
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, antioxidant activity, and density functional theory study of catechin derivatives†
Jing Wang‡
a,
Han Tang‡ a,
Bo Houc,
Pan Zhanga,
Qi Wanga,
Bang-Lei Zhanga,
Ye-Wei Huanga,
Ya Wanga,
Ze-Min Xianga,
Cheng-Ting Zi*a,
Xuan-Jun Wang*ab and
Jun Sheng*ab
a,
Bo Houc,
Pan Zhanga,
Qi Wanga,
Bang-Lei Zhanga,
Ye-Wei Huanga,
Ya Wanga,
Ze-Min Xianga,
Cheng-Ting Zi*a,
Xuan-Jun Wang*ab and
Jun Sheng*ab
aKey Laboratory of Pu-er Tea Science, Ministry of Education, Pu-er Tea Academy, Yunnan Agricultural University, Kunming 650201, PR China. E-mail: zichengting@126.com; wangxuanjun@gmail.com; shengj@ynau.edu.cn; Fax: +86-871-6522-6711; Tel: +86-159-1257-9655
bState Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Kunming 650201, PR China
cState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, PR China
First published on 24th November 2017
Abstract
Catechin derivatives were synthesized, and their structures were characterized by 1H-NMR, 13C-NMR, and mass spectrometry. The target compounds were evaluated for their antioxidant activities. Compound 2 showed the highest antioxidant activity, with an IC50 value of 136.637 μM, whereas methylated derivatives showed weak activity. Density functional theory (DFT) studies were carried out at the B3LYP/6-311++G (d, p) level of theory. According to the geometries, molecular electrostatic potential (MEP), bond dissociation enthalpy (EDE), the HOMO and LUMO, and reactivity indices (η, μ, ω, ω+, and ω−), we predicted the free radical scavenging capacity of catechins and their derivatives from their structures. We also found that the B-ring of catechins is a stronger electron donor than the A- or D-ring, and that there is a good relationship between the bond dissociation enthalpies (BDEs). These theoretical results will be helpful in the development of new or modified antioxidant compounds.
Reactive oxygen species (ROS) are continuously produced as byproducts of mitochondrial electron transport during cellular respiration in the body. ROS include the superoxide anion (O2−), hydrogen peroxide (H2O2), the hydroxyl radical (HO˙) and singlet oxygen (1O2).1 These species are unstable and react readily with other molecules to achieve stability. “Oxidative stress” is the pro-oxidative state when the ROS level exceeds the capability of defense mechanisms. This can pose a threat to cells, causing damage to DNA, lipids, and proteins, and potentially leading to conditions such as cell ageing, cardiovascular diseases and cancerous cell growth.2,3 Antioxidants provide protection against oxidative damage by scavenging free radicals and reducing ROS. For this reason, many compounds have been widely studied for their antioxidant activities, using various methodologies.4,5
Catechins are plant polyphenolic compounds. The most important and rich natural source of catechins has been identified in green tea (Camellia sinensis Theaceae),6,7 which is one of the most widely consumed beverages in the world. The major green tea catechins are (−)-epicatechin (EC), (−)-epicatechin-3-gallate (ECG), (−)-epigallocatechin (EGC), (−)-epigallocatechin-3-gallate (EGCG) (Fig. 1), which is the most important active compound. These are responsible for the beneficial effects of green tea in the prevention or treatment of various conditions such as cancer, heart diseases, diabetes, and neurodegenerative diseases. These active compounds have also been reported to possess antioxidant, anti-inflammatory, anti-microbial and antiviral effects.8,9
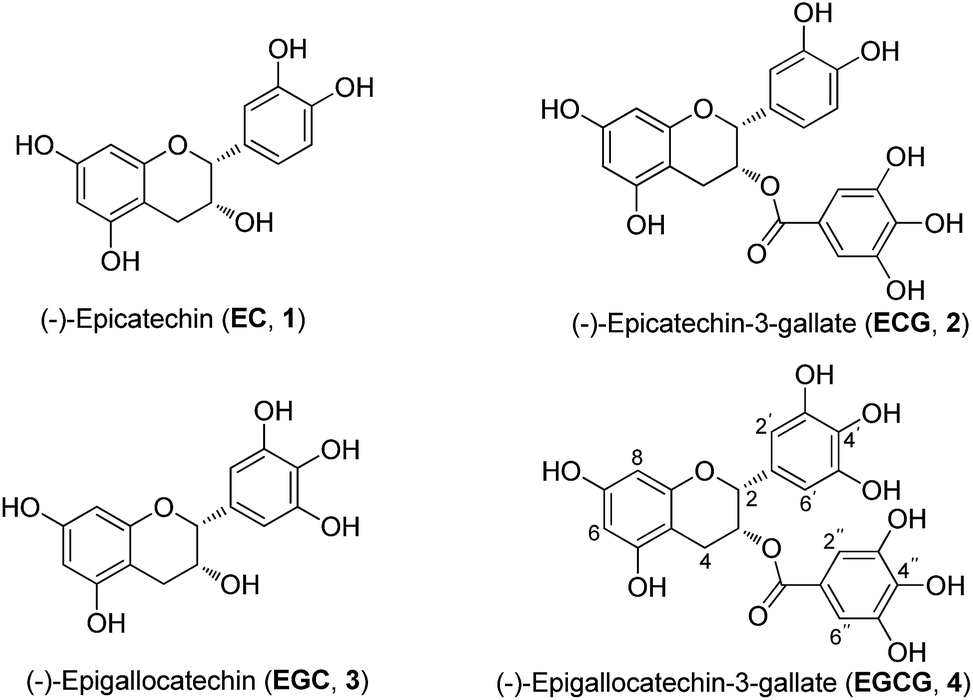 | ||
| Fig. 1 Chemical structures of catechins. | ||
However, catechins were also known to have negative effects such as pro-oxidative, cytotoxic, and phytotoxic activities, and attempts to use catechins in the treatment of human diseases have been mostly unsuccessful.9–12 It is well known that structural features, namely the number of galloyl and hydroxyl (OH) groups in catechin molecules, play an important part in their biological activities, particularly their antioxidant properties.13,14
There have been very few structure–function studies of catechins with respect to their molecular mechanisms of action. Guo et al.15 reported that the free radical scavenging activity of EGCG was stronger than that of its non-galloylated counterparts (that do not possess a galloyl D-ring, EGC). However, Valcic et al.16 provided convincing evidence that the gallate B-ring of EGCG, and not the galloyl D-ring, is the primary site for antioxidant reactions. Hence, further studies of catechins are required to give clearer insights into the structure–function relationship of the various phenolic hydroxyl groups in the A-, B-, and D-rings of catechins, and their chemopreventive mechanism.
The aim of this study was to synthesize a series of catechin derivatives, determine their radical scavenging capacities, reveal the character of interaction and evaluate the structure–activity relationships within the group of catechins i.e. the effect of the galloyl moiety on the C-ring, the number of hydroxyl groups in the B-ring, and the spatial arrangement of the substituents on the C-ring. The synthesized compounds were characterized by 1H-NMR, 13C-NMR, and mass spectrometry (MS) analyses. The antioxidant activities were experimentally verified using 2,2-diphenyl-1-picrylhydrazyl (DPPH) assays, and further rationalized based on computational studies using density functional theory (DFT).
To obtain catechin derivatives 5–8, EC (1), ECG (2), EGC (3) and EGCG (4), respectively were permethylated with dimethylsulfate and potassium carbonate in anhydrous acetone under reflux (overall yield 90%).17–20 Trimethylated EGCG (3M-EGCG, 11) and pentamethylated EGCG (5M-EGCG, 12) were prepared starting from EGCG (Scheme 1), as previously described.21,22
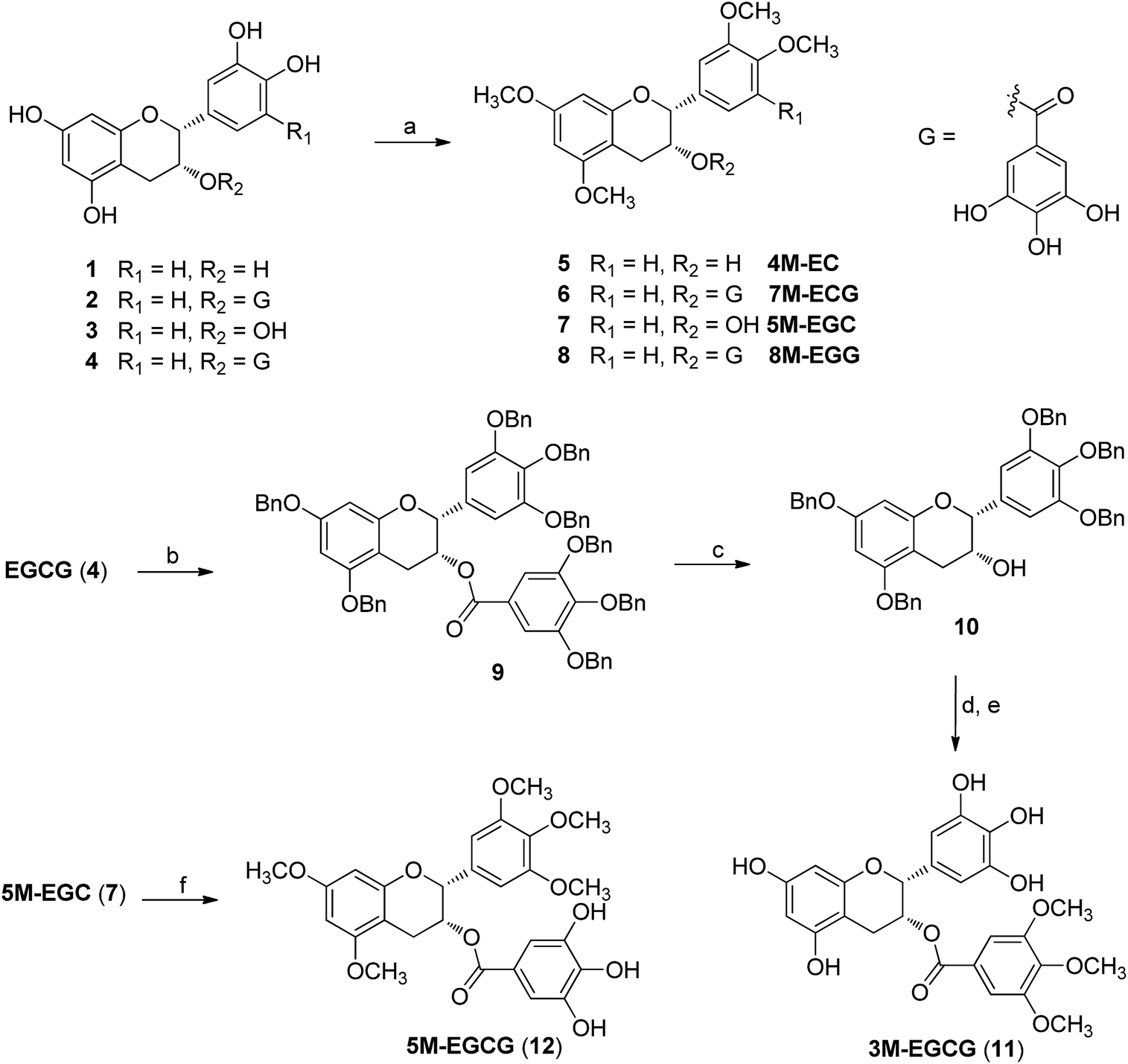 | ||
| Scheme 1 Synthesis of catechin derivatives: (a) Me2SO4, K2CO3, acetone, reflux, 2 h, ∼98%; (b) BnBr, NaH, DMF, 24 h, 96%; (c) NaOH, CH3OH, 4 h, 87%; (d) 3,4,5-trimethoxybenzoic acid, DMAP, DCC, CH2Cl2, 24 h, 51%; (e) Pd–C, H2, CH3OH, 4 h, 65%; (f) gallic acid, DMAP, DCC, CH2Cl2, 24 h, 34%. | ||
Molecular geometries obtained through theoretical methods are useful to explain the three-dimensional structures of compounds. Optimization of all compounds was carried out at the B3LYP/6-311++G (d, p) level of DFT.23 Optimized geometries of compounds 1–4, 11, and 12 are shown in Fig. 2, and important bonds lengths are listed in Table 1. Among all the compounds, only compound 1 has an X-ray structure reported in the literature.24 The differences between X-ray and calculated bond lengths were found in the range 0.001–0.017 Å for 1, and the maximum deviation for C(2)–C(11) in 1, was 0.017 Å.
 | ||
| Fig. 2 The optimized geometries of 1–4, 11, and 12 at the B3LYP/6-311++G (d, p) level of DFT. | ||
| Bond | 1a | 1 | 2 | 3 | 4 | 11 | 12 |
|---|---|---|---|---|---|---|---|
| a Experimental data of EC from ref. 24. | |||||||
| C(2)–C(3) | 1.599 | 1.547 | 1.541 | 154.6 | 1.540 | 1.545 | 1.549 |
| C(2)–C(11) | 1.506 | 1.526 | 1.519 | 152.8 | 1.520 | 1.522 | 1.523 |
| C(3)–C(4) | 1.519 | 1.520 | 1.523 | 1.521 | 1.523 | 1.520 | 1.517 |
| C(4)–C(10) | 1.498 | 1.512 | 1.511 | 1.513 | 1.511 | 1.511 | 1.509 |
| C(5)–C(6) | 1.386 | 1.392 | 1.392 | 1.393 | 1.393 | 1.393 | 1.398 |
| C(5)–C(10) | 1.405 | 1.402 | 1.403 | 1.402 | 1.402 | 1.402 | 1.402 |
| C(6)–C(7) | 1.386 | 1.393 | 1.394 | 1.394 | 1.394 | 1.394 | 1.401 |
| C(7)–C(8) | 1.389 | 1.391 | 1.391 | 1.391 | 1.391 | 1.391 | 1.391 |
| C(8)–C(9) | 1.387 | 1.393 | 1.393 | 1.393 | 1.394 | 1.394 | 1.394 |
| C(9)–C(10) | 1.396 | 1.402 | 1.403 | 1.402 | 1.403 | 1.403 | 1.399 |
| C(11)–C(12) | 1.394 | 1.399 | 1.400 | 1.399 | 1.399 | 1.398 | 1.399 |
| C(11)–C(16) | 1.389 | 1.399 | 1.397 | 1.399 | 1.396 | 1.397 | 1.394 |
| C(12)–C(13) | 1.378 | 1.387 | 1.388 | 1.390 | 1.391 | 1.391 | 1.396 |
| C(13)–C(14) | 1.391 | 1.399 | 1.400 | 1.391 | 1.392 | 1.391 | 1.407 |
| C(14)–C(15) | 1.381 | 1.389 | 1.389 | 1.391 | 1.395 | 1.395 | 1.399 |
| C(15)–C(16) | 1.385 | 1.393 | 1.393 | 1.393 | 1.392 | 1.392 | 1.395 |
| O(1)–C(2) | 1.445 | 1.439 | 1.438 | 1.439 | 1.438 | 1.437 | 1.439 |
| O(1)–C(9) | 1.386 | 1.375 | 1.371 | 1.376 | 1.371 | 1.371 | 1.373 |
| O(2)–C(3) | 1.429 | 1.425 | 1.440 | 1.425 | 1.439 | 1.438 | 1.440 |
| O(3)–C(5) | 1.366 | 1.368 | 1.368 | 1.369 | 1.369 | 1.369 | 1.367 |
| O(4)–C(7) | 1.371 | 1.369 | 1.368 | 1.369 | 1.368 | 1.368 | 1.366 |
| O(5)–C(13) | 1.376 | 1.378 | 1.378 | 1.378 | 1.377 | 1.377 | 1.366 |
| O(6)–C(14) | 1.373 | 1.364 | 1.363 | 1.372 | 1.372 | 1.372 | 1.372 |
Molecular electrostatic potential (MEP) mapping through a computer-aided method is a very useful approach to explore the reactivity of compounds. The nucleophilic and electrophilic sites in any compound are expressed in term of different color codes; a deep red color indicates an electron-rich site, whereas deep blue indicates an electron-deficient site. MEP mapping results for compounds 1–4, 11, and 12 are shown in Fig. 3. It is clear that electronic density in the compounds is concentrated in the oxygen of the 7-OH in the A-ring, which is directly attached to the benzene ring, and that protons attached to the A-ring are electron-rich sites. A similar trend was observed for 2, 4, and 11, with O-dihydroxy substitution in the B-ring, but 12 had some extra localization of electronic density on methoxy groups in B-ring. In 2, 4, and 12, the electronic density was more dispersed and was concentrated on the A-ring and B-ring, but not on the oxygen of 5′′-OH in D-ring for 2 and 4, or on 4′′-OH in D-ring for 12, while the protons attached to the D-ring were electron-deficient sites.
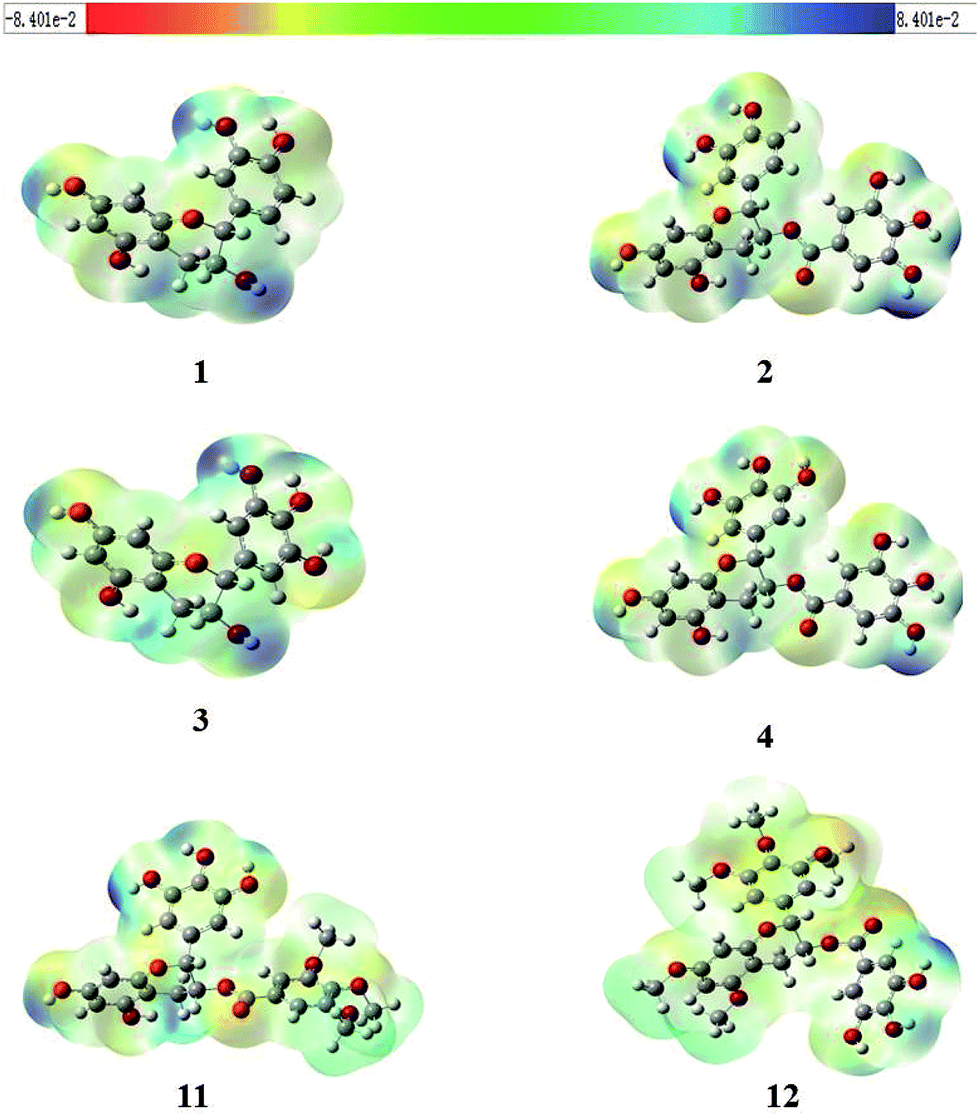 | ||
| Fig. 3 Molecular electrostatic potential (MEP) graphics of compounds 1–4, 11, and 12 (red = intense electron-rich site, yellow = medium electron-rich site, blue = electron-deficient site, light green = almost neutral site, grey = white = zero potential). | ||
Bond dissociation enthalpies (BDEs) have been calculated at the B3LYP theory level for the respective hydrogen atom elimination paths. Lower BDEs are associated with higher antioxidant activity.25 Table 2 lists the calculated BDE values for the studied compounds. As shown in Table 2, the BDE values are lower in the B-ring (3′-OH and 4′-OH) than A-ring (5-OH and 7-OH) and D-ring (3′′-OH, 4′′-OH, and 5′′-OH), except in the case of compound 12. In compound 2, the BDE (379.73 kJ mol−1) of the 3′-OH in B-ring is the lowest, and the BDE (414.89 kJ mol−1) of 7-OH in the A-ring is the highest. A similar trend was observed for compounds 1, 3, and 4, with the lowest BDE values found for 3′-OH (381.04 kJ mol−1, 385.80 kJ mol−1, and 384.98 kJ mol−1, respectively), and the highest BDE values found for 7-OH (406.50 kJ mol−1, 413.06 kJ mol−1, and 416.47 kJ mol−1, respectively). The lowest BDE of compound 11 was 381.83 kJ mol−1 for 4′-OH, while the lowest BDE of compound 12 was 390.48 kJ mol−1 for 5′′-OH. The BDE in the B-ring increased owing to the involvement of hydrogen in an intramolecular hydrogen bond with a phenolic hydroxyl, which occurs when the hydrogenation reaction produces free radicals.
| Compds | BDEsa (kJ mol−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| C5(O–H) | C7(O–H) | C3′(O–H) | C4′(O–H) | C5′(O–H) | C3′′(O–H) | C4′′(O–H) | C5′′(O–H) | |
| a 1 a.u. = 627.5095 kcal mol−1; 1 kcal mol−1 = 4.182 kJ mol−1. | ||||||||
| 1 | 400.46 | 406.50 | 381.04 | 409.12 | — | — | — | — |
| 2 | 398.10 | 414.89 | 379.73 | 408.07 | — | 405.97 | 392.06 | 389.44 |
| 3 | 400.98 | 413.06 | 385.80 | 385.50 | 407.02 | — | — | — |
| 4 | 399.41 | 416.47 | 384.98 | 384.98 | 413.58 | 407.28 | 393.11 | 390.95 |
| 11 | 395.74 | 404.66 | 382.35 | 381.83 | 408.86 | — | — | — |
| 12 | — | — | — | — | — | 390.49 | 390.75 | 390.48 |
Frontier molecular orbital analysis by computational methods is a useful way to understand the reactivity and electronic transitions within molecules.26 Frontier orbitals (HOMO and LUMO) are the main participants in electronic transitions, and their energy gap depicts the reactivity.27 The HOMO and LUMO of compounds 1–4, 11, and 12 were explored at the B3LYP/6-311++G (d, p) level of DFT.
The distribution pattern of frontier molecular orbitals (HOMOs/LUMOs along with corresponding energies) of compounds 1–4, 11, and 12 at the ground states are shown in Fig. 4. The π-cloud in the LUMOs of compounds 1 and 3 is distributed on the A-and C-rings, but the π-cloud in HOMOs of compounds 1 is distributed on the B-ring, while that of 3 is distributed on the entire skeleton (A-, B-, and C-rings). The π-cloud in the HOMOs of compounds 2, 4, 11, and 12 is distributed on the A-, B-, and C-rings, and the π-cloud in the LUMOs of compounds 2, 4, 11, and 12 is distributed on the D-ring. Introducing different groups on the benzene ring does not have much effect on the electronic cloud. The free radicals scavenging reaction of catechins is an electronic process, and the results show that the free radical reaction occurred primarily on the B-ring.
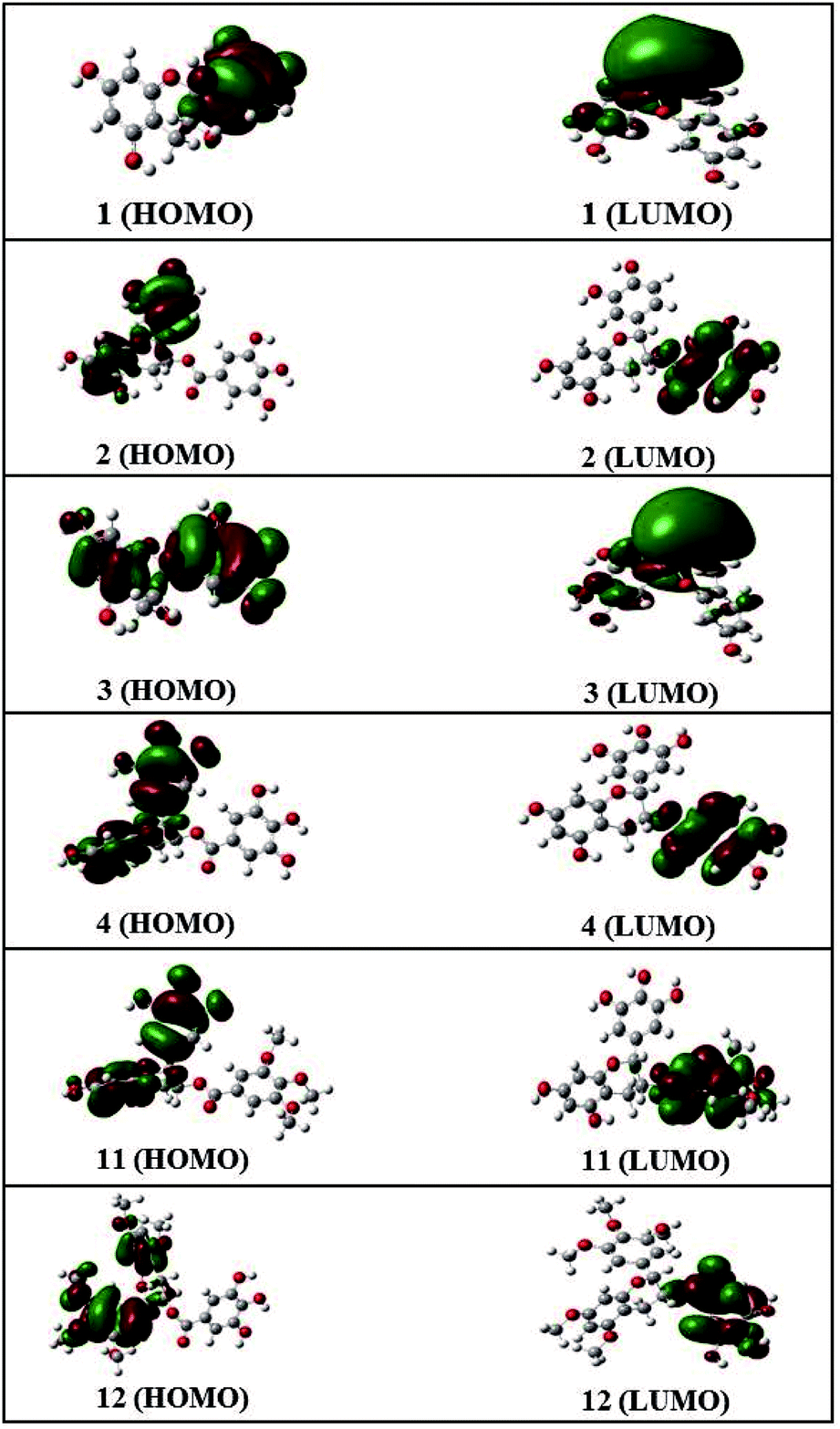 | ||
| Fig. 4 HOMO and LUMO orbitals of compounds 1–4, 11, and 12. | ||
Detailed HOMO and LUMO energies of compounds 1–4, 11, and 12, along with their gaps, are listed in Table 3. Among all compounds, 2 showed the lowest HOMO–LUMO energy gap, i.e., 0.162 eV, while 12 showed the largest energy gap (0.206 eV).
Reactivity indices such as chemical hardness (η), electrophilicity (ω), electronic chemical potential (μ), electron acceptor power (ω+), and electron donor power (ω−) are excellent tools to describe the hardness, reactivity and stability of compounds.28–32 The reactivity indices of compounds 1–4, 11, and 12 were calculated and are given in Table 4. It is clear that among all compounds, 2 has the lowest value of η, i.e., 0.081 eV, whereas 12 has the highest value (0.103 eV). These findings are consistent with the LUMO–HOMO band gaps of all compounds. Compound 2 has the highest electronic chemical potential (μ) value (0.142 eV) among compounds 1–4, 11, and 12, while 12 has the lowest chemical potential value (0.113 eV). The results indicate that 12 has the lowest electrophilicity index value and is nucleophilic in nature, whereas 2 has the highest value ω, i.e., 0.124 eV, and is strongly electrophilic in nature. In addition, among the set of compounds, 2 has the highest electron acceptor power (ω+) and electron donor power (ω−) values (0.064 eV and 0.206 eV, respectively), while 12 has the lowest electron acceptor power and electron donor power values (0.019 eV and 0.131 eV, respectively).
| Compds | Reactivity indicesa (eV) | ||||
|---|---|---|---|---|---|
| ηb | μc | ωd | ω+e | ω−e | |
| a 1 a.u. = 27.21165 eV; 1 eV = 1.60219 × 10−19 J.b η = (EHOMO − ELUMO)/2.c μ = (EHOMO + ELUMO)/2.d ω = μ2/2η.e ω+ = (I + 3A)2/16(I − A), ω− = (3I + A)2/16(I − A), I ≈ −EHOMO, A ≈ −ELUMO. | |||||
| 1 | 0.098 | 0.124 | 0.078 | 0.029 | 0.153 |
| 2 | 0.081 | 0.142 | 0.124 | 0.064 | 0.206 |
| 3 | 0.099 | 0.123 | 0.076 | 0.027 | 0.150 |
| 4 | 0.082 | 0.141 | 0.121 | 0.061 | 0.202 |
| 11 | 0.100 | 0.124 | 0.077 | 0.028 | 0.153 |
| 12 | 0.103 | 0.113 | 0.062 | 0.019 | 0.131 |
Compounds 1–8, 11, and 12 were tested against DPPH at different concentrations, and the results were compared with those of a standard natural antioxidant, ascorbic acid. All results are presented as IC50 values in Table 1. Almost all the permethylation derivatives (compounds 5–8) showed no antioxidant activity, and the highest scavenger activity was observed in compound 2, whose IC50 value was 136.637 μM. Among all the synthesized compounds, 11 and 12 with IC50 1254.901 μM and 4119.869 μM, respectively, showed antioxidant activity (Table 5).
| Compounds | DPPH IC50b (μM) |
|---|---|
| a Results are expressed as a mean ± standard deviation (n = 3).b DHHP radical scavenging activities are expressed as IC50 concentrations of the compounds (μM) required to inhibit 50% of the radicals; NA: did not reach 50% inhibition of the DPPH radicals at concentrations used in this study; VC: vitamin C. | |
| 1 | 251.028 |
| 2 | 136.637 |
| 3 | 277.005 |
| 4 | 155.646 |
| 5 | NA |
| 6 | NA |
| 7 | NA |
| 8 | NA |
| 11 | 1254.901 |
| 12 | 4119.869 |
| VC | 49.892 |
The radical scavenging capacities of catechin derivatives were interacted with structure–activity relationships, which the B-ring of catechins is a stronger electron donor than other rings. In addition, the acid–base equilibrium may affect the antioxidant activity of these compounds. Participations of hydroxyl group in acid–base equilibrium is phenolic groups, including dipole–dipole interaction and hydrogen bonging interaction could be simultaneously offered stability on the reactive oxygen species.
Conclusions
In conclusion, this study reports the synthesis of catechin derivatives, starting from green tea catechins EC, ECG, EGC, and EGCG. All compounds were screened for antioxidant activity: the highest antioxidant activity was observed in compound 2, whose IC50 value was 136.637 μM; permethylated derivatives (compounds 5–8) showed no antioxidant activity, partially methylated derivatives (compounds 11 and 12) showed weak activity. A theoretical calculation based on DFT was performed to understand the antioxidant activity of catechins and their derivatives in relation to molecular structure. On the basis of the geometries, positive charge of hydrogen, MEP, EDE, the HOMO and LUMO, and reactivity indices (η, μ, ω, ω+, and ω−), we interpreted the free radical scavenging capacity of the catechins and their derivatives in terms of their structures. We also found that the B-ring is a stronger electron donor than the A- or D-ring, and that there is a good relationship between the BDEs. These theoretical results will be helpful in the future development of antioxidant compounds.Author contributions
Cheng-Ting Zi, Xuan-Jun Wang, and Jun Sheng conceived and designed the experiments. Han Tang and Pan Zhang synthesized the compounds. Jing Wang and Qi Wang measured antioxidant activity. Bo Hou and Bang-Lei Zhang did the theoretical calculations. Yei-Wei Huang, Ya Wang, and Ze-Min Xiang did the analysis of the data. Cheng-Ting Zi and Xuan-Jun Wang wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21602196); the Natural Science Foundation of Yunnan Province (No. 2017FD084); and Yunnan Agricultural University Natural Science Foundation for Young Scientists (No. 2015ZR08).Notes and references
- M. Roesslein, C. Hirsch, J. P. Kaiser, H. F. Krug and P. Wick, Int. J. Mol. Sci., 2013, 14, 24320 CrossRef PubMed.
- P. Sharma, A. B. Jha, R. S. Dubey and M. Pessarakli, J. Bot., 2012, 2012, 1 CrossRef.
- B. Halliwell, Reactive oxygen species and the central nervous system, in Free Radicals in the Brain, Springer-Verlag, Berlin, Heidelberg, Germany, 1992 Search PubMed.
- Z. Q. Liu and D. Wu, J. Phys. Org. Chem., 2009, 22, 308 CrossRef CAS.
- H. H. Hussain, G. Babic, T. Durst, J. S. Wright, M. Flueraru, A. Chichirau and L. L. Chepelev, J. Org. Chem., 2003, 68, 7023 CrossRef CAS PubMed.
- D. J. Zeeb, B. C. Nelson, K. Albert and J. J. Dalluge, Anal. Chem., 2000, 72, 5020 CrossRef CAS PubMed.
- I. E. Dreosti and D. Sc, Nutr. Rev., 1996, 54, S51 CrossRef CAS PubMed.
- P. Velayutham, A. Babu and D. M. Liu, Curr. Med. Chem., 2008, 15, 1840 CrossRef.
- C. Cabrera, R. Artacho and R. Giménez, J. Am. Coll. Nutr., 2006, 25, 79 CrossRef CAS PubMed.
- M. Kaku and K. Nakagawa, J. Health Sci., 2009, 55, 441 CrossRef CAS.
- T. L. Weir, H. P. Bais and J. M. Vivanco, J. Chem. Ecol., 2003, 29, 2397 CrossRef CAS PubMed.
- T. Ishii, T. Mori, T. Ichikawa, M. Kaku, K. Kusaka, Y. Uekusa, M. Akagawa, Y. Aihara, T. Furuta, T. Wakimoto, T. Kan and T. Nakayama, Bioorg. Med. Chem., 2010, 18, 4892 CrossRef CAS PubMed.
- C. A. Rice-Evans, N. J. Miller and G. Paganga, Trends Plant Sci., 1997, 2, 152 CrossRef.
- P. G. Pietta, J. Nat. Prod., 2000, 63, 1035 CrossRef CAS.
- Q. Guo, B. Zhao, S. Shen, J. Hou, J. Hu and W. Xin, Biochim. Biophys. Acta, 1999, 1427, 13 CrossRef CAS.
- S. Valcic, A. Muders, N. E. Jacobsen and D. C. Liebler, Chem. Res. Toxicol., 1999, 12, 382 CrossRef CAS PubMed.
- R. Bernini, F. Crisante, P. Gentili, F. Morana, M. Pierini and M. Piras, J. Org. Chem., 2011, 76, 820 CrossRef CAS PubMed.
- C. P. Bahl, R. Murari, M. P. Parthasarathy and T. R. Seshadri, Indian J. Chem., 1974, 12, 1038 CAS.
- A. Mori, C. Nishino, N. Enoki and S. Tawata, Phytochemistry, 1988, 27, 1017 CrossRef CAS.
- S. F. Lin, Y. H. Lin, M. Lin, Y. F. Kao, R. W. Wang, L. W. Teng, S. H. Chuang, J. M. Chang, T. T. Yuan, K. C. Fu, K. P. Huang, Y. S. Lee, C. C. Chiang, S. C. Yang, C. L. Lai, C. B. Liao, P. Chen, Y. S. Lin, K. T. Lai, H. J. Huang, J. Y. Yang, C. W. Liu, W. Y. Wei, C. K. Chen, R. A. Hiipakka, S. Liao and J. J. Huang, Eur. J. Med. Chem., 2010, 45, 6068 CrossRef CAS PubMed.
- N. T. Zaveri, Org. Lett., 2001, 3, 843 CrossRef CAS PubMed.
- C. Auger, J. H. Kim, P. Chabert, M. Chaabi, E. Anselm, X. Lanciaux, A. Lobstein and V. B. Schini-Kerth, Biochem. Biophys. Res. Commun., 2010, 393, 162 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- F. R. Fronczek, G. Gannuch and W. L. Mattice, J. Chem. Soc., Perkin Trans. 2, 1984, 10, 1611 RSC.
- M. Lucarini, P. Pedrielli, G. F. Pedulli, S. Cabiddu and C. Fattuoni, J. Org. Chem., 1996, 61, 9259 CrossRef CAS.
- M. N. Arshad, T. Mahmood, A. F. Khan, M. Zia-Ur-Rehman, A. M. Asiri, I. U. Khan, K. Ayub, A. Mukhtar and M. T. Saeed, Chin. J. Struct. Chem., 2015, 34, 15 Search PubMed.
- M. N. Arshad, A. M. Asiri, K. A. Alamry, T. Mahmood, M. A. Gilani, K. Ayub and A. S. Birinji, Spectrochim. Acta, Part A, 2015, 142, 364 CrossRef CAS PubMed.
- C. A. Mebi, J. Chem. Sci., 2011, 123, 727 CrossRef CAS.
- J. R. Johns and J. A. Platts, Org. Biomol. Chem., 2014, 12, 7820 CAS.
- J. L. Gázquez, A. Cedillo and A. Vela, J. Phys. Chem., 2007, 111, 1966 CrossRef PubMed.
- R. G. Parr and L. J. Bartolotti, J. Am. Chem. Soc., 1982, 104, 3801 CrossRef CAS.
- Y. K. Zhang and W. T. Yang, Theor. Chem. Acc., 2000, 103, 346 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR and MS spectra, computational study, antioxidant activity assay. See DOI: 10.1039/c7ra11496f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |