
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of α-sulfonyloxyketones via iodobenzene diacetate (PIDA)-mediated oxysulfonyloxylation of alkynes with sulfonic acids†
Yang Zhang*,
Hua Tan and
Weibing Liu *
*
College of Chemical Engineering, Guangdong University of Petrochemical Technology, 2 Guandu Road, Maoming 525000, P. R. China. E-mail: 450692624@qq.com; lwb409@gdupt.edu.cn; Fax: +86-668-2923575; Tel: +86-668-2923956
First published on 24th November 2017
Abstract
A simple yet powerful method to synthesize a variety of α-sulfonyloxyketones has been developed. This novel method can be applied for the direct oxysulfonyloxylation of alkynes with sulfonic acids to access a variety of α-sulfonyloxyketones. Compared to the reported methods for the application of PIDA, this study expands its application scope and uses it not only as the oxidant but also as the carrier of “O” to form the carbonyl group in the products. In addition, under the established conditions, this methodology not only exhibits a broad substrate scope but also demonstrates exclusive regioselectivity with substrates of 1,2-disubstituted internal alkynes.
In recent decades, due to their low toxicity, high stability, environmental-friendliness, easy availability, clean transformation and very useful oxidizing properties, hypervalent iodine compounds have attracted great interest from the chemical synthesis community1 and emerged as a useful alternative for a wide variety of organic transformations.2 To date, the existing studies have mainly focused on the oxidation reactions which use iodine(III) and iodine(V) compounds as substitutes to replace highly toxic heavy-metal-based oxidants.3 Recently, the application scope of hypervalent iodine reagents has been expanded, and a dual role has emerged for iodine(III) and iodine(V) compounds in organic oxidation transformations which employ them not only as organic oxidants, but also as carriers of some organic functional groups, such as –O2CR,4 –CF3,5 –OTs,6 –NR2,7 –C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–R,8 –N3,9 and –Ar,10 in the “atom-transfer” oxidative coupling reactions that lead to the formation of new C–C and C–hetero bonds. However, reports involving the use of hypervalent iodine reagents as the “O” source of a carbonyl group are very rare to date. Since our protracted interest in the community of hypervalent iodine reagents involves oxidative reactions,11 in this report we will describe one such process using PIDA as the oxidant and the “O” source for carbonyl group, along with alkynes and sulfonic acids to access α-sulfonyloxyketones in a one-step procedure (Scheme 1). α-Sulfonyloxyketones are important organic intermediates12 for α-azidation of ketones13 and the synthesis of a variety of heterocyclic compounds.14 To date, a variety of synthetic approaches have been reported for their synthesis, however, they have mainly been focused on the oxidative transformations between ketones and sulfonic acids.15 The present study describes the synthesis of α-sulfonyloxyketones via direct difunctionalization of alkynes with sulfonic acids in the presence of PIDA to produce α-sulfonyloxyketones, which is an approach that to the best of our knowledge has not been reported until now.
C–R,8 –N3,9 and –Ar,10 in the “atom-transfer” oxidative coupling reactions that lead to the formation of new C–C and C–hetero bonds. However, reports involving the use of hypervalent iodine reagents as the “O” source of a carbonyl group are very rare to date. Since our protracted interest in the community of hypervalent iodine reagents involves oxidative reactions,11 in this report we will describe one such process using PIDA as the oxidant and the “O” source for carbonyl group, along with alkynes and sulfonic acids to access α-sulfonyloxyketones in a one-step procedure (Scheme 1). α-Sulfonyloxyketones are important organic intermediates12 for α-azidation of ketones13 and the synthesis of a variety of heterocyclic compounds.14 To date, a variety of synthetic approaches have been reported for their synthesis, however, they have mainly been focused on the oxidative transformations between ketones and sulfonic acids.15 The present study describes the synthesis of α-sulfonyloxyketones via direct difunctionalization of alkynes with sulfonic acids in the presence of PIDA to produce α-sulfonyloxyketones, which is an approach that to the best of our knowledge has not been reported until now.
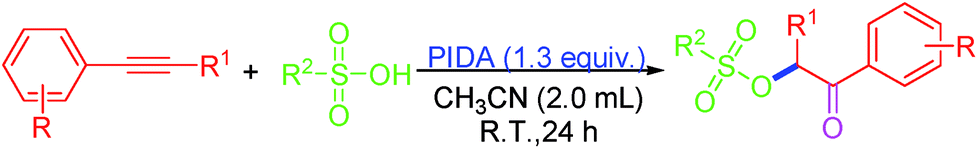 | ||
| Scheme 1 PIDA-mediated oxysulfonyloxylation of alkynes. | ||
This study was started by using the model substrates phenylacetylene (1a) and p-toluenesulfonic acid (PTSA) (2a) to probe the feasibility of this reaction (Table 1). Initially, the reaction was performed with 1 equiv. of PIDA in 1,2-dichloroethane (DCE) at room temperature for 12, 24 and 36 h. We were satisfied to find that the desired product 1-oxo-1-phenylethyl 4-methylbenzenesulfonate 3aa, was obtained with a 39%, 54% and 54% yield, respectively (entries 1–3). These moderate yields motivated us to improve the yield of the desired product 3aa. On switching from DCE to other solvents like toluene, dioxane, CH3CN, CH3OH and Et3N (entries 4–8), the best result was obtained in CH3CN (80% yield) (entry 6). Intriguingly, the reaction performed poorly when the reaction was carried out at 70 °C, and the yield of the product 3aa decreased to 57% (entry 9). In contrast, a slight increase in the yield was obtained by varying the equivalents of PIDA from 1.0 to 1.3 (entry 10).
|
|||
|---|---|---|---|
| Entry | Solvent | Time (h) | Yield%b |
| a Reaction conditions: 1a (1.0 mmol), 2a (1.0 mmol), PIDA (1.0 equiv.), solvent (2.0 mL).b GC yield.c Reaction temperature: 0 °C.d PIDA: 1.3 equivalent. | |||
| 1 | DCE | 12 | 39 |
| 2 | DCE | 24 | 54 |
| 3 | DCE | 36 | 54 |
| 4 | Toluene | 24 | 61 |
| 5 | Dioxane | 24 | 75 |
| 6 | CH3CN | 24 | 80 |
| 7 | CH3OH | 24 | 21 |
| 8 | Et3N | 24 | 27 |
| 9c | CH3CN | 24 | 57 |
| 10d | CH3CN | 24 | 85 |

The scope of the reaction was investigated by varying the substrates of 1 and 2 under the established conditions (Scheme 2). Initially, the reaction of a variety of terminal arylethynylenes bearing various substituents was investigated first. It was found that the electronic properties of the substituents on the phenyl ring did not affect the yield too much. According to the results of the yields, terminal arylethynylenes bearing electron-donating substituents (i.e., alkyl and methoxy) performed slightly better in this transformation than those bearing electron-withdrawing substituents (i.e., F−, Br− and Cl−), and produced the desired product in relatively high yields. For instance, terminal arylethynylenes bearing electron-donating substituents, such as Me (1b), Et (1d), propyl (1e), tBu (1f) and MeO (1j) produced the desired products 3ba, 3da, 3ea, 3fa and 3ja in good yields, while electron-withdrawing substituents, such as the F (1g) and Cl (1h) groups led to decreased yields. Also, we were disappointed to find that the position of a given substituent on the phenyl ring of terminal arylethynylenes did impact the yield significantly, and para-substituted products were more favorable than ortho- and meta-substituted ones. For example, 1-bromo-2-ethynylbenzene (1i) produced 3ia with the lowest yield (59%). To our satisfaction, this conversion proceeded smoothly with high regioselectivity when 1,2-disubstituted internal alkynes like methyl 3-phenylpropiolate (1k) and 1-(pent-1-ynyl)benzene (1l) were used as the substrates and produced their corresponding product (3ka and 3la) with excellent yield. In order to further expand the scope of this protocol, several sulfonic acid derivatives like methanesulfonic acid (2b), ethanesulfonic acid (2c), phenylsulfonic acid (2d) and naphthalene sulfonic acid (2e) were used as substrates under the established conditions. The experimental results confirmed that all these aliphatic sulfonic acids and arylsulfonic acids are excellent partner of PTSA (2a), and all the tested reactions proceeded smoothly and produced their corresponding product with excellent yield (3ab: 87%, 3ac: 85%, 3ad: 82% and 3ae: 81%).
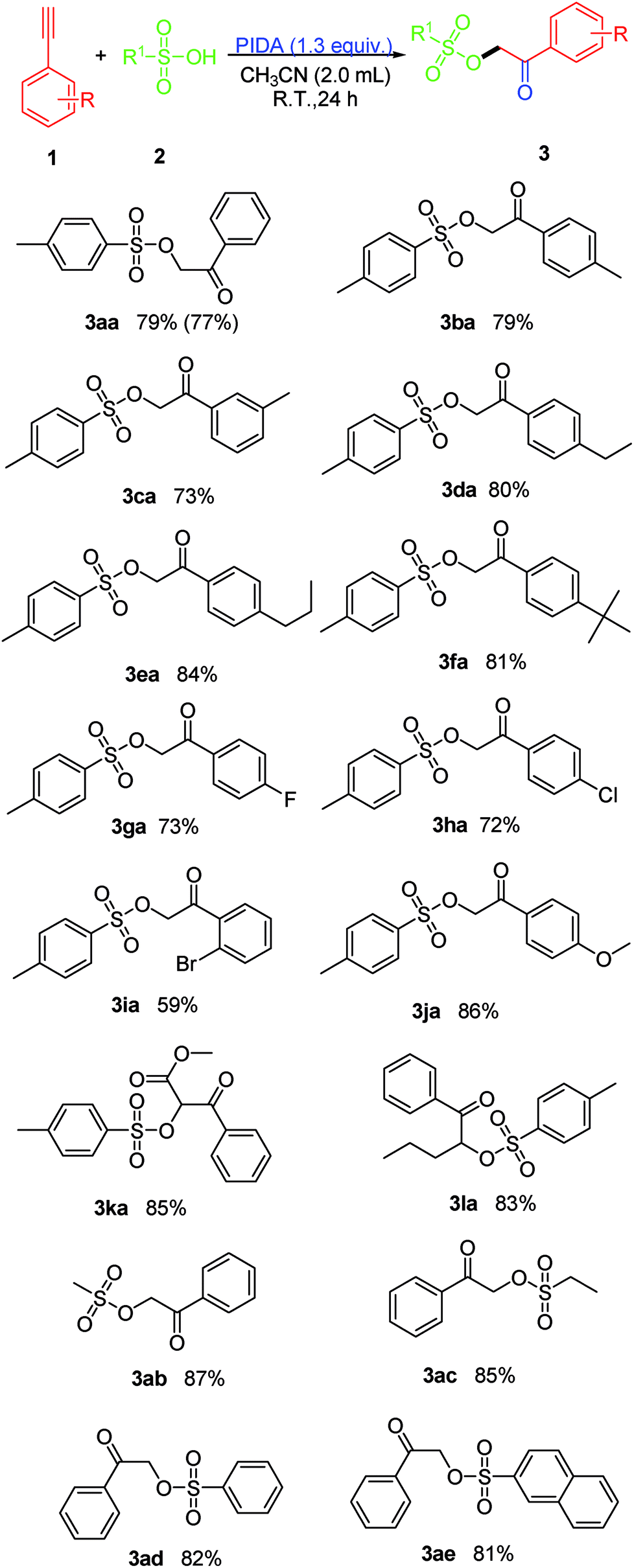 | ||
| Scheme 2 Exploring of the substrate scope (all reactions are carried out on a 2.0 mmol scale using CH3CN (2.0 mL) as the solvent and all the listed yields are isolated yield. The number in parentheses is the isolated yield of propiophenone 1a (10.0 mmol scale) after purification by column chromatography). | ||
To clarify the source of oxygen in the generated carbonyl group in this work, we conducted two control experiments (Scheme 3). Firstly, we noticed that the yield of 3aa was not affected when the reaction was conducted under nitrogen atmospheres, which ruling out the possibility of the involvement of atmospheric oxygen for this transformation. Besides, when the reaction was carried out in the presence of excess H218O, 3aa was obtained with a 79% yield without 18O-labeled in the product, thereby excluding the possibility of the “O” being derived from H2O in the reaction system. Taken together, we believe that the “O” in the carbonyl group in this work is derived from PIDA.
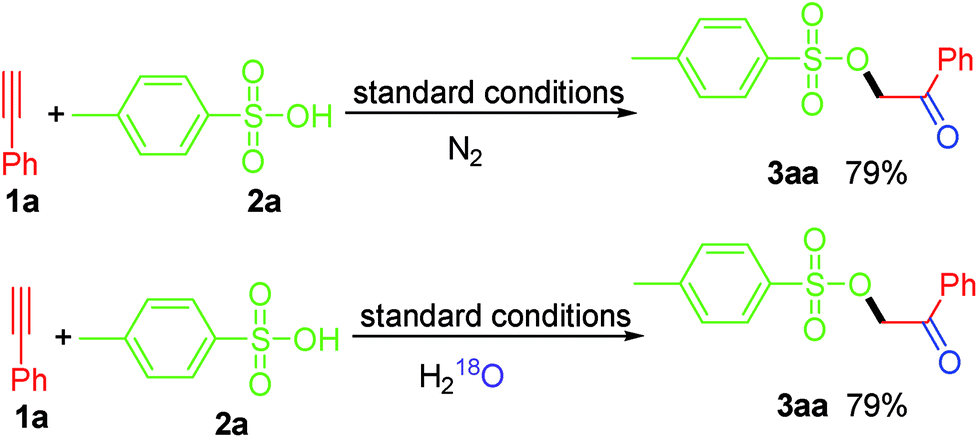 | ||
| Scheme 3 Control experiments. | ||
Based on the results of the experiments described above, a plausible mechanism is proposed, as shown in Scheme 4 and exemplified by the production of 3aa. The reaction begins with the formation of bridged iodonium ion 4 through the activation of 1a by PhI(OAc)2 together with the spontaneous dissociation of the acetoxy anion from the iodine center.7 The second step involves the direct nucleophilic oxysulfonyloxylation of 4 and yields intermediate 5. Subsequently, the reduction elimination of 5 through the release of PhI leads to the formation of intermediate 6 which undergoes hydrolysis to produce the enol 7.16 Finally, the tautomerization of enol 8 liberates the stable α-sulfonyloxyketone 3aa. Alternatively, the reaction maybe begins with the combination of 1a with PIDA to activate the C–C triple bond to give electrophilic intermediate 8, which then reacts with the nucleophilic acetate anion to generate intermediate 9.17 The release of one molecule of acetate anion from intermediate 9 yields iodonium ylide 10. The following hydrolysis of the ylide 10 gives ketone intermediate 11.18 Then the nucleophilic attack of sulphonate anion on the electrophilic iodonium cation affords the final product α-sulfonyloxyketone 3aa.
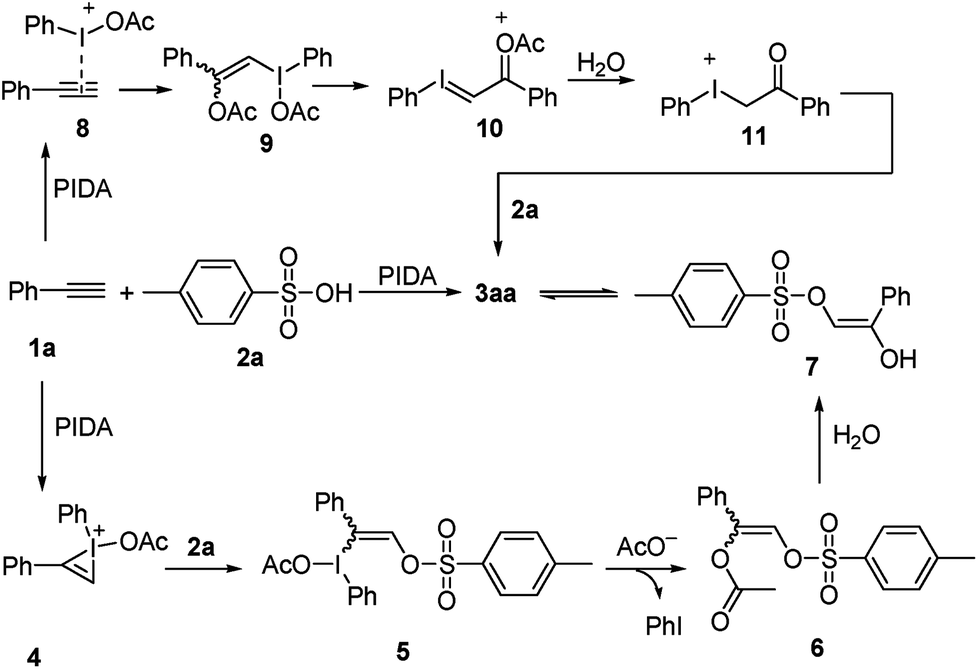 | ||
| Scheme 4 Possible reaction mechanism. | ||
In conclusion, we have developed a novel hypervalent iodine(III)-promoted method for oxysulfonyloxylation of alkynes using sulfonic acids to access α-sulfonyloxyketones. Compared to the reported methods involving the application of PIDA, the present study expands the application scope of PIDA as an oxidant and O-source of carbonyl group. In general, this approach exhibited a broad substrate scope for the synthesis of α-sulfonyloxyketones under milder conditions. In addition, the easy availability of reactants as well as the use of simple oxidant and O-source makes the present method a useful new option for α-sulfonyloxyketone synthesis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Nakamura, S. Tanaka, A. Imamiya, R. Takane, C. Ohta, K. Fujimura, T. Maegawa and Y. Miki, Org. Biomol. Chem., 2017, 15, 6702 Search PubMed
; R. Sakamoto, T. Inada, S. Selvakumar, S. A. Moteki and K. Maruoka, Chem. Commun., 2016, 52, 3758 RSC
- E. Deruer and S. Canesi, Org. Biomol. Chem., 2017, 15, 3736 CAS
- C. Yang, G. Y. Cheng, B. F. Huang, F. T. Xue and C. Jiang, RSC Adv., 2016, 6, 87134 RSC
- Z. J. Wang, M. Kanai and Y. Kuninobu, Org. Lett., 2017, 19, 2398 CrossRef CAS PubMed
- X. Wang, Y. X. Ye, S. N. Zhang, J. J. Feng, Y. Xu, Y. Zhang and J. B. Wang, J. Am. Chem. Soc., 2011, 133, 16410 CrossRef CAS PubMed
- G. F. Koser, A. G. Relenyi, A. N. Kalos, L. Rebrovic and R. H. Wettbach, J. Org. Chem., 1982, 47, 2487 CrossRef CAS
- K. Kiyokawa, S. Yahata, T. Kojima and S. Minakata, Org. Lett., 2014, 16, 4646 CrossRef CAS PubMed
- A. Utaka, L. N. Cavalcanti and L. F. Silva, Chem. Commun., 2014, 50, 3810 RSC
- Y. P. Fan, W. Wan, G. B. Ma, W. Gao, H. Z. Jiang, S. Z. Zhu and J. Hao, Chem. Commun., 2014, 50, 5733 RSC
- H. Y. Niu, C. Xia, C. R. Qu, Q. Zhang, Y. Jiang, Y. Z. Mao, Z. Y. Lia and H. M. Guo, Org. Biomol. Chem., 2011, 9, 5039 CAS
- W. B. Liu, P. Zhou, C. Chen, Q. Zhang and Z. B. Zhu, Org. Biomol. Chem., 2013, 11, 542 CAS
- B. T. Cho, W. K. Yang and O. K. Choi, J. Chem. Soc., Perkin Trans. 1, 2001, 1204 RSC
- R. S. Varma, K. P. Naicker and D. Kumar, J. Mol. Catal. A: Chem., 1999, 149, 153 CrossRef CAS
- K. C. Nicolaou, T. Montagnon, T. Ulven, P. S. Baran, Y. L. Zhong and F. Sarabia, J. Am. Chem. Soc., 2002, 124, 5718 CrossRef CAS PubMed
- B. Basdevant and C. Y. Legault, J. Org. Chem., 2015, 80, 6897 CrossRef CAS PubMed
- J. P. Wan, Y. F. Lin, X. J. Cao, Y. Y. Liu and L. Wei, Chem. Commun., 2016, 52, 1270 RSC
- J. Deng and J. Luo, Tetrahedron, 2013, 69, 5937 CrossRef
- D. Zhang-Negrerie and Y. Du, Chem.–Eur. J., 2015, 21, 5193 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11875a |
| This journal is © The Royal Society of Chemistry 2017 |