
Reaction engineering of biocatalytic (S)-naproxen synthesis integrating in-line process monitoring by Raman spectroscopy†
M.
Aßmann
a,
A.
Stöbener
a,
C.
Mügge
 b,
S. K.
Gaßmeyer
b,
L.
Hilterhaus
a,
R.
Kourist
bc,
A.
Liese
*a and
S.
Kara
*a
b,
S. K.
Gaßmeyer
b,
L.
Hilterhaus
a,
R.
Kourist
bc,
A.
Liese
*a and
S.
Kara
*a
aInstitute of Technical Biocatalysis, Hamburg University of Technology, Denickestr. 15, 21073 Hamburg, Germany. E-mail: liese@tuhh.de; selin.kara@tuhh.de
bJunior Research Group for Microbial Biotechnology, Ruhr-University Bochum, Universitätstr. 150, 44780 Bochum, Germany
cInstitute of Molecular Biotechnology, Graz University of Technology, Petersgasse 14, A8010 Graz, Austria
First published on 19th May 2017
Abstract
Biocatalytic (S)-naproxen synthesis using an (S)-selective arylmalonate decarboxylase mutant (AMDase G74C/M159L/C188G/V43I/A125P/V156L, AMDase-CLGIPL) exposes a promising environmentally friendly alternative to conventional chemical synthesis strategies. The reaction progress of naproxen synthesis catalyzed by AMDase-CLGIPL covalently immobilized onto a robust acrylate carrier was investigated with respect to reaction engineering. Kinetic characterization of the immobilized enzyme reveals a KM value of 22.1 ± 0.1 mM in the naproxen malonate conversion and an inhibiting effect of the produced naproxen with a Ki of 26.3 ± 1.4 mM. However, an effective process can be realized without in situ product removal yielding (S)-naproxen with an ee of 99%. By optimizing the product work-up, an isolated yield of 92% was achieved with total turnover numbers between 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 107000 in five repetitive batches. Furthermore, process monitoring with in-line Raman spectroscopy was successfully applied to analyze the reaction progress with a root mean square error of prediction of 0.8 mM (corresponding to 4%).
000 and 107000 in five repetitive batches. Furthermore, process monitoring with in-line Raman spectroscopy was successfully applied to analyze the reaction progress with a root mean square error of prediction of 0.8 mM (corresponding to 4%).
Introduction
The application of biotransformations as an environmentally friendly alternative for the production of fine chemicals and pharmaceuticals has increased considerably in the last few decades.1,2 In particular, in the sector of chiral products, for example in the production of enantiopure pharmaceuticals, the integration of enzymes is of high interest.3 Indeed, more than 50% of the industrially realized biotransformation processes are found in the pharmaceutical industry4,5 and furthermore in many emerging application areas, for example in biocatalytic polymerization in materials science.6An interesting target class of chiral pharmaceuticals are non-steroidal anti-inflammatory drugs (NSAID). Widely used examples of NSAID are α-arylpropionate derivatives (so-called profens), like ibuprofen or naproxen.7 The enantiopure synthesis of these chiral molecules can be realized in a biocatalytic way using an enantioselective enzyme named arylmalonate decarboxylase (AMDase, EC 4.1.1.76). The natural (R)-selectivity of the wild-type AMDase was reversed to the (S)-selective AMDase variant G74C/M159L/C188G/V43I/A125P/V156L (AMDase-CLGIPL) through directed enzyme engineering,8 which has been successfully applied in the synthesis of flurbiprofen.9 Recently, we demonstrated the use of this designed AMDase-CLGIPL for the synthesis of (S)-naproxen with enhanced stability achieved through immobilization.10
Biocatalysts can be applied in a process in various forms, for example (i) as whole cells (enzyme overexpressed in a microbial host), (ii) as a cell lysate containing an overexpressed enzyme or (iii) as a purified enzyme – all in their solubilized free as well as immobilized forms. As biocatalyst purification is material-, energy- and time-consuming, the aim of the present study is to operate the process with the crudest form of the biocatalyst that still allows efficient conversion. Often, whole cells are used for bioproduction applications to keep the cost of the upstream process low. Nevertheless, the application of whole cells has some disadvantages, such as low tolerance to high concentrations of reactants, unwanted side reactions catalyzed by other cells' internal enzymes or low organic solvent tolerance, and is therefore not appropriate for every process. Processes with purified enzymes are the simplest to implement; however, the cost of enzyme isolation needs to be compensated for by reusing the biocatalyst.11 For this purpose, membrane filtration12,13 or immobilization of the enzyme onto a robust support offers an efficient solution for biocatalyst recycling.14
The feasibility of biocatalyst integration into a process is determined by the availability of a suitable enzyme and the choice of an optimal reactor concept.15 For development of the best possible bioprocess, besides the selection of pH, temperature, buffer type and ionic strength, it is necessary to determine the thermodynamics and kinetics of the conversion of the substrate to the target product. Based on this, an optimal type of reactor and its respective process operation window can be identified.16
Online analytical techniques offer the possibility to complete bioprocess optimization, since the real-time availability of process data permits process control with pinpoint accuracy. In particular, spectroscopic techniques are frequently employed in this regard, since they are non-destructive and provide data in real time.17 Vibrational spectroscopy in combination with multivariate calibration is a versatile tool to monitor the concentrations of organic compounds. Established methods in vibrational spectroscopy are infrared (near, mid, and far IR range)18,19 and Raman spectroscopy.20 While infrared spectroscopy in aqueous solutions has limitations due to its broad and intense water absorbance bands, Raman spectroscopy is less prone to interferences from water. Nonetheless, not every biocatalytic reaction carried out in aqueous solution can be easily monitored by Raman spectroscopy. Due to a low-to-medium Raman activity of many small molecules, typical product concentrations in a bioprocess may be below the limit of detection, if reasonable integration times are used. In spite of the low detection sensitivity, Raman spectroscopy has been successfully applied to in-process monitoring of pharmaceutical production and allows rapid and easy measurements without sample preparation.21
In this study, we demonstrate the biocatalytic synthesis of (S)-naproxen using immobilized AMDase-CLGIPL. Making use of detailed reaction engineering studies, the operational window for efficient and selective (S)-naproxen synthesis is selected. The feasibility of in-line process monitoring by Raman spectroscopy is additionally highlighted.
Experimental
Protein expression
Recombinant Escherichia coli BL21 (DE3) containing an IPTG (isopropyl-β-D-thiogalactopyranoside) inducible plasmid with a gene for arylmalonate decarboxylase G74C/M159L/C188G/V43I/A125P/V156L (AMDase-CLGIPL) was cultivated as described previously.9 After protein expression, the cells were harvested by centrifugation (20 min, 4 °C, and 17700 × g) and stored at −80 °C unless directly used. For cell lysis, a cell pellet was resuspended in water to a concentration of 0.2 gcells mL−1 and ultrasonication was performed using a Sonopuls HD2070 sonicator (Berlin, Germany) (90% intensity, 3 × 3 min, and interjacent cooling on ice). The cell lysate was separated by centrifugation from the solid cell components (30 min, 4 °C, and 74000 × g). The freshly prepared cell lysate was then used for enzyme immobilization. The concentration of the obtained protein solution was determined by the Bradford assay using the reported method of Zor and Selinger.22
Enzyme immobilization
Amino C2 acrylate (Iris Biotech GmbH, Marktredwitz, Germany) was used as the carrier (pore size = 120 nm, particle diameter = 423 μm) for the immobilization of AMDase-CLGIPL. The amine-functionalized carrier was pre-activated with a 2% (v/v) glutaraldehyde solution (212 mM, ratio = 1:4 (wcarrier/v, 250 mgcarrier mL−1), 1 h, overhead shaker, room temperature, and 8 rpm), filtered with a sintered glass filter (pore size 2, 40–100 μm) and then carefully washed with water. For immobilization, the carrier material was incubated with the cell lysate containing AMDase-CLGIPL (100 mgcarrier mL−1, 39.6 mgprotein mL−1, water pH of 8.0 adjusted with 1 M NaOH, 17 h, and overhead shaker at 8 rpm and room temperature). Afterwards, the carrier was washed twice with one volume of water (pH 8.0) and with one volume of 0.5 M NaCl solution. Fractions of the supernatant and the washings were collected for further determination of protein concentration. The enzyme loading of the carrier was determined to be 8.6 mgAMDase-CLGIPL gcarrier−1. The target AMDase-CLGIPL content was determined to be ∼30% of the total protein amount of the cell lysate according to an activity assay performed for the purified enzyme and the cell lysate under the same reaction conditions. Reaction assay contained 20 mM phenylmalonate, 10% (v/v) purified enzyme or cell lysate (both previously diluted 100-fold and the protein concentration measured via the Bradford assay) in water at pH 8.0 adjusted with 1 M NaOH, 30 °C, and 500 rpm on a 0.5 mL scale.
Regarding the activity of the free cell lysate and its immobilized form, a volumetric activity of 50 U mL−1 was observed with the cell lysate, whereas 3 U mgimmo−1 was found for the immobilized enzyme based on a standard activity assay (20 mM naproxen malonate, pH 8 at 30 °C and 500 rpm).
Kinetic characterization
The kinetic investigation of immobilized AMDase-CLGIPL on amino C2 acrylate was carried out in 1 mL scale experiments. To analyze the initial rates of naproxen malonate conversion, different amounts of the immobilized enzyme preparation (3.6 mg to 21.8 mg) were placed in round-bottom microreaction tubes in a thermoshaker at 900 rpm. The reaction was started by the addition of 1 mL of pre-tempered (30 °C) naproxen malonate solution in water (pH 8.0) at different concentrations (0.2 mM to 98.4 mM) to evaluate the enzyme's saturation kinetics.The investigation of product inhibition was carried out with 20 mg immobilized enzyme preparation in 1 mL reaction volume. The naproxen malonate concentration was fixed to 45 mM while using different amounts of product naproxen (0.5 mM to 67.4 mM) in water at a pH of 8.0. The reaction mixture was tempered to 30 °C and added to the immobilized enzyme to initiate the reactions. Samples were taken in duplicate over the initial course of the reaction (below 5% conversion) for kinetic and product inhibition analysis and the samples were analyzed by HPLC.
Investigation of mass transfer limitations
The mass transfer limitation was investigated in two packed-bed columns with different packing volumes (1.3 mL and 3.3 mL). The amount of the carrier per volume of the packed column was determined to be 0.49 g mL−1. For diffusion analysis, a 5 mM flurbiprofen malonate solution in water with a pH of 8.0 was used. Residence times (τ) were fixed to 1, 2, 5 and 6 min in both packed-bed columns. The conversion values were analyzed after five residence times to ensure that steady-state conditions were achieved. Samples were taken in duplicate and analyzed by HPLC.(S)-Naproxen synthesis in repeated batch experiments
The synthesis of (S)-naproxen in repeated batch experiments was carried out in two 100 mL stirred tank reactor systems. The pH of the reaction solution (prepared in water) containing the starting material was adjusted to 8.0 using 1 M NaOH. The maximum solubility of naproxen malonate at pH 8.0 and 30 °C was determined to be 202 ± 9 mM. 45 g L−1 amino C2 acrylate with immobilized AMDase-CLGIPL was applied for the conversion of a 48.6 mM (13.3 g L−1) naproxen malonate solution in water at pH 8.0, 30 °C and 700 rpm in a conventional stirred tank reactor (STR) as well as in a rotating bed reactor (RBR) system (SpinChem AB, Umeå, Sweden). During the course of the biotransformation to (S)-naproxen in the unbuffered system yielding full conversion, a pH shift from 8.0 to 8.3 was detected; thus, the sodium salt of the product was formed. To monitor the reaction course as a function of time, samples were withdrawn and analyzed via HPLC. Between the batches, the immobilized enzyme preparation was washed with 100 mL water, filtered (filtration was required only in the case of STR) and stored at 4 °C until next usage.Analysis of the precipitation efficiency
The influence of different hydrochloric acid concentrations on the precipitation efficiency of naproxen was analyzed. To 1 mL of a 105 mM naproxen solution (pH 9.0), 200 μL of hydrochloric acid solutions (0.25 M to 10 M HCl) were added. Subsequently, the solution with the precipitate of naproxen was mixed and centrifuged (30 min, room temperature, and 12100 × g). The supernatant was analyzed via HPLC to determine the remaining naproxen concentration after precipitation.
Product isolation
After the conversion of naproxen malonate to (S)-naproxen, the product solution was separated by decantation from the immobilized enzyme preparation. The remaining product on the carrier (outer and inner surfaces) was washed with 100 mL water and the product-containing washing solution was added to the collected reaction solution (pH range of 8.3 to 8.6). The resulting 200 mL reaction mixture containing the product (>99.7% conversion to naproxen) was acidified with 40 mL of 6 M hydrochloric acid solution to an end concentration of 1 M HCl. As a consequence, naproxen was transformed from the water-soluble naproxen sodium salt to the insoluble protonated acid. The precipitated naproxen was separated from the solution with filter paper (Macherey-Nagel, type 640 d) over a Buechner funnel with a vacuum pump, washed with hydrochloric acid (100 mL, 1 M) and dried in a vacuum oven (45 °C for 8 h). The white crystalline naproxen was quantified and the quality of the obtained product was analyzed with respect to enantiopurity by GC using a chiral column. Samples of ∼1 mg of the product were resuspended in 600 μL MTBE, supplemented with 100 μL MeOH and derivatized with 50 μL trimethylsilyldiazomethane (TMS) (TCI Germany, 0.6 M in hexane) for 30 min at room temperature. The derivatization reaction was stopped by the addition of 5 μL of pure acetic acid and the product was subsequently dried. For analysis, the samples were resuspended in 200 μL ethyl acetate. The samples were analyzed by GC on a Shimadzu GC Plus 2010 (Duisburg, Germany) using an FS Hydrodex-β-6TBDM column (Macherey-Nagel, Düren, Germany), with a method operated isothermally at 160 °C with an injection split of 1/20. The elution order was (S)-naproxen (35.7 min) and (R)-naproxen (36.3 min) with baseline separation (cf. the ESI† for representative chromatograms). The ee values were determined from the relative peak areas according to standard procedures.HPLC analytics
HPLC analysis was realized with a C18 reversed phase column (Nucleodur C18 pyramid 250/4.6, Macherey-Nagel, Düren, Germany) in an Agilent 1100 HPLC system (Frankfurt, Germany) using an isocratic eluent of ACN:H2O:TFA (59.025:39.025:0.05). A flow rate of 0.8 mL min−1 was maintained for 9 min. The detection was carried out with a diode array detector (DAD) at a wavelength of 245 nm. Typical retention times were naproxen malonate = 3.8 min, naproxen = 5.8 min, flurbiprofen malonate = 4.4 min, and flurbiprofen = 7.6 min.
Raman spectroscopy for in-line process monitoring
The feasibility of in-line Raman spectroscopy for the given reaction was evaluated on a small scale (25 & 50 mL). A lightproof stirred tank reactor was set up with an aqueous solution of naproxen malonate (25 mM), and free AMDase-CLGIPL was added to provide a concentration of 80 mg L−1. A MultiSpec® Raman process spectrometer (tec5 AG, Oberursel, Germany) with a fiber-coupled titanium/sapphire probe (excitation wavelength = 785 nm, power = 500 mW, integration time = 30 s) was used to record spectra. The probe was immersed directly in the liquid reaction mixture. Raman spectra were measured at a time interval of 15 minutes, and offline samples for HPLC were drawn at the same times to obtain the corresponding concentrations of substrates and products. Raman spectra were cut to the spectral range from 370 to 1450 cm−1, and the baseline was corrected applying a concave rubberband method (OPUS 7.0, Bruker Optik GmbH, Ettlingen, Germany; 64 baseline points, 10 iterations). For chemometrics, the spectral range included in analysis was further narrowed (1350 to 1430 cm−1) to include only the two most intense Raman bands. The Unscrambler X (CAMO Software, Oslo, Norway) was used for multivariate regression. Twelve training samples (Raman spectra and corresponding HPLC data) were available for multivariate calibration. The multivariate regression was calculated with the non-iterative partial least squares algorithm. The model was then applied to predict the concentrations of naproxen in seven test samples not included in the calibration data set.Results and discussion
Kinetic characterization of the immobilized enzyme
First, we analyzed the immobilized enzyme with respect to kinetics of naproxen formation from naproxen malonate (Fig. 1). The enzyme shows Michaelis–Menten-type saturation kinetics. With increasing naproxen malonate concentration, the initial activity of the immobilized enzyme increases to a maximal conversion rate of 3.6 ± 0.1 U mgimmo−1. In the analyzed substrate concentration range up to 100 mM naproxen malonate, no excess substrate inhibition was observed. As a consequence, it is not required to establish a fed-batch reactor or a continuously operated stirred tank reactor (CSTR) to maintain the substrate concentrations at a low level. A standard batch reactor system is applicable.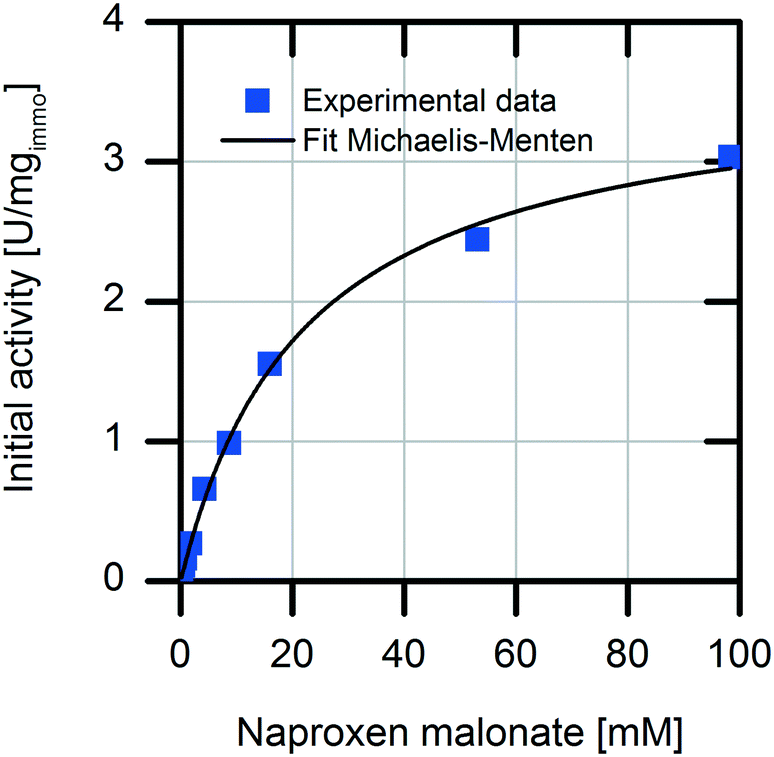 | ||
| Fig. 1 Kinetic characterization of the (S)-selective AMDase-CLGIPL for naproxen malonate conversion. Reaction conditions: 1 mL volume, varying amounts of immobilized enzyme on amino C2 acrylate from cell lysate in water, at 30 °C, pH 8, 900 rpm, enzyme loading: 8.6 mgAMDase-CLGIPL gcarrier−1. | ||
The immobilized enzyme preparation demonstrates an apparent Michaelis–Menten constant of KM,NM immo = 22.1 ± 0.1 mM in contrast to KM,NM free = 0.08 ± 0.03 mM for the free enzyme (data not shown) accounting for possible diffusion limitations. Many immobilized enzyme preparations show a decreased affinity to the substrate.23 However, in some cases, even an enhancement of the conversion rate due to stabilization of the active enzyme conformation was observed.24 Here, a decreased enzymatic affinity with a shift of the Michaelis–Menten constant to an approx. 300-fold higher substrate concentration was determined. The enzyme could be immobilized onto the carrier surface as well as inside the porous carrier material (pore volume: 1.14 mL gcarrier−1). Due to immobilization, the protein structure is fixed in a more rigid form and conversion to the product depends on diffusion of the substrate to the enzyme located within the carrier material. Hence, the observed affinity shift could be explained by a diffusion limitation.25 Such an internal diffusion limitation could be investigated by breaking the immobilized enzyme into smaller fragments and analyzing the fragmented immobilized enzyme with respect to activity changes which are dependent on the particle sizes,26,27 but this investigation was not a part of this current study. Furthermore, immobilization of an enzyme might change its conformation as well as its microenvironment. This may induce changes in the apparent kinetic parameters. In most cases, the maximal conversion rate is decreased by enzyme immobilization.28 In our system, we observed about 40% reduction in the maximal enzymatic activity due to enzyme immobilization (kcat free = 151 min−1 and kcat immo = 90 min−1). The overall increased productivity of immobilized enzymes often compensates for these effects.
For selection of the optimal reactor for biocatalytic profen synthesis, knowledge of the influence of the product on the enzyme is important. In particular, in the case of continuously operated reactor concepts, like the plug-flow reactor (PFR) or CSTR, strong product inhibition could hamper the full conversion. In our reaction system, the formed product naproxen might cause an inhibiting effect on the enzymatic conversion, as it is known that the activity of AMDase decreases significantly in the presence of the formed product.29 Therefore, detailed kinetic investigations were carried out with respect to possible competitive product inhibition by naproxen. The result clearly shows the inhibiting effect of the product (Fig. 2). The immobilized enzyme is competitively inhibited by naproxen with an inhibition constant Ki,Nap immo of 26.3 ± 1.4 mM. For an efficient reaction, the quotient of the affinity constant to the inhibition constant should be KM/Ki < 1. In reaction systems with values above one for this quotient, no full conversion is possible without removal of the formed product.30,31
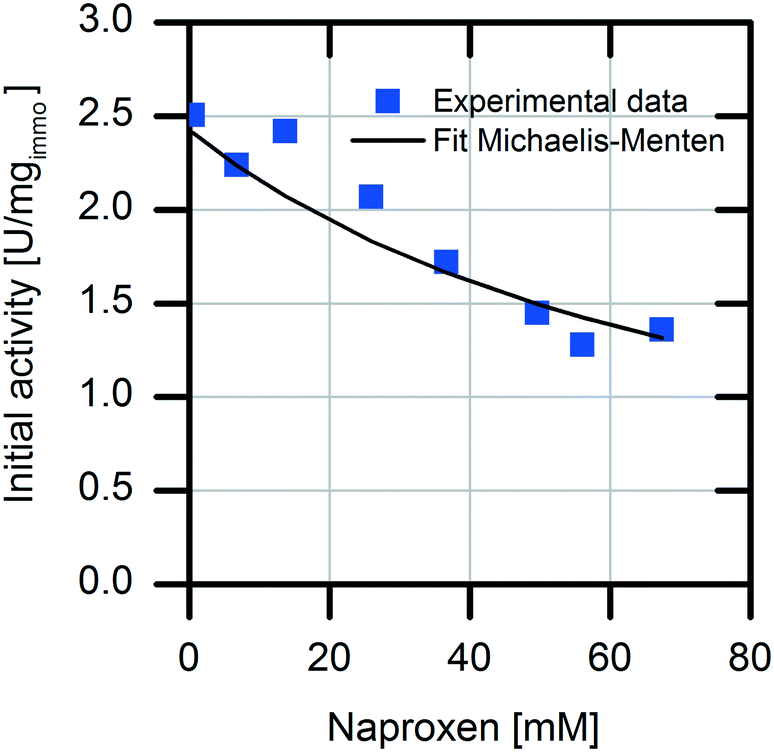 | ||
| Fig. 2 Investigation of competitive product inhibition of (S)-selective AMDase-CLGIPL for naproxen malonate conversion with different amounts of naproxen. Reaction conditions: 1 mL volume, 20 mg immobilized enzyme on amino C2 acrylate from cell lysate in water, at 30 °C, pH 8, 900 rpm and with 45 mM naproxen malonate, enzyme loading: 8.6 mgAMDase-CLGIPL gcarrier−1. | ||
The immobilized AMDase-CLGIPL on the amino C2 acrylate carrier reveals a KM/Ki value of 0.84; thus, an efficient conversion is possible without in situ naproxen removal.
The obtained values in the kinetic investigation of the immobilized enzyme (Vmax, KM,NM and Ki,Nap) can be used to generate a model for the prediction of the reaction rate of naproxen synthesis based on the Michaelis–Menten kinetics for competitive product inhibition (eqn (1)).
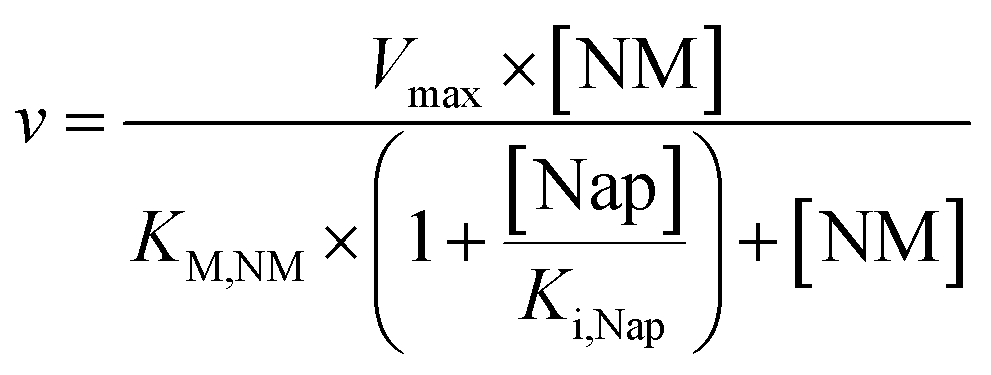 | (1) |
Investigation of external mass transfer limitations
Another important factor for choosing the optimal reactor for a production process is the possible diffusion limitation of the immobilized enzyme. The diffusion limitation was determined using a simple experimental setup with two packed-bed columns with different packing volumes (1.3 mL and 3.3 mL) of the immobilized enzyme.25 For this purpose, the flow rate of the substrate solution was adjusted to establish the same residence times (τ = 1, 2, 5 and 6 min) in the packed-bed columns in both experimental setups. A difference in the conversion rates of the two packed columns was determined (Fig. 3). When the packed column having the smaller volume was used, lower conversions were detected. This result indicates an external mass transfer limitation by the acrylic carrier material. Because of higher convection at higher flow rates, the diffusion limitation at the carrier is reduced.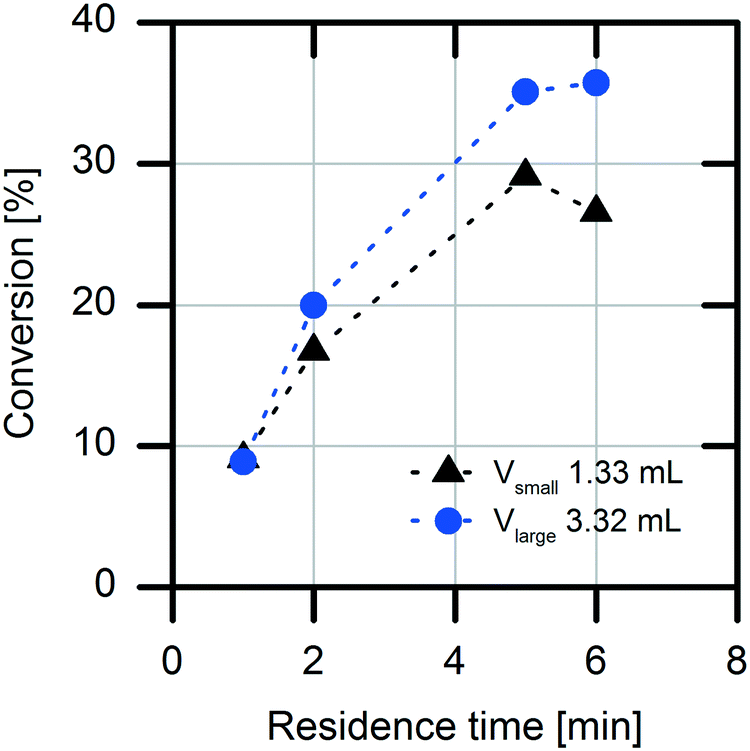 | ||
| Fig. 3 Analysis of diffusion limitation of the immobilized AMDase-CLGIPL on amino C2 acrylate in two packed-bed columns with different packing volumes (1.3 mL and 3.3 mL). The determination of the diffusion was measured at different flow rates with fixed residence times (1, 2, 5 and 6 min). Reaction conditions: 5 mM flurbiprofen malonate in water, at pH 8.0 and room temperature. | ||
Overall, characterization of the immobilized enzyme revealed a high KM value for the substrate naproxen malonate, an inhibiting effect of the formed product naproxen, and a decreased enzymatic activity. The inhibiting effect of the formed product is more pronounced at high conversions, which is the case for a fed-batch reactor as well as for a CSTR operating under outflow conditions. Hence, a batch concept was chosen based on these results: (i) the high KM value and (ii) the observed product inhibition.
In our study, we demonstrate the possibility for naproxen synthesis in two different stirred tank batch reactor concepts: a conventional stirred tank reactor (STR) and a rotating bed reactor (RBR). In the latter, the biocatalyst is located inside a stirrer element separated from the reaction solution, establishing a higher flux passing through the carrier which can (i) reduce the diffusion limitation through the carrier, (ii) increase the stability of the biocatalyst and (iii) enable easy separation as well as recycling of the biocatalyst from the reaction solution.32
Biocatalytic (S)-naproxen synthesis in batch experiments
The scaling-up from 1 mL experiments to 100 mL reactions with a 50 mM substrate was performed with the above-described two stirred tank reactor systems for the conversion of naproxen malonate with AMDase-CLGIPL immobilized on the amino C2 acrylate carrier.In both reactor concepts, complete conversion of naproxen malonate to (S)-naproxen in five repeated batch experiments was achieved. The two reactor configurations show similar performance with respect to product formation (Fig. 4). The kinetic model developed for the immobilized enzyme, with the Ki and KM values determined (eqn (1)), fits well to the experimental data of both batch reactions run in two reactors in parallel (Fig. 5).
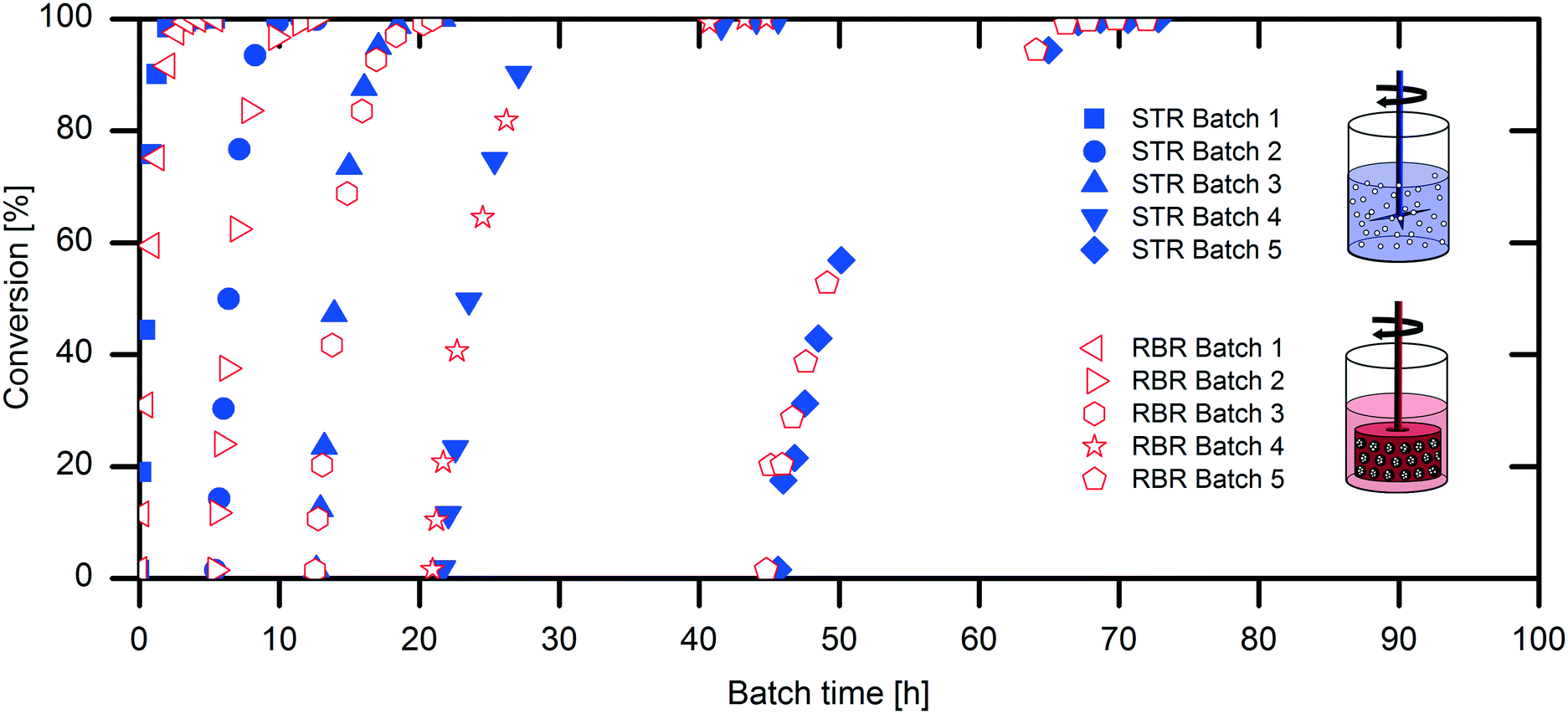 | ||
| Fig. 4 Repeated batch experiments of the naproxen malonate conversion in STR and RBR for comparison. Reaction conditions: 100 mL reaction volume in water, at pH 8.0, 700 rpm, 30 °C, 45 g L−1 AMDase-CLGIPL on amino C2 acrylate, enzyme loading: 8.6 mgAMDase-CLGIPL gcarrier−1, 48.6 mM naproxen malonate. Between the batches, the immobilized enzyme preparation was washed with 100 mL water, filtered (in the case of STR) and stored at 4 °C until next usage. | ||
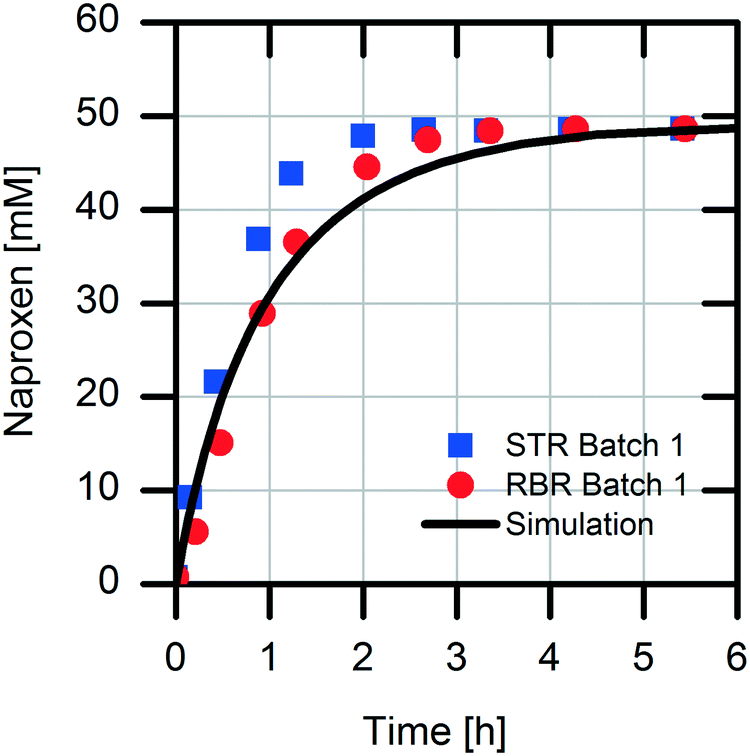 | ||
| Fig. 5 Simulation and experimental data of the naproxen synthesis in the first batch. Reaction conditions: 100 mL volume in water, 4.5 g L−1 immobilized AMDase-CLGIPL from cell lysate on the amino C2 acrylate carrier, at 30 °C, pH 8.0, 700 rpm. Enzyme loading: 8.6 mgAMDase-CLGIPL gcarrier−1. Simulation based on kinetic data of the immobilized AMDase-CLGIPL with the parameters KM,NM: 22.1 mM, Vmax: 3.6 U mgimmo enzyme−1 and Ki,NA: 26.3 mM. | ||
There are several different parameters influencing a biocatalytic process. A decisive parameter is the enzymatic stability under process conditions, which can be described with the half-life time (t1/2) in which 50% of the enzymatic activity is lost by enzyme deactivation. The half-life time can be determined by exponential fitting of enzymatic activity data. Another parameter to characterize process productivity is the total turnover number (TTN, eqn (2)), which can be calculated using the ratio of the catalytic efficiency constant (kcat (s−1)) to the deactivation constant (kdes (s−1)) under process conditions.25,33
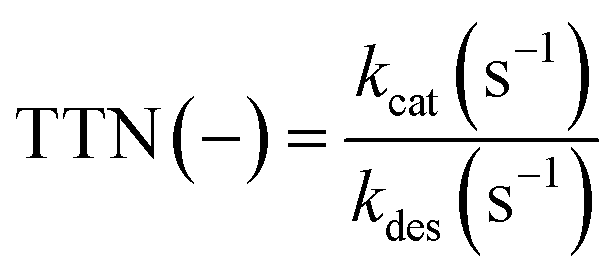 | (2) |
Analysis of the specific activities in the conventional STR and RBR reveals a 20% reduced specific activity during the batch experiments in the RBR compared to the results obtained with the conventional STR (0.5 U mgimmo−1 less specific activity) (Fig. 6). However, calculation of the enzyme deactivation constant reveals a slightly increased half-life time in the RBR (t1/2,RBR = 23 h and t1/2,STR = 21 h) (Table 1).
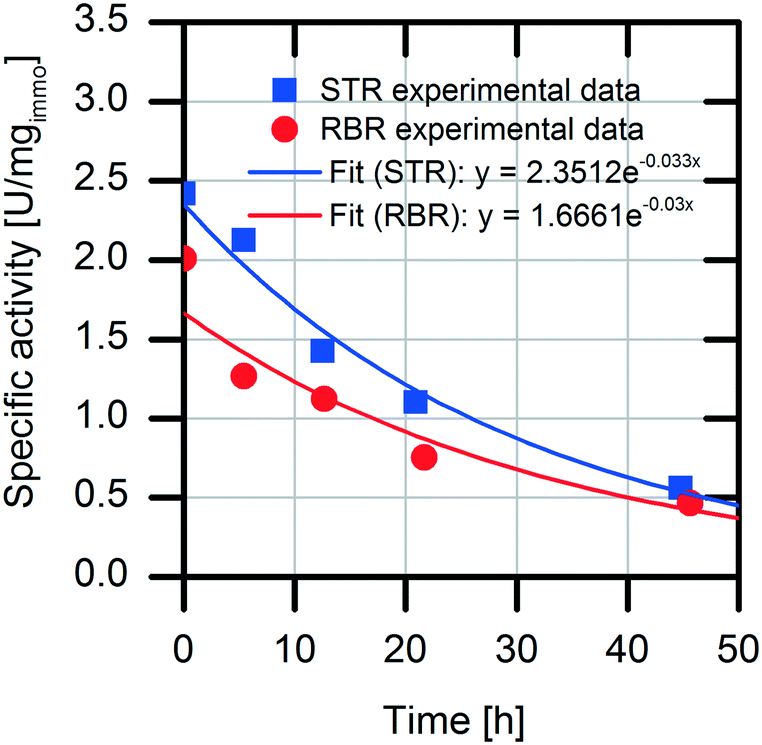 | ||
| Fig. 6 Investigation of the stability of the immobilized AMDase-CLGIPL in STR and RBR. Reaction conditions: 100 mL volume in water, at pH 8.0, 700 rpm, 30 °C, 45 g L−1 immobilized AMDase-CLGIPL on amino C2 acrylate, enzyme loading: 8.6 mgAMDase-CLGIPL gcarrier−1, 48.6 mM naproxen malonate. | ||
| Parameter | STR | RBR |
|---|---|---|
| t 1/2 [h] | 21.0 ± 0.3 | 23.1 ± 1.5 |
| k cat [h−1] | 3526.8 ± 53.6 | 2499.2 ± 158.2 |
| k des [h−1] | 0.033 ± 0.0005 | 0.03 ± 0.002 |
| TTN [–] | 106873 ± 1625 |
83306 ± 5273 |
From the maximal activity (kcat) observed during the respective first batch experiments and enzyme deactivation (kdes) observed over the repeated batches, the TTN values were calculated to be 83306 for the RBR and 106873 for the STR system. These values are in a range of TTN values described for some representative biocatalysts (104 to 107).33 Nevertheless, the calculation of the TTN is an estimation for the applicability of the biocatalysts to product synthesis. A few influencing parameters, like the reduction of the conversion rate by product inhibition or the influence of the enzymatic affinity on the substrate, are neglected. With both reactor setups, (S)-naproxen was successfully synthesized by means of enzymatic catalysis.
The rotating bed reactor has various advantages compared to the conventional stirred tank reactor: reduced shear forces on the immobilized enzyme preparation, enhanced mass transfer and finally practicability. The handling of the immobilized enzyme is easier and faster through the fast separation of the immobilized enzyme preparation from the reaction solution, avoiding an additional filtration step and facilitating washing away of the immobilized enzyme from the reaction solution. Based on the above given advantages and the similar process parameters compared to the traditional STR in AMDase-catalyzed profen synthesis, the rotating bed reactor concept can be regarded as a promising option for industrial applications.
(S)-Naproxen isolation
For downstream processing (DSP), product precipitation was chosen as an easy and economical way for isolation compared to extraction followed by solvent evaporation. This is possible due to the differences in the solubility of (S)-naproxen in its protonated form and sodium salt form (0.07 mg mL−1, protonated; 266 mg mL−1, sodium salt).34 According to these literature data, the sodium salt of naproxen is 3800-fold more soluble in aqueous medium than the protonated acid form.For the investigation of product precipitation efficiency, different hydrochloric acid strengths were tested (Fig. 7). The naproxen content in the supernatant increases clearly with decreasing amount of hydrochloric acid. With threefold molar excess of hydrochloric acid in the reaction solution, >99.5% of the (S)-naproxen is located in the precipitate. Accordingly, an excess concentration of 1 M hydrochloric acid in the reaction solution was chosen for the DSP.
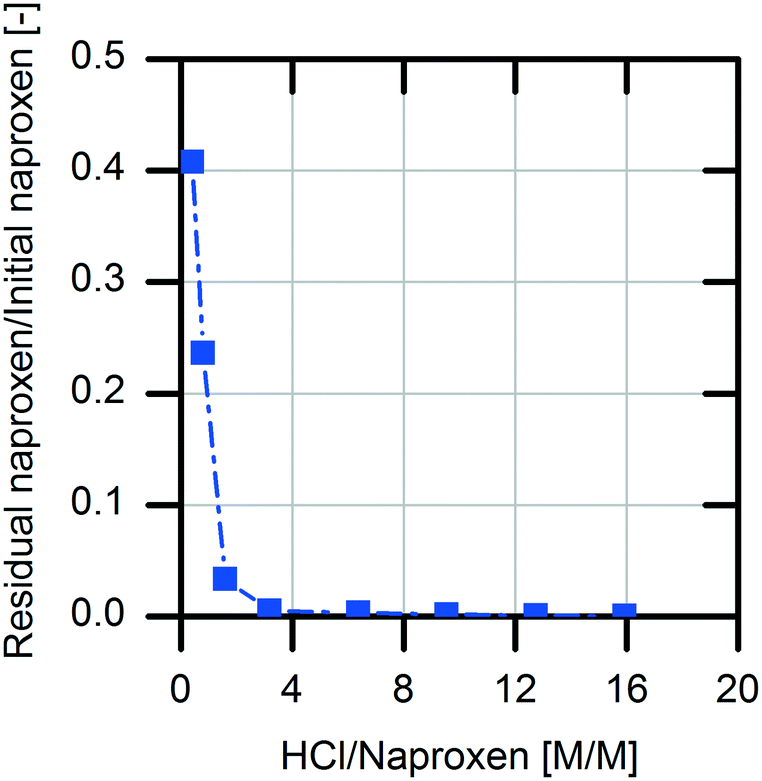 | ||
| Fig. 7 Analysis of the precipitation efficiency for the product isolation of naproxen. For this investigation, a 100 mM naproxen product solution with a pH of 9.0 was treated with hydrochloric acid of different strengths. The precipitated product was separated by centrifugation and the supernatant was analyzed with respect to the remaining product of the soluble part. | ||
The precipitated (S)-naproxen was separated using paper filter and dried in a vacuum oven at 40 °C for 8 hours. The amount of isolated naproxen was 10.3 g, corresponding to an isolated yield of 92% (according to 11.2 g max. naproxen yield). The product was analyzed by chiral gas chromatography and 1H NMR spectroscopy (see the ESI,† Fig. S2 and S4). The enantiomeric excess (ee) value of (S)-naproxen was determined to be 99%.
For the biocatalytic synthesis of (S)-naproxen, a productivity of about 140 kgproduct kgenzyme−1 is reached. This result is higher than the given minimum productivity of enzyme applicability to pharmaceutical production of 50–100 kgproduct kgenzyme−1.35 The environmental (E) assessment of the reaction itself revealed an E-factor (kgwaste kgproduct−1) value of 89 kgwaste kg(S)-naproxen−1, based on a complete conversion of the substrate naproxen malonate in aqueous solution leading to (S)-naproxen as the target product and CO2 as the by-product. Thus, the E-factor value of the process reported herein was found to be in the expected range for the production of pharmaceuticals with values of 25–100 gwaste gproduct−1.36 However, it has to be noted that the downstream steps for isolation and purification of the product were not included in the assessment. Nevertheless, considering the substrate scope of AMDase, the demonstrated biocatalytic profen synthesis utilizing the immobilized enzyme represents a promising alternative to conventional chemical syntheses.
In-line monitoring with Raman spectroscopy
The chromatographic concentration determination with HPLC involves the inherently time-delayed availability of results. If the process is to be automatized or monitored in real time, a spectroscopic technique is necessary for in-line concentration determination. The feasibility of in-line Raman spectroscopy was studied for the biocatalytic naproxen synthesis catalyzed by free AMDase-CLGIPL with a probe immersed inside a stirred tank reactor. During the progression of the reaction, Raman spectra in the range from 1450 to 800 cm−1 show various bands with variations in intensity and Raman shifts (see the ESI,† Fig. S3). The two most prominent Raman bands are located in the spectral range from 1350 to 1430 cm−1 (Fig. 8). These bands were chosen to test the feasibility of in-line reaction monitoring. They are interpreted as a symmetric deformation vibration δs(CH3) and a valence vibration of the C–OH group of the carboxylic acid ν(C–OH). Both vibrations are expected in the given spectral range and known to be of negligible importance in infrared spectroscopy.37 Vibrations with minor IR activity (requiring a change of the dipole moment during vibration) typically show major Raman activity (due to changes in polarizability).38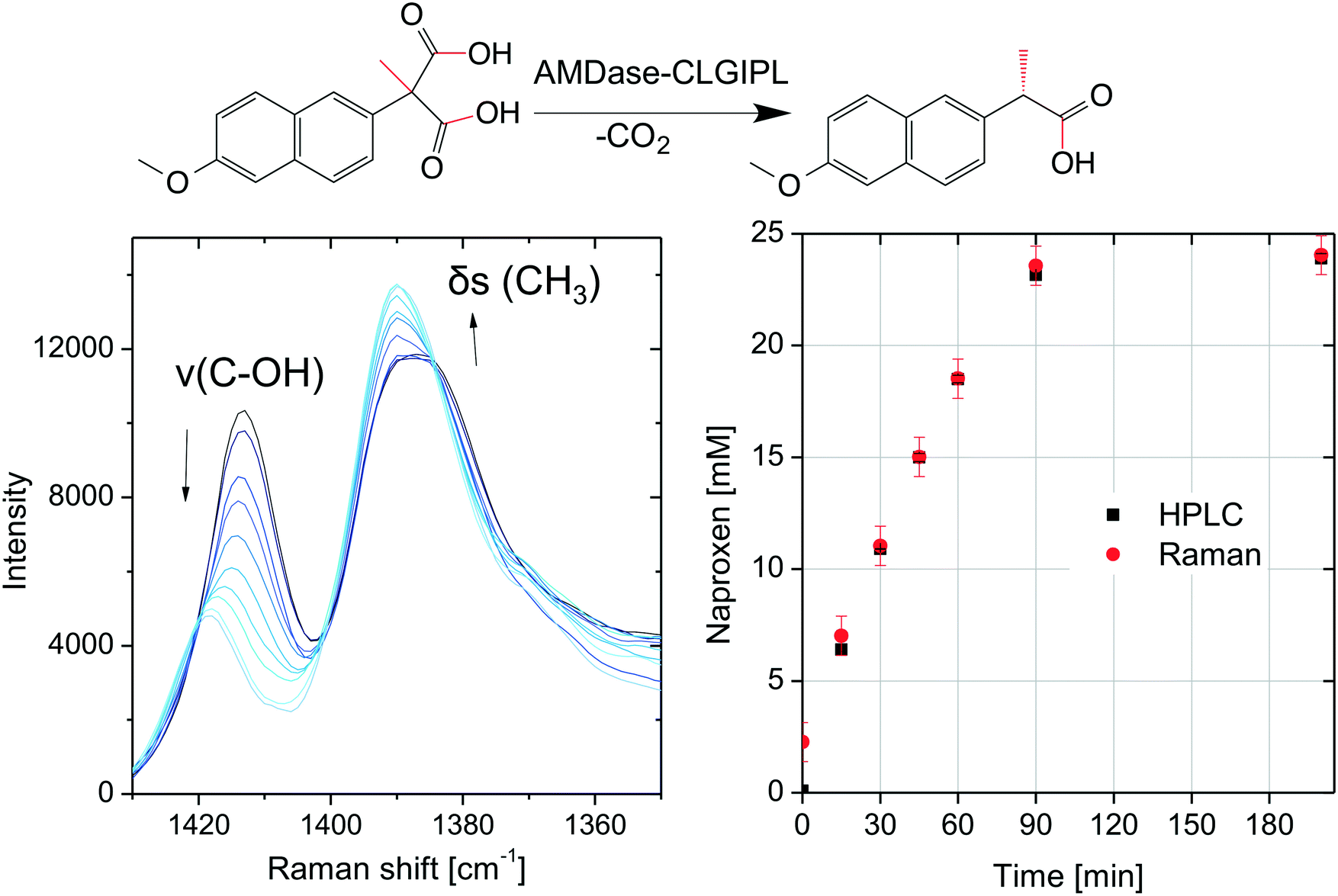 | ||
| Fig. 8 Left: Raman spectra measured during naproxen synthesis, arrows indicate the temporal course of the reaction; right: naproxen concentration determined with HPLC and Raman spectroscopy (RMSEP indicated by error bars); reaction conditions: 30 °C, starting material naproxen malonate (25 mM) in aqueous solution, pH 8, free AMDase-CLGIPL (80 mg L−1). | ||
The minor change in the intensity of the CH3 vibration can be explained by the transformation of the adjacent carbon atom from a quaternary to a tertiary binding mode. For the C–OH group, a stronger change can be expected, as the decarboxylation removes one of the carboxylic acid groups and thus changes the binding strength of the second one. Consistently, a decrease in intensity and an energy shift are observed.
Multivariate calibration was necessary for quantitative concentration determination since the observed Raman bands are not baseline separated. The chemometric modeling was performed with the partial least squares (PLS) method. The data were fit well using the calculated PLS model with three factors, which was characterized by a coefficient of determination of 0.99 and a root mean square error (RMSE) of calibration of 0.3 mM corresponding to a relative RMSE of 1% (normalized to the concentration range). In spite of the very low number of twelve spectra used for calibrating the model, it was applicable to predict concentrations from seven spectra not included in the calibration data set with a root mean square error of prediction of 0.8 mM corresponding to a relative RMSE of 4%. Although the PLS model was calibrated with a small data set and therefore lacks robustness, it is possible to monitor the course of the reaction as shown in Fig. 8. The very low root mean square errors indicate an excellent applicability of Raman spectroscopy to the monitoring of naproxen synthesis with free AMDase. Furthermore, Raman measurements were performed for the biocatalytic naproxen synthesis with AMDase-CLGIPL immobilized on amino C2 acrylate. In this case, far fewer meaningful variations are visible in the spectra throughout the reaction (see the ESI,† Fig. S4). This is not surprising as a high amount of carrier particles is present in the focal volume irradiated by the laser. The Raman bands in the spectral range from 1350 to 1430 cm−1 are attenuated and their shape is influenced, but can be distinguished in spite of the carrier. The suggested reactor for the biocatalytic naproxen synthesis is the RBR. In this reactor type, such disturbances can be overcome completely, since enzyme-free optical conditions are present. Thus, Raman spectroscopy is a promising in-line analytical technique for the biocatalytic synthesis of naproxen. The proof-of-principle of in-line concentration determination completes the process design for naproxen synthesis.
Conclusions
Kinetic investigations of the immobilized AMDase-CLGIPL reveal a 300-fold higher Michaelis–Menten constant (KM) in the naproxen malonate conversion and about 40% reduced specific activity compared to the free enzyme. Furthermore, AMDase-CLGIPL is inhibited by the formed naproxen. Nevertheless, naproxen synthesis can be realized without in situ product removal. Thus, the most appropriate reactor types are the batch and plug-flow-reactors as continuous variants.We could successfully scale-up the enzymatic process from 1 mL to 100 mL in two different stirred tank reactor concepts. The productivities of biocatalytic enantiopure (S)-naproxen synthesis are in a range for applicability of the biocatalyst to pharmaceutical production; thus, immobilized AMDase-CLGIPL is proven to be a promising option for (S)-naproxen synthesis. We are confident that this concept can be extended to the synthesis of (R)-naproxen with an appropriate enzyme variant, or even to other substrates, given that similar enzymatic parameters apply to these compounds.
Furthermore, the applicability of Raman spectroscopy as an in-line analytical method was evaluated for the biocatalytic naproxen synthesis. In spite of a small data set, a chemometric model with low root mean square errors of calibration and prediction was calculated, indicating the high performance capacity of Raman spectroscopy as an online analytical method for the given reaction.
Acknowledgements
Financial support from the Deutsche Bundesstiftung Umwelt (DBU, eng. German Federal Environmental Foundation), grant no. AZ30818-32, is gratefully acknowledged. We thank Emil Byström (SpinChem AB, Umeå, Sweden) for the kind provision of the rotating bed reactor, and Björn Neuer and Prof. Dr. Gerrit A. Luinstra from the Institute of Technical and Macromolecular Chemistry (University of Hamburg) for 1H NMR purity analysis of (S)-naproxen. We thank tec5 AG (Oberursel, Germany) for providing the Raman spectrometer for the feasibility study and Tim Ramelow for performing the Raman measurements.References
- Applied Biocatalysis: From Fundamental Science to Industrial Applications, ed. L. Hilterhaus, A. Liese, U. Kettling and G. Antranikian, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016 Search PubMed.
- J. Tao, G.-Q. Lin and A. Liese, Biocatalysis for the pharmaceutical industry: Discovery, Development, and Manufacturing, John Wiley Sons (Asia) Pte Ltd, Singapore, 2009 Search PubMed.
- R. Wohlgemuth, I. Plazl, P. Znidarsic-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33(5), 302–314 CrossRef CAS PubMed.
- A. J. Straathof, S. Panke and A. Schmid, Curr. Opin. Biotechnol., 2002, 13(6), 548–556 CrossRef CAS PubMed.
- Industrial Biotransformations, ed. A. Liese, K. Seelbach and C. Wandrey, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- A. van den Wittenboer, L. Hilterhaus and A. Liese in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- R. Kourist, P. Domínguez de María and K. Miyamoto, Green Chem., 2011, 13(10), 2607–2618 RSC.
- S. Yoshida, J. Enoki, R. Kourist and K. Miyamoto, Biosci., Biotechnol., Biochem., 2015, 79(12), 1965–1971 CrossRef CAS PubMed.
- S. K. Gaßmeyer, J. Wetzig, C. Mügge, M. Aßmann, J. Enoki, L. Hilterhaus, R. Zuhse, K. Miyamoto, A. Liese and R. Kourist, ChemCatChem, 2016, 8(5), 916–921 CrossRef.
- M. Aßmann, C. Mügge, S. Gaßmeyer, J. Enoki, L. Hilterhaus, R. Kourist, A. Liese and S. Kara, Front. Microbiol., 2017, 8(448), 1–8 Search PubMed.
- D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25(2), 66–73 CrossRef CAS PubMed.
- A. Liese and S. Lütz, Nonimmobilized Biocatalysts in Industrial Fine Chemical Synthesis, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim, 2006 Search PubMed.
- R. Yuryev, S. Strompen and A. Liese, Beilstein J. Org. Chem., 2011, 7, 1449–1467 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42(15), 6223–6235 RSC.
- P. Tufvesson, W. Fu, J. S. Jensen and J. M. Woodley, Food Bioprod. Process., 2010, 88(1), 3–11 CrossRef CAS.
- R. H. Ringborg and J. M. Woodley, React. Chem. Eng., 2016, 1(1), 10–22 CAS.
- S. Kara, J. J. Mueller and L. Liese, Chim. Oggi, 2011, 29(2), 38–41 CAS.
- J. J. Mueller, S. Baum, L. Hilterhaus, M. Eckstein, O. Thum and A. Liese, Anal. Chem., 2011, 83(24), 9321–9327 CrossRef CAS PubMed.
- J. Gebhard, D. Sellin, L. Hilterhaus and A. Liese, Chem. Ing. Tech., 2013, 85(7), 1016–1022 CrossRef CAS.
- K. A. Bakeev, Process analytical technology, Blackwell Publishing, Oxford, 2005 Search PubMed.
- T. de Beer, A. Burggraeve, M. Fonteyne, L. Saerens, J. P. Remon and C. Vervaet, Int. J. Pharm., 2011, 417(1–2), 32–47 CrossRef CAS PubMed.
- T. Zor and Z. Selinger, Anal. Biochem., 1996, 236(2), 302–308 CrossRef CAS PubMed.
- M. Sardar, I. Roy and M. N. Gupta, Biotechnol. Prog., 2003, 19(6), 1654–1658 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40(6), 1451–1463 CrossRef CAS.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42(15), 6236–6249 RSC.
- E. Durand, J. Lecomte, B. Baréa, G. Piombo, E. Dubreucq and P. Villeneuve, Process Biochem., 2012, 47(12), 2081–2089 CrossRef CAS.
- A. Petrenz, P. D. D. María, A. Ramanathan, U. Hanefeld, M. B. Ansorge-Schumacher and S. Kara, J. Mol. Catal. B: Enzym., 2015, 114, 42–49 CrossRef CAS.
- J. M. Guisan, Immobilization of enzymes, Humana Press Inc., New Jersey, 2006 Search PubMed.
- R. Obata and M. Nakasako, Biochemistry, 2010, 49(9), 1963–1969 CrossRef CAS PubMed.
- L. G. Lee and G. M. Whitesides, J. Org. Chem., 1986, 51(1), 25–36 CrossRef CAS.
- A. Liese, M. Karutz, J. Kamphuis, C. Wandrey and U. Kragl, Biotechnol. Bioeng., 1996, 51(5), 544–550 CrossRef CAS PubMed.
- H. Mallin, J. Muschiol, E. Byström and U. T. Bornscheuer, ChemCatChem, 2013,(5), 3529–3532 CrossRef CAS.
- T. A. Rogers and A. S. Bommarius, Chem. Eng. Sci., 2010, 65(6), 2118–2124 CrossRef CAS PubMed.
- R. Liu, Water-Insoluble Drug Formulaton, Internpharm/CRC Press, Florida, 2000 Search PubMed.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15(1), 266–274 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9(12), 1273 RSC.
- E. Pretsch, P. Bühlmann and M. Badertscher, Structure Determination of Organic Compounds, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009 Search PubMed.
- B. Schrader, Infrared and Raman Spectroscopy, VCH Verlagsgesellschaft mbH, Weinheim, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, Raman and GC data for process analysis. See DOI: 10.1039/c7re00043j |
| This journal is © The Royal Society of Chemistry 2017 |