
Tandem μ-reactor-GC/MS for online monitoring of aromatic hydrocarbon production via CaO-catalysed PET pyrolysis†
S.
Kumagai
 *a,
R.
Yamasaki
a,
T.
Kameda
a,
Y.
Saito
a,
A.
Watanabe
b,
C.
Watanabe
b,
N.
Teramae
bc and
T.
Yoshioka
a
*a,
R.
Yamasaki
a,
T.
Kameda
a,
Y.
Saito
a,
A.
Watanabe
b,
C.
Watanabe
b,
N.
Teramae
bc and
T.
Yoshioka
a
aGraduate School of Environmental Studies, Tohoku University, 6-6-07 Aoba, Aramaki-aza, Aoba-ku, Sendai, Miyagi 980-8579, Japan. E-mail: kumagai@env.che.tohoku.ac.jp; Fax: +81 22 795 7212; Tel: +81 22 795 7212
bFrontier Laboratories Ltd., 4-16-20, Saikon, Koriyama, Fukushima 963-8862, Japan
cDepartment of Chemistry, Graduate School of Science, Tohoku University, Aoba-ku, Sendai, Miyagi 980-8578, Japan
First published on 1st September 2017
Abstract
The present work demonstrates the online monitoring of aromatic hydrocarbon production via two-step CaO catalysed pyrolysis of poly(ethylene terephthalate) (PET), employing tandem μ-reactor-gas chromatography/mass spectrometry (TR-GC/MS). PET produces high-boiling terephthalic acid (TPA) during pyrolysis, which hinders the online monitoring of PET pyrolysis. In this work, TR allowed for independent control of the PET pyrolysis and CaO catalytic reaction with a very small sample loading (<1 mg) and split injection into the GC/MS (split ratio 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) system; thus, fatal line clogging by TPA could be avoided. Thus, we successfully demonstrated the effect of CaO basicity on the time- and temperature-dependent dynamic production of aromatic hydrocarbons. Strongly basic CaO accelerated the decarboxylation of PET pyrolysates to afford useful aromatic hydrocarbons such as benzene, toluene, and styrene with 99.7% selectivity in the oil. In contrast, weakly basic CaO enhanced benzophenone production in preference to benzene formation. The poor deoxygenation ability of the weakly basic CaO increased the concentration of oxygen-containing compounds in the oil. Finally, the time- and temperature-dependent dynamic pathways and the mechanism involving strongly basic/weakly basic CaO were established. These findings allow for a clearer understanding of the nature of PET catalytic pyrolysis, which will be helpful for advancing PET recycling. Furthermore, the novel methodology—online monitoring of a two-step pyrolysis–catalytic upgrading process involving high-boiling compounds—will gain the highest demand in the fields of green chemistry and reaction engineering.
:1) system; thus, fatal line clogging by TPA could be avoided. Thus, we successfully demonstrated the effect of CaO basicity on the time- and temperature-dependent dynamic production of aromatic hydrocarbons. Strongly basic CaO accelerated the decarboxylation of PET pyrolysates to afford useful aromatic hydrocarbons such as benzene, toluene, and styrene with 99.7% selectivity in the oil. In contrast, weakly basic CaO enhanced benzophenone production in preference to benzene formation. The poor deoxygenation ability of the weakly basic CaO increased the concentration of oxygen-containing compounds in the oil. Finally, the time- and temperature-dependent dynamic pathways and the mechanism involving strongly basic/weakly basic CaO were established. These findings allow for a clearer understanding of the nature of PET catalytic pyrolysis, which will be helpful for advancing PET recycling. Furthermore, the novel methodology—online monitoring of a two-step pyrolysis–catalytic upgrading process involving high-boiling compounds—will gain the highest demand in the fields of green chemistry and reaction engineering.
Introduction
Plastic is a necessary material for technological innovation in diverse fields because it has desirable properties such as light weight, flexibility, and transparency; further, various functionalities can be incorporated into plastic by combining it with polymers, additives, and metals. However, complex plastics are hard to recycle and cannot be treated by current recycling methods due to technical and economic reasons. Therefore, the development of a new technique that can advance plastic recycling technology to the next level is desired.Poly(ethylene terephthalate) (PET) is the most commonly used polyester across the world and is often combined with additives and metals for modification of its properties to suit the targeted applications. Therefore, effective recycling of PET waste into reprocessed resin or chemical feedstock, except for clear bottles, hasn't been achieved yet. Pyrolysis is widely used to break down polymers into smaller molecules by heat under an inert atmosphere, wherein both plastics and organic additives are converted into gases, liquids, and solids.1 However, pyrolysis of PET produces significant amounts of high-boiling terephthalic acid (TPA; b.p. ∼400 °C),2,3 which causes corrosion and clogging of pipes.4,5 We previously developed a process for the simultaneous recovery of benzene (an important petrochemical feedstock)-rich aromatic oils and complete TPA suppression by using calcium oxide (CaO).6–8 This process has been extended to mixed plastics,9,10 metal-PET composites,6 and PET-based waste carpets.11 Furthermore, a CaO-based two-step process (Scheme 1) has been developed to set optimized independent temperatures for the PET degradation and CaO reaction,12,13 and applied for the simultaneous recovery of benzene and metals from PET-metal composites.14 Some other research groups have reported that FeOOH,15,16 TiO2/SiO2,17 and ZSM-511 suppress TPA formation during PET pyrolysis.
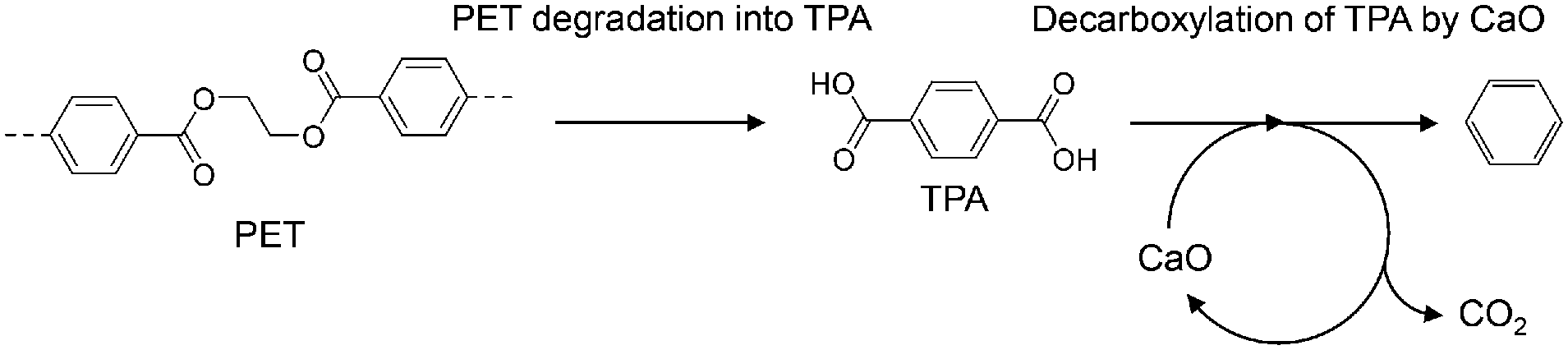 | ||
| Scheme 1 Two-step PET degradation and decarboxylation of TPA by CaO. | ||
Thus, pyrolysis of PET is an important and attractive topic for advances in plastic recycling, and CaO is one of the most inexpensive catalysts that can aid the conversion of waste PET into benzene-rich aromatic oil.
Online product analysis is the best method to reveal the dynamic behaviour of product emission. Thermogravimetry-mass spectrometry (TG-MS) and TG-Fourier transform infrared spectrometry (TG-FTIR) are well known techniques for online product analysis during the pyrolysis of various types of plastics.18–23 These methods are often carried out by mixing plastics and solid catalysts or adsorbents to evaluate the performance of the solids and the product emission behaviour.24–27 However, the drawbacks of these analyses are as follows: (1) high-boiling compounds (b.p. <300 °C) are deposited inside the long transfer tube connecting the TG and MS or FTIR components. (2) Plastics and mixed solids are heated via the same temperature programme, while the optimum temperatures for the pyrolysis of plastics and reaction with solids are different in most cases. The first drawback is more serious in the case of PET pyrolysis as it produces significant amounts of acidic high-boiling TPA that leads to fatal line clogging and corrosion.4,5 Regarding the second drawback, the optimum temperatures for PET pyrolysis and various catalysts including CaO are different.13
To overcome these obstacles, tandem μ-reactor-GC/MS (TR-GC/MS) (Fig. 1)28 was employed for the two-step PET pyrolysis–CaO process, for the first time. The independently controlled furnaces facilitate the pyrolysis of PET and the reaction with CaO at respective optimum temperatures. TR-GC/MS can set small sample loading (<1 mg) with high split injection into the GC/MS instrument, TPA is carried to the MS chamber without column blocking. Since TR-GC/MS is a relatively new technique,28 it has only been used for hydrogenation,28 transformation,28 and aromatization29,30,50 to treat comparatively low boiling compounds. Further, the effect of CaO basicity on the upgrading of PET pyrolysates, especially with respect to online monitoring of aromatic hydrocarbon production, has not been investigated before owing to technical hurdles. Therefore, this work presents a pioneering investigation into the online monitoring of processes involving high-boiling compounds.
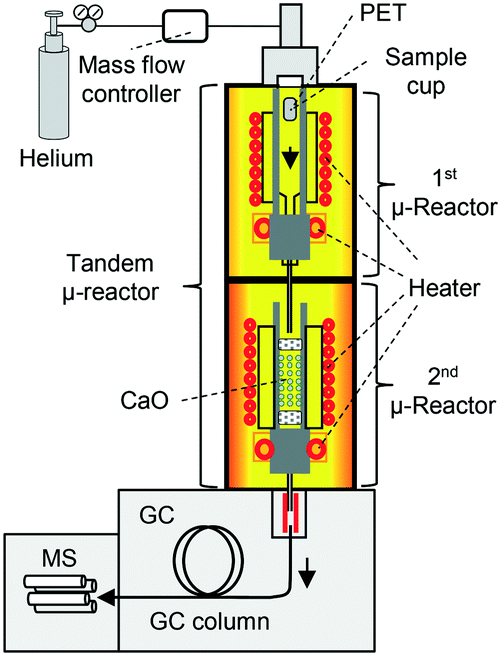 | ||
| Fig. 1 Tandem μ-reactor-GC/MS system. | ||
Herein, a two-step process involving PET pyrolysis and upgrading of PET pyrolysates by CaO (PET/CaO process) was carried out using TR-GC/MS with four types of CaO having different basicities. Two types of TR-GC/MS experiments were performed. The first was a simple two-step experiment involving PET pyrolysis at 450 °C and subsequent upgrading of PET pyrolysates by CaO at 700 °C employing a separation GC column, to reveal the effect of CaO basicity on product distribution. The second involved online monitoring of the emission behaviour of temperature-dependent products from the CaO fixed bed. The PET pyrolysates produced at 450 °C were carried to the CaO bed heated to 450 °C to form a CaO–PET pyrolysate complex. Next, the complex was gradually degraded by heating at the rate of 10 °C min−1. Products generated from the CaO fixed bed were monitored by online GC/MS. Based on the combination of two types of TR-GC/MS studies, the overall process from PET pyrolysis to the time- and temperature-dependent CaO-catalysed pathway and the underlying mechanisms were established.
Experimental
Materials
Powdered PET pellets, ground to a particle size below 250 μm, were used in the present work. The C, H, and O compositions of this sample were 62.4 wt%, 4.3 wt%, and 33.4 wt%, respectively, which is consistent with the theoretical chemical structure of PET.Four types of CaO with different basicities were manufactured via calcination of CaCO3 by Okutama Industries Co., Ltd. Each CaO sample was ground and sieved to a particle size of 0.3–1.0 mm. The characteristics of the prepared CaO are summarized in Table 1. The abbreviations CaOA, CaOB, CaOC, and CaOD indicate the order of basicity of CaO, where CaOC and CaOD show comparable basicity. Thus, CaO samples with different levels of porosity and basicity were successfully prepared. The effect of calcination conditions on the CaO properties is not discussed because it is beyond the scope of this investigation. Detailed characterization procedures, conditions, and results are summarized in the ESI.†
| Name | BET surface area [m2 g−1] | Pore volume [cm3 g−1] | Lattice parameter, aa [Å] | Crystallite sizeb [Å] | Base site densityc [μmol g−1] | Base site density on CaO surfaced [μmol m−2] | CO2 desorption temperaturee [°C] |
|---|---|---|---|---|---|---|---|
| a Average of a determined from (111), (200), (220), (311), (222), (400), and (331) planes. b Determined by the Halder–Wagner method.32 c Determined using the area of the CO2 temperature-programmed desorption (CO2-TPD) profile. d Determined using base site density [μmol g−1] and BET surface area [m2 g−1]. e Peak temperature of CO2 desorption determined by CO2-TPD. | |||||||
| CaOA | 6.4 | 2.7 × 10−2 | 4.805 | 432 | 315 | 49 | 601 |
| CaOB | 1.0 | 6.5 × 10−3 | 4.806 | 528 | 44 | 44 | 524 |
| CaOC | 1.7 | 6.5 × 10−3 | 4.809 | 557 | 30 | 18 | 489 |
| CaOD | 1.4 | 5.2 × 10−3 | 4.809 | 597 | 25 | 18 | 489 |
TR-GC/MS experiments
TR-GC/MS (Fig. 1; TR: Rx-3050 TR, Frontier Laboratories Ltd.; GC: 7890A, Agilent Technology, He gas flow: 1 mL min−1, split injection (100:1); MS: 5975C, Agilent Technology, MSD source: 230 °C, MS quadrupole: 150 °C, acquisition mode: scan, scanning range: m/z = 10–800, MS library: NIST08, MSD ChemStation E.02.01.1177) was employed in all experiments. The details of TR are described elsewhere.28,31 Experiments were repeated as appropriate to ensure reliability of the results.
TR-GC/MS employing a separation column
In order to investigate the effect of CaO basicity on the product distribution, TR-GC/MS experiments were performed using an Ultra ALLOY® metal capillary column UA+-1 (Frontier Laboratories Ltd.). 29.2 mg CaO was filled in a quartz tube reactor inside the 2nd μ-reactor; the air inside the reactor was replaced with helium. The 2nd μ-reactor was heated to 900 °C and kept at this temperature for 1 h to convert CaCO3 and Ca(OH)2 into CaO. XRD data confirmed that both Ca(OH)2 and CaCO3 were successfully removed under these calcination conditions (Fig. S1†). The temperatures of the 1st and 2nd μ-reactors were set to 450 and 700 °C, respectively, which are the optimal temperatures for the pyrolysis of PET, and benzene recovery and CaO regeneration.13,14,33 When the reactor temperature was stabilized, the sample holder filled with PET (1 mg) was placed into the 1st μ-reactor, where the PET was pyrolysed. We confirmed that PET pyrolysis is terminated within 10 min at 450 °C. Therefore, the temperatures of the 1st (450 °C) and 2nd μ-reactors (700 °C) were maintained for 20 min (10 min for PET pyrolysis completion, with an additional 10 min). Products generated from the TR for 20 min were trapped at −196 °C using a cryotrap placed between the GC injection port and the separation column. After terminating the reaction in the TR, the cryotrap was removed and the GC program was promptly initiated (column oven temperature: 40 °C (5 min) → 10 °C min−1 → 300 °C (10 min)). For comparison, pyrolysis of PET without CaO was also carried out.TR-GC/MS employing a deactivated column
In order to reveal time- and temperature-dependent product emission during the temperature-programmed degradation of the CaO–PET pyrolysate complex, in situ product monitoring was carried out using TR-GC/MS with a deactivated column, UADTM (Frontier Laboratories Ltd.). The GC oven temperature was kept at 300 °C to prevent condensation of the products. Temperatures of the 1st and 2nd μ-reactors were first set to 450 °C.7,34 When the reactor temperature was stabilized, the sample holder filled with PET (1 mg), was placed into the 1st μ-reactor, where the PET was pyrolysed. The PET pyrolysates were carried to the 2nd μ-reactor, where they reacted with CaO to form a complex.7,34 20 min following PET injection, the temperature of the 2nd μ-reactor was increased to 800 °C at the rate of 10 °C min−1. MS data were recorded from the instant of PET injection to the end of the heating program in the 2nd μ-reactor. The amounts of the PET sample and CaO, and the calcination conditions were the same as those mentioned in the previous section.Results and discussion
Effect of CaO basicity on product distribution
The GC/MS profiles obtained from the two-step PET pyrolysis–CaO process with CaO having different basicities are summarized in Fig. 2. Major liquid products such as benzene (3), biphenyl (10), toluene (4), and styrene (5) in descending order of intensity were observed in the presence of CaOA, consistent with the results of our previous work using a CaOA filled two-step reactor.14 Therefore, the TR successfully mimics a separately built two-step reactor system.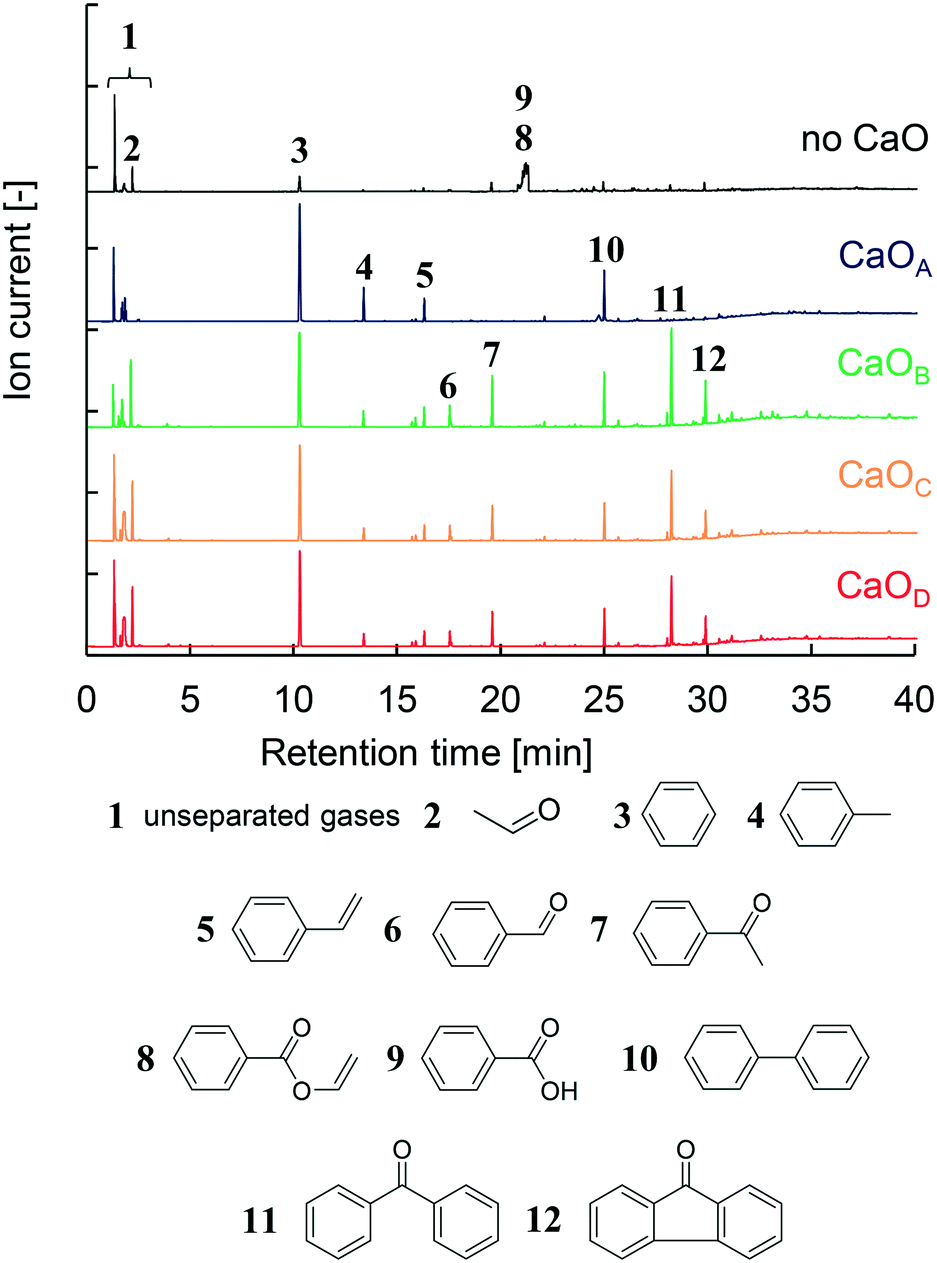 | ||
| Fig. 2 GC/MS spectra obtained in the presence and absence of CaO with different basicities and identification of major products. | ||
The compositions of the major oil compounds (2–12) shown in Fig. 2 are calculated based on the peak area in the GC spectrum and summarized in Table 2. Unseparated gases (1) were not used for the peak area calculation. Based on our previous work,14 these gases are CO2, CH4, and C2–C4 hydrocarbons, which are collected by liquid N2. In the absence of CaO, benzoic acid (8) accounts for 69.1% of the products, which includes unseparated vinyl benzoate (9), and hydrocarbons comprise 14.2%. Aldehydes such as acetaldehyde (2) and benzaldehyde (6), and ketones such as acetophenone (7), benzophenone (11), and fluorenone (12), are also observed in 16.6% composition. In addition, CO is the major gaseous product in PET pyrolysis14 but it is not captured by liquid N2. Under the present conditions, TPA is not detected because it is rapidly decarboxylated into benzoic acid and benzene at 700 °C in the 2nd μ-reactor.
| Compound | No CaO | CaOA | CaOB | CaOC | CaOD |
|---|---|---|---|---|---|
| Hydrocarbons | 14.2 | 99.7 | 46.8 | 51.4 | 46.2 |
| Benzene | 8.0 | 63.0 | 31.9 | 35.7 | 29.7 |
| Toluene | 0.6 | 11.9 | 3.2 | 3.1 | 3.2 |
| Styrene | 1.3 | 7.4 | 3.0 | 3.7 | 3.5 |
| Biphenyl | 4.3 | 17.4 | 8.7 | 8.9 | 9.8 |
| Aldehydes & ketones | 16.6 | 0.3 | 53.2 | 48.6 | 53.8 |
| Acetaldehyde | 6.8 | 0.0 | 12.5 | 14.7 | 11.8 |
| Benzaldehyde | 0.6 | 0.2 | 4.1 | 0.9 | 3.9 |
| Acetophenone | 3.7 | 0.0 | 8.8 | 8.8 | 9.5 |
| Benzophenone | 2.2 | 0.0 | 19.7 | 17.2 | 20.4 |
| Fluorenone | 3.3 | 0.0 | 8.0 | 7.0 | 8.2 |
| Benzoic acid & vinyl benzoate | 69.1 | 0.0 | 0.0 | 0.0 | 0.0 |
| Total/area% | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
In the presence of CaOA, the hydrocarbon content was increased to 99.7%, with the major component being benzene (63.0%). Other oxygen-containing compounds except for aldehydes were prevented in the presence of CaOA. Unexpectedly, in the presence of CaOB, the composition of aldehydes and ketones increased to 53.2% while that of hydrocarbons decreased to 46.8%. In particular, the benzophenone content was notably enhanced to 19.7%.
CaOA and CaOB have comparable base site densities on the surface, which suggests that the base strength on the CaO surface strongly influences the product selectivity. The product distribution in the presence of CaOC and CaOD showed a similar trend to that in the presence of CaOB (46.2–51.4% hydrocarbons and 48.6–53.8% aldehydes & ketones). Benzoic acid and vinyl benzoate were completely prevented in the presence of all the CaO samples. Thus, it could be concluded that the CaO basicity strongly influences the product distribution; strongly basic CaO selectively afforded hydrocarbons, while weakly basic CaO enhanced the formation of aldehydes and ketones.
Product emission behaviour at the instant of PET injection
The mass spectra of the selected major compounds obtained in the presence and absence of CaO with different basicities are summarized in Fig. 3. In this section, the behaviour in the first 20 min following PET injection at 450 °C is discussed. The high-boiling TPA exerted no adverse effect during the online monitoring of products. Considerable production of CO2 (m/z 44, Fig. 3(b)), benzene (m/z 78, Fig. 3(c)), and acetaldehyde (m/z 29, Fig. 3(f)) is confirmed, while the peak intensities of the other hydrocarbons (Fig. 3(d) and (e)) and ketones (Fig. 3(h) and (i)) are very low.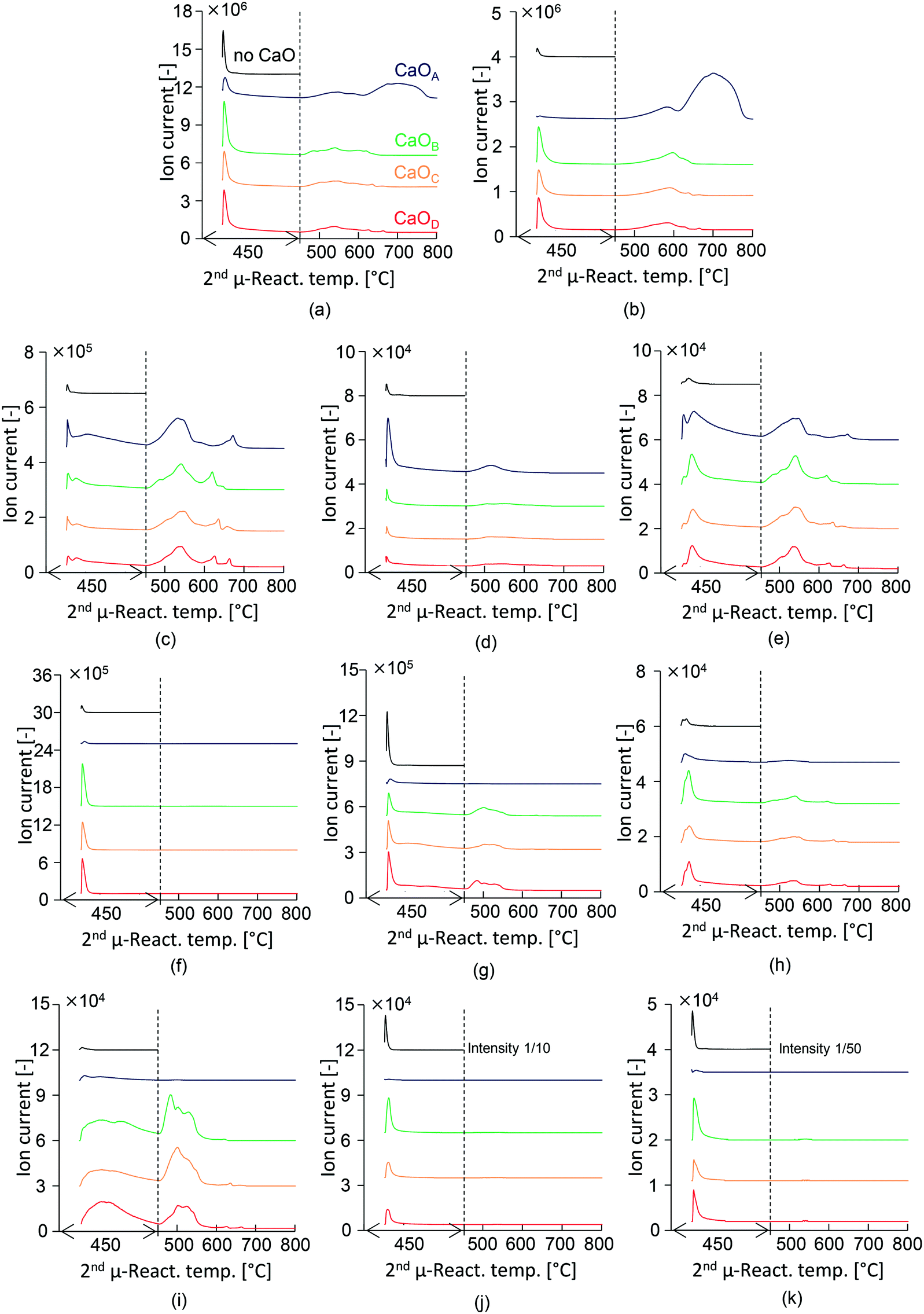 | ||
| Fig. 3 Time- and temperature-dependent mass spectra obtained in the absence and presence of CaO with different basicities: (a) total ion chromatogram; (b) m/z 44 (molecular ion of CO2); hydrocarbons: (c) m/z 78 (molecular ion of benzene), (d) m/z 92 (molecular ion of toluene), and (e) m/z 154 (molecular ion of biphenyl); aldehydes and ketones: (f) m/z 29 (main fragment ion of acetaldehyde), (g) m/z 105 (the sharp peak at the instant of PET injection is mainly derived from the main fragment ion of acetophenone, benzoic acid, and vinylbenzoate; the broad peak at ∼500 °C is derived from benzophenone), (h) m/z 180 (molecular ion of fluorenone), and (i) m/z 182 (molecular ion of benzophenone); acids: (j) m/z 122 (molecular ion of benzoic acid) and (k) m/z 149 (the strongest fragment ion of TPA). | ||
A sharp TIC peak is observed at the instant of PET injection, in the presence of each type of CaO, while the intensity of CaOA is significantly lower than that of other CaO types. This is due to the effective capture of CO2, benzoic acid, and TPA because CaOA has the strongest basicity. Aromatic hydrocarbons such as benzene (m/z 78, Fig. 3(c)), toluene (m/z 92, Fig. 3(d)), and biphenyl (m/z 154, Fig. 3(e)) in the presence of CaOA are enhanced as compared to the experiments with the other types of CaO. These results support those discussed in the previous section. Benzene is produced at the instant of PET injection and it continues for 20 min. Toluene is produced at the instant of PET injection. The generation of benzene and biphenyl is synchronized, suggesting that the produced benzene is immediately consumed for biphenyl formation.
The total ion chromatogram (TIC) of the products is shown in Fig. 3(a). In the absence of CaO, a sharp peak is observed at the instant of PET injection, which is stabilized within 10 min. PET pyrolysis is thus completed within 10 min. Highly intense peaks due to TPA (m/z 149, Fig. 3(k)) and benzoic acid (m/z 122, Fig. 3(j)); and acetophenone and benzoic acid (m/z 105, Fig. 3(g)) are observed.
The intensity of the m/z 105 peak, which corresponds to the main fragment ion of acetophenone and benzoic acid, is substantially decreased in the presence of CaOA, suggesting that CaOA with the strongest basicity enhanced the removal of acetophenone and benzoic acid. CO2 is not detected in the presence of CaOA, suggesting that it is immediately absorbed as CaCO3.35,36
In contrast, CaOB, CaOC, and CaOD enhanced the formation of ketones such as fluorenone (m/z 180, Fig. 3(h)) and benzophenone (m/z 182, Fig. 3(i)), as mentioned in the previous section. Fluorenone is produced at the instant of PET injection, while benzophenone is slowly generated with time, similar to the case of benzene. This observation suggests that the production of benzene and benzophenone is slow and competitive, with the selectivity of these products being strongly influenced by the CaO basicity. In addition, the low-basicity CaO increased the formation of acetaldehyde (m/z 29, Fig. 3(f)), which is consistent with the results reported in the previous section. The incomplete adsorption of benzoic acid, TPA, and CO2 is due to the lower basicity of CaO.
Temperature-dependent product emission behaviour
The 2nd μ-reactor was heated at the rate of 10 °C min−1 to investigate the temperature-dependent product emission behaviour. Benzene production gradually increased with increasing temperature; the peak top temperatures were 532, 541, 545, and 541 °C in the presence of CaOA, CaOB, CaOC, and CaOD, respectively, suggesting that the strongly basic CaOA lowers the benzene production temperature. In addition, multistep benzene production was observed in the presence of all CaO types. The second peak of toluene is observed at ∼500 °C in the presence of CaOA. Biphenyl production was synchronized with benzene production, as explained in the previous section. Fluorenone also showed a similar emission behaviour to benzene, suggesting that it is a secondary product of benzene as well as biphenyl. In the presence of CaOB, CaOC, and CaOD, benzophenone production increased with increasing temperature; the corresponding peak top temperatures were comparable: 503, 501, and 502 °C. Benzophenone production occurred at a temperature ∼40 °C lower than that for benzene. CO2 emission increased with increasing temperature; the peak top temperatures in the presence of CaOA, CaOB, CaOC, and CaOD were 580, 589, 587, and 596 °C, respectively, which were higher than those for benzene and benzophenone production. This result suggested that benzene and benzophenone are first released from the CaO surface, and then, the carboxylate unit is released as CO2 by heating. Strong second CO2 emission (699 °C) from CaOA was observed, which corresponded to the decomposition of CaCO3. Acetophenone was released at temperatures beyond 450 °C.Suggested reaction pathway and mechanism in the two-step PET pyrolysis–CaO process
Based on the product distribution and the time- and temperature-dependent product emission behaviour, the pyrolytic pathway for the major products in the 1st μ-reactor is summarized in Fig. 4.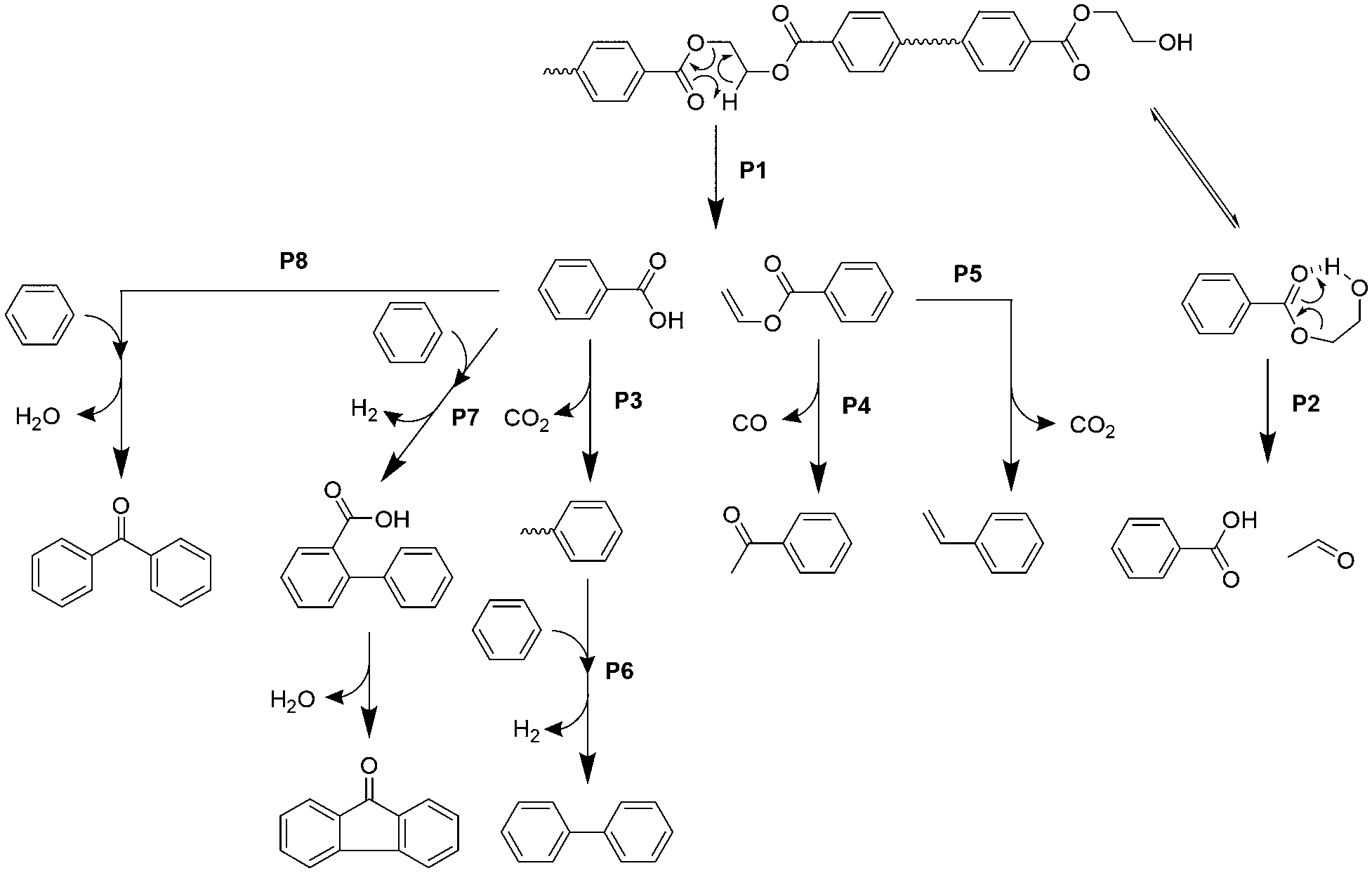 | ||
| Fig. 4 Suggested pathway for major products during the PET pyrolysis. | ||
Carboxyl and vinyl end groups are formed by ester bond cleavage via β-hydrogen transfer to the carbonyl carbon (pathway P1).37,38 Iengar and Ritchie39 reported that the hydroxyethyl end group, which is commonly present in PET, is pyrolysed into a carboxyl end group simultaneously with acetaldehyde production via a seven-membered ring transition state (pathway P2). CO2 production is mainly due to the thermal decarboxylation of TPA and benzoic acid (pathway P3).37 Vinyl benzoate is further converted into acetophenone by rearrangement of the vinyl ester group with successive CO elimination (pathway P4), which is supported by Bednas et al.40 and Allan et al.41 Although P4 is the major pathway for vinyl benzoate, it is converted into styrene via decarboxylation (pathway P5),41 which is consistent with the styrene production in the absence of CaO. On the other hand, production of coupling compounds such as biphenyl, fluorenone, and benzophenone is not significant at 450 °C in the absence of CaO (Fig. 3), while biphenyl, fluorenone, and benzophenone are thermally formed via pathways P6, P7, and P8, respectively, in the 2nd μ-reactor at 700 °C in the absence of CaO.42 The effective suppression of fluorenone and benzophenone in the presence of CaOA is due to the complete adsorption of benzoic acid, which in turn is attributable to the strong basicity.
The pyrolysates are carried into the 2nd μ-reactor, where they react with CaO. The suggested mechanism for the CaO-catalysed reaction and the time- and temperature-dependence of each reaction are summarized in Fig. 5. Multistep production of benzene and CO2 observed in the online monitoring, two decarboxylation pathways, decarboxylation on the CaO surface (pathway C1) and decarboxylation via salt decomposition (pathway C2) are considered. Benzene production at the instant of PET injection mainly progresses at the strongly basic sites via pathway C1. The H atom of the carboxyl group is acidic, and therefore, benzoic acid is easily deprotonated. Strong interaction between the CaO surface and the carboxylate induces homolysis of the C–C bond between the carbonyl C and the benzene ring. In the case of CaOA, the residual carboxylate is taken up as CaCO3 due to the strong basicity,35,36 while the weakly basic CaO releases the carboxylate as CO2 at 450 °C. The main benzene production stage at temperatures higher than 450 °C occurs by pathways C1 and C2. At the moderately basic sites, a higher temperature is required for decarboxylation (pathway C1). In fact, the benzene production temperature is lowered with increasing CaO basicity, as explained in the previous sections. The residual carboxylate is released as CO2 at temperatures higher than that for benzene release. Beyond 550 °C, decomposition of the salt formed from CaO and BA or TPA34 occurs at the instant of PET injection. CaCO3 is decarbonated at temperatures higher than 600 °C, while CaCO3 formation is favoured by the strongly basic CaOA.
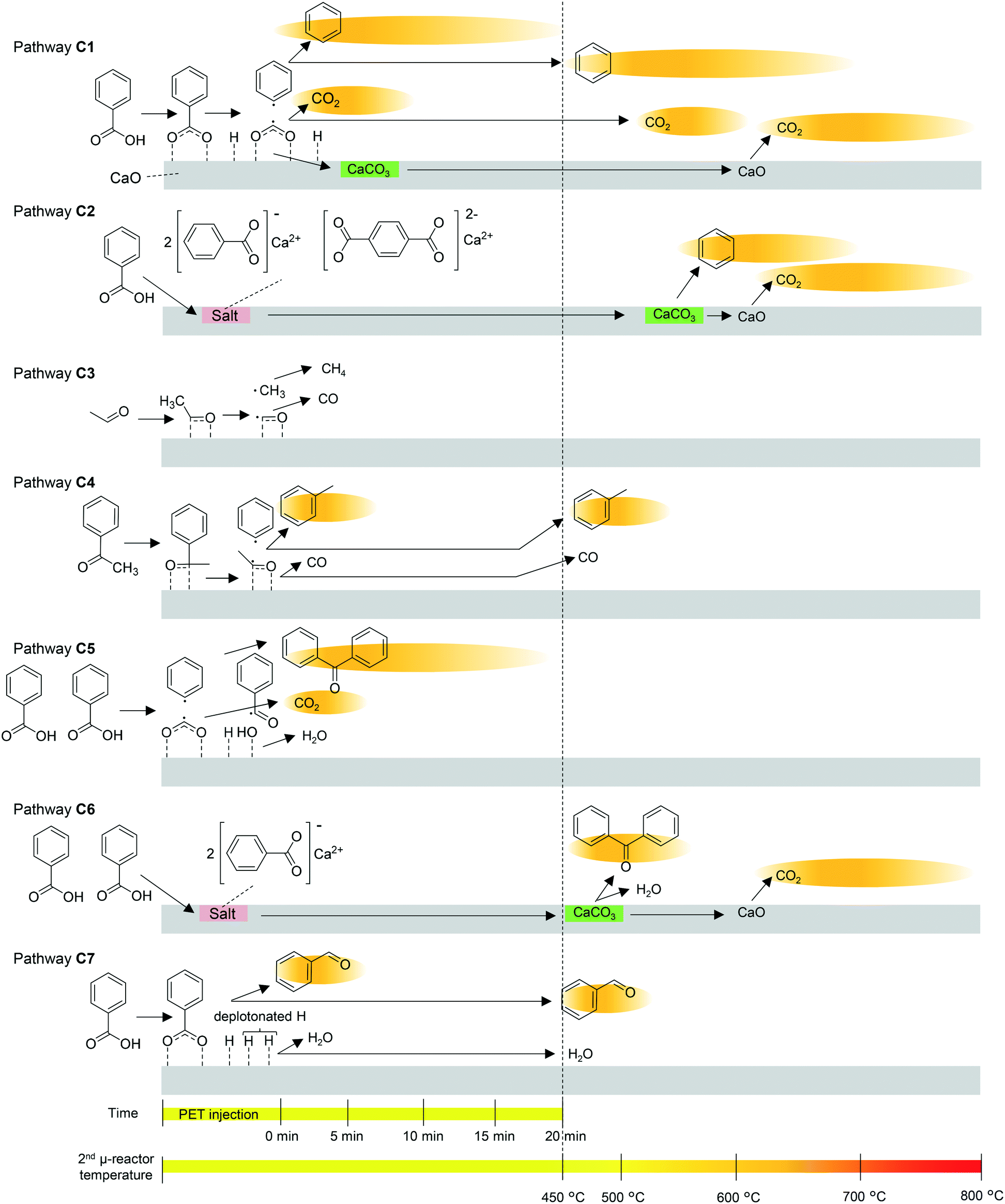 | ||
| Fig. 5 Proposed time- and temperature-dependent CaO-catalysed pathways. The background of each product indicates the time- and temperature range of production observed in online monitoring. | ||
Thus, it is concluded that CaO functions as a decarboxylation catalyst and an absorbent for carboxyl acids and CO2. It is considered that the CO2 absorbent has a negative impact on the reduction of basic sites for the adsorption of carboxyl acids. In contrast, Mckee43 and Cazorla-Amorós et al.44 reported that carbon deposited on the CaCO3 surface is gasified at the interface between CaCO3 and carbon by CO2, which has a positive effect in preventing coke deposition. CO2 absorption might also affect the Boudouard equilibrium (2CO ⇄ CO2 + C),45 and influence the CO/CO2 composition in the products. Thus, it is confirmed that CaCO3 is regenerated to CaO by heating, although the durability of CaO as a catalyst and the competitive effects by CO2 in this system need to be investigated in detail in the future.
The strongly basic CaO enhances acetaldehyde decomposition via pathway C3. The aldehyde oxygen and carbon are strongly attracted to the Ca2+ and O2− sites on CaOA, resulting in methyl radical formation via homolysis of the C–C bond. The methyl radical abstracts hydrogen from the aldehyde, and then, CH4 and CO are released. Although these products are not traced in this work, CH4 generation is reported to be enhanced by CaO addition during PET pyrolysis.6,14
Similarly, acetophenone is absorbed on the CaO surface, and then, a phenyl radical is released via homolytic scission of the C–C bond. The residual acetaldehyde is further decomposed via pathway C3, resulting in a methyl radical. When the phenyl radical and methyl radical are coupled, toluene is formed (pathway C4). Toluene production is significantly enhanced in the presence of CaOA, while acetaldehyde and acetophenone are completely inhibited (Table 2). In addition, simultaneous production of benzene and toluene, and inhibition of acetaldehyde and acetophenone are observed during online monitoring. The presence of a second small peak of toluene at ∼500 °C might be attributed to pathway C4, i.e., the moderately basic sites in CaOA. On the other hand, the weakly basic CaO decomposes acetophenone via pathway C3, but the next acetaldehyde decomposition via pathway C4 is insufficient. Therefore, enhanced production of acetaldehyde and suppression of acetophenone are observed.
The weakly basic CaO enhances the production of benzophenone via a free-radical mechanism involving phenyl and benzoyl radicals (pathways C5 and C6).46 CaOB, CaOC, and CaOD have much lower basicity than CaOA, and the Ca2+ ions show Lewis acid properties,47 which enhance the formation of benzoyl radicals. At the same time, a phenyl radical is produced via pathway C1, which is not significant compared with CaOA. Coupling of the benzoyl and phenyl radicals then forms benzophenone. The two-step production of benzophenone indicated during online monitoring implies that benzophenone is produced on the CaO surface at lower temperatures (pathway C5) and via salt decomposition at higher temperatures (pathway C6) in a similar manner to benzene being produced via pathways C1 and C2.
A slight increase in benzaldehyde formation in the presence of the weakly basic CaO could be explained by the hydrogenation of benzoic acid (pathway C7). Yokoyama et al.48 and Hölderich and Tjoe49 reported that heterogeneous catalysts that have very weakly acidic sites and relatively strongly basic sites as well as a small specific surface area (∼2 m2 g−1) are suitable for the hydrogenation of benzoic acid. CaOB, CaOC, and CaOD have much lower basicity than CaOA, and the Ca2+ ions show Lewis acid properties.47 In the present work, the selectivity for hydrogenation is very low when using CaOB, CaOC, and CaOD, and they are still selective for decarboxylation, which is due to their low lattice energy and high basicity for hydrogenation.48 In pathway C1, benzene is produced via abstraction of hydrogen from the CaO surface by the phenyl radical. However, if the phenyl radical is consumed to form biphenyl (pathway P6) or fluorenone (pathway P7), H adsorbed on the CaO surface is retained. In addition, the unshared electron pair on oxygen on the CaOA surface could abstract hydrogen from the benzene ring as a Lewis base, which enhances the coupling reaction and retains H on the CaO surface. In fact, biphenyl and fluorenone are substantially enhanced in the presence of any CaO; therefore, the amount of hydrogen was apparently enough for slight enhancement of benzaldehyde.
Thus, the present work clearly demonstrated that the product selectivity and time- and temperature-dependent product emission behaviour are significantly influenced by the basicity of CaO using a TR-GC/MS system.
Conclusions
Online TR-GC/MS was successfully applied for the first time to monitor the formation of aromatic hydrocarbons via a two-step PET/CaO process, without line clogging by the high-boiling TPA. The effects of CaO basicity on the product selectivity in the oil were evaluated, and the maximum (99.7%) formation of aromatic hydrocarbons such as benzene, toluene, and styrene was observed in the presence of strongly basic CaO. In contrast, weakly basic CaO resulted in decreased (to ∼50%) product selectivity, while benzophenone production was significantly enhanced. The pyrolytic pathway and the time- and temperature-dependent CaO-catalysed pathways and mechanisms were also established based on the online monitoring of the products; strongly basic CaO selectively enhanced decarboxylation and reduced the required temperature. On the other hand, benzoyl radical formation via dehydroxylation competes against decarboxylation in the presence of weakly basic CaO. Thus, strong basicity is required for the highly selective production of aromatic hydrocarbons.The novel TR-GC/MS methodology proposed herein allows for elucidation of the complicated time- and temperature-dependent pyrolysis/catalysis behaviour, as well as catalyst evaluation. Our approach can be used for the development of new sustainable chemical processes focusing on various combinations of polymeric waste and catalysts, which would be beneficial for reaction engineering and advancing green chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JSPS KAKENHI (grant number 16H05893).Notes and references
- S. Kumagai and T. Yoshioka, Sekiyu Gakkaishi, 2016, 59, 243–253 CrossRef CAS.
- M. Dzieciol and J. Trzeszczynski, J. Appl. Polym. Sci., 2001, 81, 3064–3068 CrossRef CAS.
- T. Yoshioka, G. Grause, C. Eger, W. Kaminsky and A. Okuwaki, Polym. Degrad. Stab., 2004, 86, 499–504 CrossRef CAS.
- M. Fukushima, M. Shioya, K. Wakai and H. Ibe, J. Mater. Cycles Waste Manage., 2009, 11, 11–18 CrossRef CAS.
- M. Fukushima, B. Wu, H. Ibe, K. Wakai, E. Sugiyama, H. Abe, K. Kitagawa, S. Tsuruga, K. Shimura and E. Ono, J. Mater. Cycles Waste Manage., 2010, 12, 108–122 CrossRef CAS.
- S. Kumagai, G. Grause, T. Kameda and T. Yoshioka, J. Mater. Cycles Waste Manage., 2014, 16, 282–290 CrossRef CAS.
- S. Kumagai, G. Grause, T. Kameda and T. Yoshioka, Chem. Lett., 2014, 43, 637–639 CrossRef CAS.
- T. Yoshioka, E. Kitagawa, T. Mizoguchi and A. Okuwaki, Chem. Lett., 2004, 33, 282–283 CrossRef CAS.
- G. Grause, S. Matsumoto, T. Kameda and T. Yoshioka, Ind. Eng. Chem. Res., 2011, 50, 5459–5466 CrossRef CAS.
- S. Kumagai, I. Hasegawa, G. Grause, T. Kameda and T. Yoshioka, J. Anal. Appl. Pyrolysis, 2015, 113, 584–590 CrossRef CAS.
- S. Du, J. A. Valla, R. S. Parnas and G. M. Bollas, ACS Sustainable Chem. Eng., 2016, 4, 2852–2860 CrossRef CAS.
- G. Grause, T. Handa, T. Kameda, T. Mizoguchi and T. Yoshioka, J. Appl. Polym. Sci., 2011, 120, 3687–3694 CrossRef CAS.
- G. Grause, T. Handa, T. Kameda, T. Mizoguchi and T. Yoshioka, Chem. Eng. J., 2011, 166, 523–528 CrossRef CAS.
- S. Kumagai, G. Grause, T. Kameda and T. Yoshioka, Environ. Sci. Technol., 2014, 48, 3430–3437 CrossRef CAS PubMed.
- T. Masuda, Y. Miwa, K. Hashimoto and Y. Ikeda, Polym. Degrad. Stab., 1998, 61, 217–224 CrossRef CAS.
- T. Masuda, T. Kushino, T. Matsuda, S. R. Mukai, K. Hashimoto and S.-i. Yoshida, Chem. Eng. J., 2001, 82, 173–181 CrossRef CAS.
- E. Obuchi, M. Suyama and K. Nakano, J. Mater. Cycles Waste Manage., 2001, 3, 88–92 CAS.
- G. Liu, Y. Liao and X. Ma, Waste Manage., 2017, 61, 315–326 CrossRef CAS PubMed.
- W. Jin, D. Shen, Q. Liu and R. Xiao, Polym. Degrad. Stab., 2016, 133, 65–74 CrossRef CAS.
- W. J. Liu, K. Tian, H. Jiang and H. Q. Yu, J. Hazard. Mater., 2016, 310, 217–225 CrossRef CAS PubMed.
- J. Wu, T. Chen, X. Luo, D. Han, Z. Wang and J. Wu, Waste Manage., 2014, 34, 676–682 CrossRef CAS PubMed.
- Z. Czegeny, E. Jakab, M. Blazso, T. Bhaskar and Y. Sakata, J. Anal. Appl. Pyrolysis, 2012, 96, 69–77 CrossRef CAS.
- G. Grause, D. Karakita, J. Ishibashi, T. Kameda, T. Bhaskar and T. Yoshioka, Chemosphere, 2011, 85, 368–373 CrossRef CAS PubMed.
- Z. Sebestyén, E. Barta-Rajnai, J. Bozi, M. Blazsó, E. Jakab, N. Miskolczi, J. Sója and Z. Czégény, Appl. Energy, 2017 DOI:10.1016/j.apenergy.2017.06.032.
- S. Kumagai, G. Grause, T. Kameda and T. Yoshioka, J. Mater. Cycles Waste Manage., 2017, 19, 282–293 CrossRef CAS.
- T. Bhaskar, M. Tanabe, A. Muto and Y. Sakata, J. Anal. Appl. Pyrolysis, 2006, 77, 68–74 CrossRef CAS.
- J. Xu, C. Liu, H. Qu, H. Ma, Y. Jiao and J. Xie, Polym. Degrad. Stab., 2013, 98, 1506–1514 CrossRef CAS.
- C. Watanabe, T. Ramus, R. Meijboom and B. Freeman, Environ. Prog. Sustainable Energy, 2014, 33, 688–692 CrossRef CAS.
- Y. Xue, A. Kelkar and X. Bai, Fuel, 2016, 166, 227–236 CrossRef CAS.
- Y.-M. Kim, J. Jae, H. W. Lee, T. U. Han, H. Lee, S. H. Park, S. Kim, C. Watanabe and Y.-K. Park, Energy Convers. Manage., 2016, 125, 277–289 CrossRef CAS.
- R. R. Freeman, A. Watanabe, C. Watanabe, N. Teramae and K. Wang, J. Anal. Appl. Pyrolysis, 2015, 111, 41–46 CrossRef CAS.
- S. Kumagai, Y. Morohoshi, G. Grause, T. Kameda and T. Yoshioka, RSC Adv., 2015, 5, 61828–61837 RSC.
- N. C. Halder and C. N. J. Wagner, Acta Crystallogr., 1966, 20, 312–313 CrossRef CAS.
- S. Kumagai, G. Grause, T. Kameda, T. Takano, H. Horiuchi and T. Yoshioka, Ind. Eng. Chem. Res., 2011, 50, 1831–1836 CrossRef CAS.
- V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2008, 42, 4170–4174 CrossRef CAS PubMed.
- V. Manovic, E. J. Anthony and D. Loncarevic, Chem. Eng. Sci., 2009, 64, 3236–3245 CrossRef CAS.
- L. H. Buxbaum, Angew. Chem., Int. Ed. Engl., 1968, 7, 182–190 CrossRef CAS.
- I. C. McNeill and M. Bounekhel, Polym. Degrad. Stab., 1991, 34, 187–204 CrossRef CAS.
- H. V. R. Iengar and P. D. Ritchie, J. Chem. Soc., 1956, 3563–3570 RSC.
- M. E. Bednas, M. Day, K. Ho, R. Sander and D. M. Wiles, J. Appl. Polym. Sci., 1981, 26, 277–289 CrossRef CAS.
- R. J. P. Allan, R. L. Forman and P. D. Ritchie, J. Chem. Soc., 1955, 2717–2725 RSC.
- N. Montoya Sánchez and A. de Klerk, Energy Fuels, 2015, 29, 7910–7922 CrossRef.
- D. W. Mckee, Fuel, 1980, 59, 308–314 CrossRef CAS.
- D. Cazorla-Amorós, A. Linares-Solano and C. Salinas-Maltínez de Lecca, Carbon, 1991, 29, 361–369 CrossRef.
- P. L. J. Walker, F. Rusinko, Jr. and L. G. Austin, Gas Reactions of Carbon, New York, 1959 Search PubMed.
- R. A. Hites and K. Biemann, J. Am. Chem. Soc., 1972, 94, 5772–5777 CrossRef CAS.
- M. Calatayud, A. M. Ruppert and B. M. Weckhuysen, Chemistry, 2009, 15, 10864–10870 CrossRef CAS PubMed.
- T. Yokoyama, T. Setoyama, N. Fujita, M. Nakajima, T. Maki and K. Fujii, Appl. Catal., A, 1992, 88, 149–161 CrossRef CAS.
- W. F. Hölderich and J. Tjoe, Appl. Catal., A, 1999, 184, 257–264 CrossRef.
- S. Karagöz, T. Kawakami, A. Kako, Y. Iiguni and H. Ohtani, RSC Adv., 2016, 6(52), 46108–46115 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00097a |
| This journal is © The Royal Society of Chemistry 2017 |