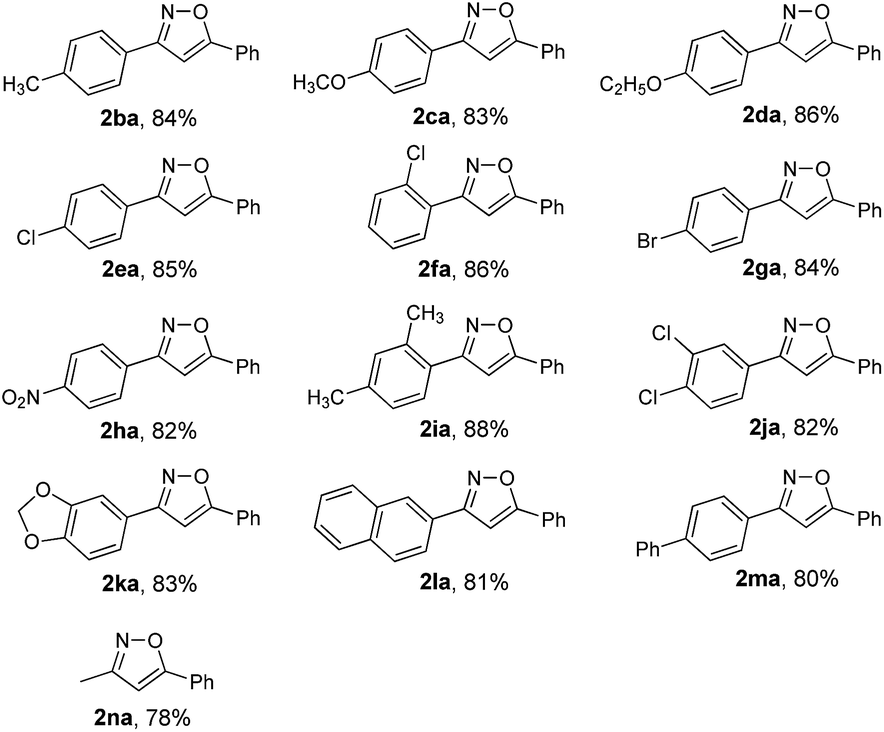
To expand the scope of this methodology, a serious of enone oximes derived from arylacetones were also used under the optimized conditions (Table 3). A variety of enone oximes, bearing electron-donating groups (–Me, –OMe, and –OEt) or electron-withdrawing groups (–Cl, –Br, and –NO2) on the benzene ring, reacted smoothly to afford the corresponding 3,5-substituted isoxazoles 2ba–2ha in good yields. Nevertheless, enone oximes bearing multi-substituents (2,4-dimethyl, 3,4-dichloro, and 3,4-methylenedioxy) at the benzene ring were also effective substituents in this transformation furnishing the corresponding products (2ia, 2ja, and 2ka) in good yields. To our delight, enone oximes originating from other aromatic ketones such as naphthalene and biphenyl were also investigated and found to form the desired products 2la and 2ma in 81% and 80% yields, respectively. Moreover, enone oxime derived from other aliphatic ketones such as acetone can also be successfully transformed into the desired isoxazole product 2na in good yield.
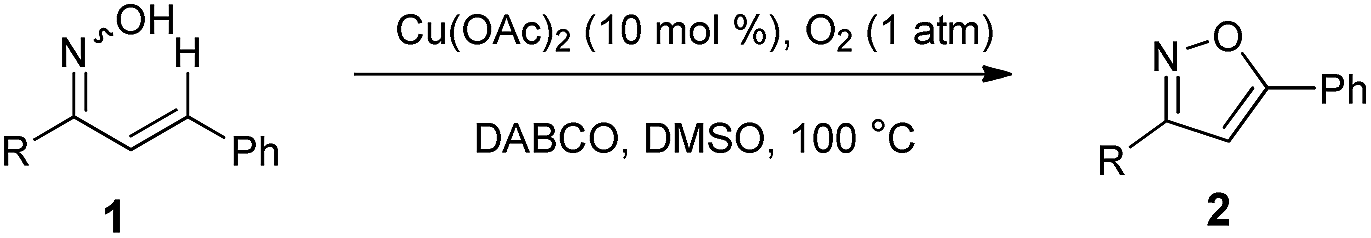
To further demonstrate the synthetic application of our developed protocol, we performed experiments using 2aa as the starting materials for diversification of isoxazole derivatives (Scheme 2). We first examined the electrophilic aromatic bromination of 2aa at the 4-position, and 2aa react smoothly with NBS to give corresponding bromination product 3.49 Additionally, we were able to affect Sonogashira and Suzuki–Miyaura cross-couplings on 4-bromo-3,5-diphenylisoxazole 3. By allowing compound 3 to react with phenyl acetylene under standard Sonogashira conditions, 3,5-diphenyl-4-(phenylethynyl)isoxazole 4 was obtained in 76% yield. Also, allowing compound 3 to react with phenylboronic acid under Suzuki–Miyaura reaction conditions provided the desired arylation product 5 in excellent yield.
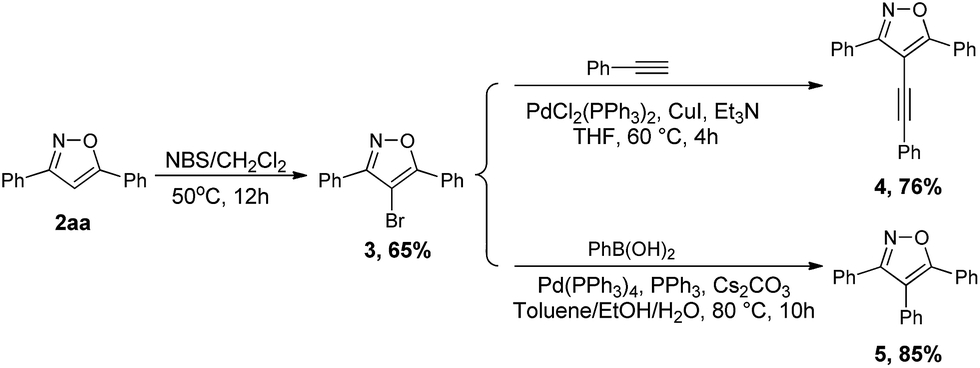 | ||
| Scheme 2 Synthetic application of the isoxazole products. | ||
Base on our experimental data and previous report,50–52 a plausible reaction mechanism for formation of 3,5-disubstituted isoxazoles 2 is proposed (Scheme 3). In route I, the reaction of enone oxime 1 with copper catalyst presumably led to the formation of Cu–O species A. Then, Cu–O species A underwent concurrent deprotonation/metalation at the sp2 C–H bond to give a metallacycle B, followed by reductive elimination and C–O bond formation leading desired isoxazoles 2 with the generation of reduced copper species,53–55 which could be oxidized by oxygen to achieve catalytic cycle. In route II, enone oxime 1 coordinated with CuII presumably led to intermediate C, which should induce the intramolecular nucleophilic addition to generate intermediate D. The subsequent deprotonation and hydrolysis of intermediate D gave intermediate E. Finally, intermediate E was oxidized by oxygen or CuII to the final product 2.
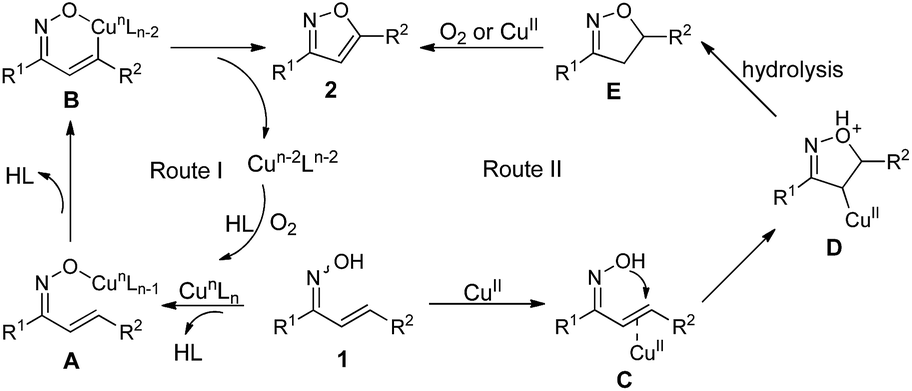 | ||
| Scheme 3 Possible reaction mechanism. | ||
In summary, we have developed a novel copper-catalyzed intramolecular oxidative C–O bond coupling reaction for the synthesis of 3,5-disubstituted isoxazoles that complements the commonly used strategies for the synthesis of isoxazoles. Advantages of our procedure include simplicity of operation, diverse substrates, the inexpensive catalytic system, oxygen as the environmentally oxidant and water as nontoxic byproduct. Further investigation of reaction mechanism and synthetic application are ongoing in our group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Xinjiang Natural Science Foundation (2015211C264) for financial support.Notes and references
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed.
- C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed.
- G. N. Hermann, P. Becker and C. Bolm, Angew. Chem., Int. Ed., 2015, 54, 7414–7417 CrossRef CAS PubMed.
- K. Matsumoto, M. Yoshida and M. Shindo, Angew. Chem., Int. Ed., 2016, 55, 5272–5276 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929–6932 CrossRef CAS PubMed.
- Z. M. Chen, M. J. Hilton and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 1146–11464 CrossRef PubMed.
- K. J. Xiao, L. Chu and J. Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 2856–2860 CrossRef CAS PubMed.
- M. Liu and C. J. Li, Angew. Chem., Int. Ed., 2016, 55, 10806–10810 CrossRef CAS PubMed.
- A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- K. V. N. Esguerra, Y. Fall, L. Petitjean and J.-P. Lumb, J. Am. Chem. Soc., 2014, 136, 7662–7668 CrossRef CAS PubMed.
- Y. Yang, W. Dong, Y. Guo and R. M. Rioux, Green Chem., 2013, 15, 3170–3175 RSC.
- C.-Y. Liu, Y. Li, J.-Y. Ding, D.-W. Dong and F.-S. Han, Chem.–Eur. J., 2014, 20, 2373–2381 CrossRef CAS PubMed.
- Q. Feng and Q. Song, J. Org. Chem., 2014, 79, 1867–1871 CrossRef CAS PubMed.
- J. Over and F. Maseras, Chem. Commun., 2013, 49, 10486–10488 RSC.
- E. Boess, D. Sureshkumar, A. Sud, C. Wirtz, C. Farés and M. Klussmann, J. Am. Chem. Soc., 2011, 133, 8106–8109 CrossRef CAS PubMed.
- X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636–3646 CrossRef CAS PubMed.
- H. He, Z. Wang and W. Bao, Adv. Synth. Catal., 2010, 352, 2905–2912 CrossRef CAS.
- M. M. Guru, M. A. Ali and T. Punniyamurthy, Org. Lett., 2011, 13, 1194–1197 CrossRef CAS PubMed.
- G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932–1934 CrossRef CAS PubMed.
- S. Ueda and H. Nagasawa, Angew. Chem., Int. Ed., 2008, 47, 6411–6413 CrossRef CAS PubMed.
- R. M. Kumbhare, U. B. Kosurkar, M. Janaki Ramaiah, T. L. Dadmal, S. N. Pushpavalli and M. Pal-Bhadra, Bioorg. Med. Chem. Lett., 2012, 22, 5424–5427 CrossRef CAS PubMed.
- J. Liu, L.-F. Yu, J. B. Eaton, B. Caldarone, K. Cavino, C. Ruiz, M. Terry, A. Fedolak, D. Wang, A. Ghavami, D. A. Lowe, D. B. Runner, R. J. Lukas and A. P. Kozikowski, J. Med. Chem., 2011, 54, 7280–7288 CrossRef CAS PubMed.
- P. Cali, L. Nærum, S. Mukhija and A. Hjelmencrantz, Bioorg. Med. Chem. Lett., 2004, 14, 5997–6000 CrossRef CAS PubMed.
- G. Daidone, D. Raffa, B. Maggio, F. lescia, V. M. C. Cutuli, N. G. Mangano and A. Caruso, Arch. Pharm., 1999, 332, 50–54 CrossRef CAS PubMed.
- M. Tanaka, T. Haino, K. Ideta, K. Kubo, A. Mori and Y. Fukazawa, Tetrahedron, 2007, 63, 652–665 CrossRef CAS.
- T. Morita, S. Fuse and H. Nakamura, Angew. Chem., Int. Ed., 2016, 55, 13580–13584 CrossRef CAS PubMed.
- C. Kesornpun, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Angew. Chem., Int. Ed., 2016, 55, 3997–4001 CrossRef CAS PubMed.
- G. R. Kumar, Y. K. Kumar and M. S. Reddy, Chem. Commun., 2016, 52, 6589–6592 RSC.
- S. U. Dighe, S. Mukhopadhyay, S. Kolle, S. Kanojiya and S. Batra, Angew. Chem., Int. Ed., 2015, 54, 10926–10930 CrossRef CAS PubMed.
- S. Pusch, D. Kowalczyk and T. Opatz, J. Org. Chem., 2016, 81, 4170–4178 CrossRef CAS PubMed.
- R. D. Rosa, H. D. M. Moraes, L. A. Zimmermann, E. P. Schenkel, M. Steindel and L. S. C. Bernardes, Eur. J. Med. Chem., 2017, 128, 25–35 CrossRef PubMed.
- R. Harigae, K. Moriyama and H. Togo, J. Org. Chem., 2014, 79, 2049–2058 CrossRef CAS PubMed.
- A. Yoshimura, K. R. Middleton, A. D. Todora, B. J. Kastern, S. R. Koski, A. V. Maskaev and V. V. Zhdankin, Org. Lett., 2013, 15, 4010–4013 CrossRef CAS PubMed.
- A. M. Jawalekar, E. Reubsaet, F. P. J. T. Rutjes and F. L. Van Delft, Chem. Commun., 2011, 47, 3198–3200 RSC.
- A. Speranca, B. Godoi and G. Zeni, J. Org. Chem., 2013, 78, 1630–1637 CrossRef CAS PubMed.
- C. Praveen, A. Kalyanasundaram and P. T. Perumal, Synlett, 2010, 777–781 CAS.
- S. Tang, J. He, Y. Sun, L. He and X. She, Org. Lett., 2009, 11, 3982–3985 CrossRef CAS PubMed.
- J. P. Waldo and R. C. Larock, J. Org. Chem., 2007, 72, 9643–9647 CrossRef CAS PubMed.
- X. Zhu, Y. F. Wang, W. Ren, F. L. Zhang and S. Chiba, Org. Lett., 2013, 15, 3214–3217 CrossRef CAS PubMed.
- K. V. G. C. Sekhar, T. V. N. V. T. Sasank, H. N. Nagesh, N. Suresh, K. M. Naidu and A. Suresh, Chin. Chem. Lett., 2013, 24, 1045–1048 CrossRef.
- V. G. Desai, S. R. Naik and K. L. Dhumaskar, Synth. Commun., 2014, 44, 1453–1460 CrossRef CAS.
- V. G. Desai and S. G. Tilve, Synth. Commun., 1999, 29, 3017–3020 CrossRef CAS.
- R. F. Kurangi, R. Kawthankar, S. Sawal, V. G. Desai and S. G. Tilve, Synth. Commun., 2007, 37, 585–587 CrossRef CAS.
- K. Maeda, T. Hosokawa, S. I. Murahashi and I. Moritani, Tetrahedron Lett., 1973, 14, 5075–5076 CrossRef.
- S. Guin, T. Ghosh, S. K. Rout, A. Banerjee and B. K. Patel, Org. Lett., 2011, 13, 5976–5979 CrossRef CAS PubMed.
- Y. Sun, A. Abdukader, D. Lu, H. Zhang and C. Liu, Green Chem., 2017, 19, 1255–1258 RSC.
- T. Sakakibara, T. Kume and T. Hase, Chem. Express, 1989, 4, 85–88 CAS.
- S. Ueda and H. Nagasawa, J. Org. Chem., 2009, 74, 4272–4277 CrossRef CAS PubMed.
- L. Zhang, G. Y. Ang and S. Chiba, Org. Lett., 2010, 12, 3682–3685 CrossRef CAS PubMed.
- J. J. Neumann, M. Suri and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7790–7794 CrossRef CAS PubMed.
- X. Li, L. Huang, H. Chen, W. Wu, H. Huang and H. Jiang, Chem. Sci., 2012, 3, 3463–3467 RSC.
- S. Ueda and H. Nagasawa, J. Am. Chem. Soc., 2009, 131, 15080–15081 CrossRef CAS PubMed.
- M. M. Guru, M. A. Ali and T. Punniyamurthy, J. Org. Chem., 2011, 76, 5295–5308 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11436b |
| This journal is © The Royal Society of Chemistry 2017 |