
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe GTPase hGBP1 converts GTP to GMP in two steps via proton shuttle mechanisms†
Ravi
Tripathi
*,
Rachel
Glaves‡
and
Dominik
Marx
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, 44780 Bochum, Germany. E-mail: ravi.tripathi@theochem.rub.de
First published on 22nd August 2016
Abstract
GTPases play a crucial role in the regulation of many biological processes by catalyzing the hydrolysis of GTP into GDP. The focus of this work is on the dynamin-related large GTPase human guanine nucleotide binding protein-1 (hGBP1) which is able to hydrolyze GTP even to GMP. Here, we studied the largely unknown mechanisms of both GTP and GDP hydrolysis steps utilizing accelerated ab initio QM/MM metadynamics simulations to compute multi-dimensional free energy landscapes. We find an indirect substrate-assisted catalysis (SAC) mechanism for GTP hydrolysis involving transfer of a proton from the water nucleophile to a nonbridging phosphoryl oxygen via a proton relay pathway where the rate-determining first step is concerted-dissociative nature. A “composite base” consisting of Ser73, Glu99, a bridging water molecule, and GTP was found to activate the nucleophilic water, thus disclosing the complex nature of the general base in hGBP1. A nearly two-fold reduction in the free energy barrier was obtained for GTP hydrolysis in the enzyme in comparison to bulk solvent. The subsequent GDP hydrolysis in hGBP1 was also found to follow a water-mediated proton shuttle mechanism. It is expected that the proton shuttle mechanisms unravelled for hGBP1 apply to many classes of GTPases/ATPases that possess an optimally-arranged hydrogen bonding network, which connects the catalytic water to a proton acceptor.
Guanosine triphosphate (GTP) hydrolysis catalyzed by enzymes belonging to the GTPase family is vital to a myriad of biological processes such as cell signaling, cell motility, protein synthesis/translocation, etc.1–3 These enzymes, when in the GTP-bound state, are able to transmit a cellular signal by interacting with downstream effectors. Hydrolysis of GTP into guanosine diphosphate (GDP), however, triggers substantial conformational changes within the protein resulting in impaired cellular signaling.4,5 Hence, the whole process can be understood as a switching mechanism,6,7 which involves cycling between a GTP-bound “on” state and a GDP-bound “off” state. The switching between these two states is mandatory for efficient cellular function and a slight modulation of this process is found as one of the leading causes of many forms of cancer and other infectious diseases.8,9 A detailed understanding of the functioning of these processes is crucial to recognize how these enzymes achieve their efficiency, which, in turn, can be useful in developing the therapeutic agents required when they malfunction.
An enormous number of studies have been performed in an attempt to shed light on the nature of the mechanism and the reaction pathway of phosphoryl-transfer reactions in various GTPases/ATPases.1–3 However, despite major biochemical and structural breakthroughs, no universal conclusion has yet been drawn about the detailed molecular mechanism of most of these enzymes. Among various GTPases, the catalytic mechanisms of Ras and EF-Tu have been studied extensively. Recent work on these enzymes supports a substrate-assisted catalysis (SAC) pathway,10–12 in which the substrate catalyzes its own hydrolysis, either directly or indirectly, via a proton shuttle. The GTP hydrolysis is supposed to follow one of three possible paths depending on the bonding patterns of attacking nucleophile (Nu) and leaving group (LG)1,3 (see Fig. 4(b)): in the dissociative pathway, designated DN + AN in the IUPAC nomenclature, bond breaking between the γ-phosphate and leaving oxygen leading to a stable metaphosphate ion is followed by addition of nucleophilic water to the γ-phosphate. On the other hand, the associative (AN + DN) pathway is characterized by formation of a pentacoordinated phosphate intermediate after addition of the nucleophilic water. Inbetween these two cases, a concerted pathway (ANDN) exists where breaking of the Pγ–OLG bond takes place together with formation of the Pγ–ONu bond. Overall, the dissociative and associative pathways represent two extremes of a nucleophilic substitution reaction, whereas the concerted mechanism pursues the path inbetween the two. Unfortunately, the prognosis of the path by experimental studies is not straightforward, as multiple, different pathways can be predicted by the same experimental observable.1 Carefully designed theoretical studies complementing experimental efforts are, therefore, mandatory to resolve such mechanistic insights.
In the present study, we chose the interferon-γ inducible hGBP1, a large GTPase belonging to the family of dynamin-related proteins.13,14 These enzymes show high-turnover GTPase activity and are considered to play a crucial role in host pathogen defense by exerting antibacterial properties.15 hGBP1 is of particular interest as it shares some unique biochemical features such as nucleotide-dependent oligomerization and concentration-dependent GTPase activity that are not observed in other classes of GTPases.16,17 Moreover, the most interesting feature of hGBP1 is its ability to hydrolyze GTP, not only to GDP, but also one step further to GMP, i.e. a single enzyme catalyzes two subsequent reactions.18,19 These distinct features make this enzyme an interesting prototype for both theoretical and experimental studies. Also, hGBP1 has been shown to possess antiangiogenic properties by inhibiting the expression of matrix metalloproteinase-1 (MMP-1) in endothelial cells20 and antiviral activity by hindering the replication of the hepatitis C virus (HCV).21 It was found that the GTPase activity of hGBP1 is paramount to carrying out these actions, thus adding a further incentive to investigate the catalytic mechanism of GTP hydrolysis in more detail. Several studies (mostly experimental) have already been devoted to unravelling the exact function and cellular location of hGBP1,16,17,19,22,23 however, a detailed molecular understanding of its catalytic mechanism has still not been attained.
The active site residues considered to be crucial in GTP/GDP hydrolysis in hGBP1 are Arg48, Lys51, Ser52, Ser73, Thr75 and Glu99 (as indicated in Fig. 1).19,23 The crystal structure of hGBP1 in complex with GppNHp19 (non-hydrolyzable GTP analogue) shows a direct interaction of Ser73 with the nucleophilic water (Nu), thus giving the impression that Ser73 acts as a base in hGBP1. Ser73, however, can not play this role alone as it remains protonated at physiological pH, which implies a possible role of Ser73 in a proton relay mechanism. Hence, Ser73 could act as a mediator in transferring the proton from the Nu to a γ-phosphate oxygen of GTP (indirect SAC). The same crystal structure shows that Ser73 is also connected to Glu99 via a bridging water (WAT), which indicates an additional possibility of proton relay from Ser73 to Glu99 via WAT. Furthermore, the pathway corresponding to a direct transfer of the proton from the Nu to a γ-phosphate oxygen is also feasible (direct SAC). All these possibilities, taken together, make GTP hydrolysis in hGBP1 a complex reaction to explore. A computational model and methodology capable of determining the free energy landscape that accounts for all these options on equal footing, including the minimum free energy path out of all these possible pathways, is required to unveil the catalytic mechanism of GTP hydrolysis.
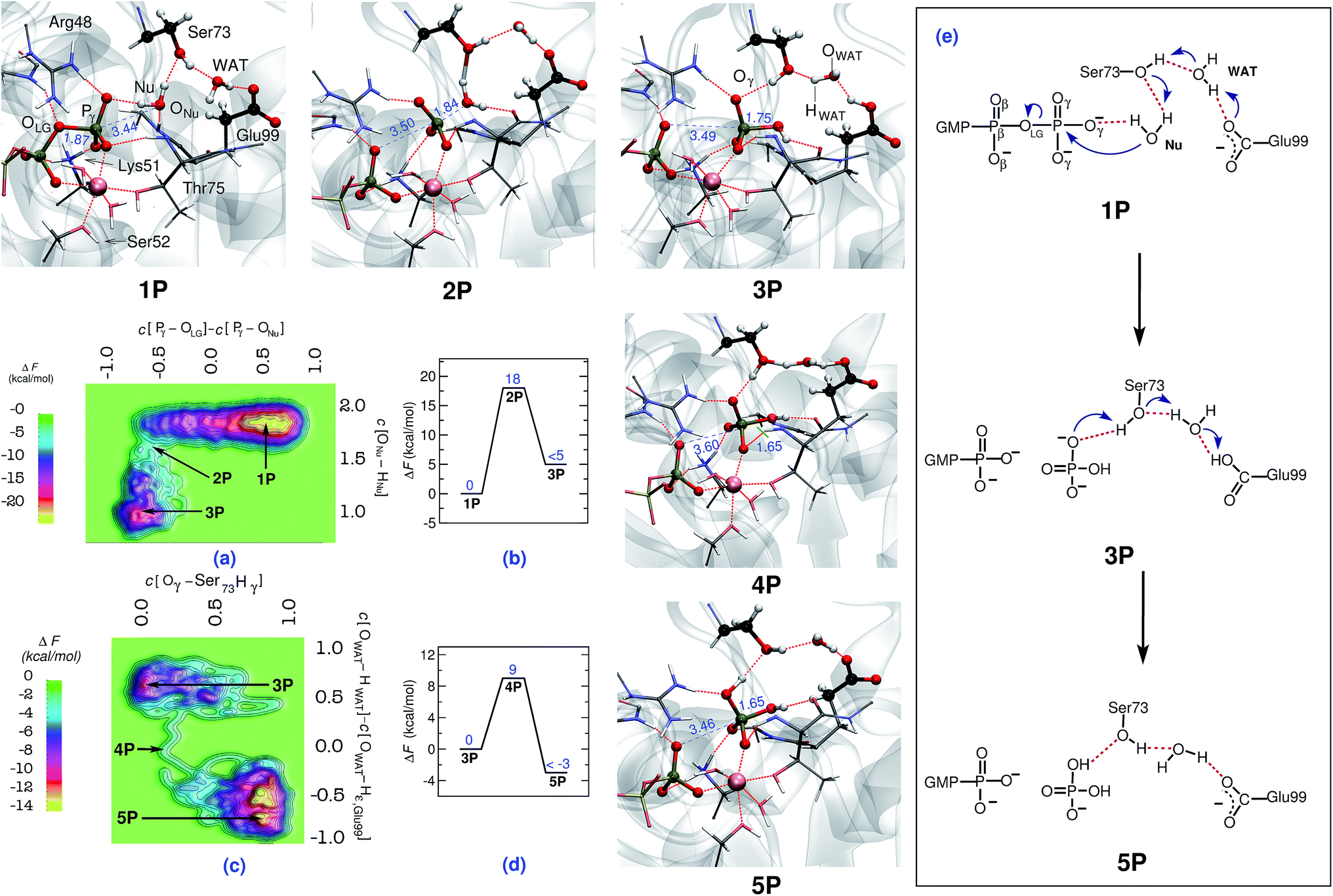 | ||
| Fig. 1 Mechanism of GTP to GDP hydrolysis by hGBP1. Free energy surfaces, (a) and (c), and corresponding free energy profiles, (b) and (d), obtained for the preferred two-step “indirect SAC” hydrolysis mechanism; note that all relative free energy in the profiles are reported w.r.t. the reactant state, that a metadynamics sampling error of activation free energies of about 2 kcal mol−1 w.r.t. the reactant state has been estimated (see ESI Section 1.4†), and that only an upper bound of the product free energy difference relative to the respective reactant state is provided which applies to all reported free energy profiles. The reactant (GTP), intermediate, and product (GDP) minima 1P, 3P, and 5P as well as the intervening TS structures 2P and 4P are depicted using representative configuration snapshots (where P signifies the protein); the P⋯O distances for leaving group and nucleophile (see also panel (e)) being relevant to this hydrolysis step are provided (blue numbers) in these snapshots. The mechanism of GTP hydrolysis in hGBP1 according to these results is summarized schematically in panel (e). The color code used here is as follows: O (red); C (black); N (blue); H (white); Mg (pink). | ||
The aim of this study was to elucidate the molecular level details of the reaction mechanism of GTP and the subsequent GDP hydrolysis by hGBP1 based on multi-dimensional free energy landscapes generated by advanced (accelerated ab initio QM/MM) molecular dynamics simulations.24 Moreover, the hydrolysis reaction of methyl triphosphate (MeTP) in bulk water, taken as a reference system, was also studied using the corresponding ab initio MD method24 in order to achieve a one-to-one comparison between the enzymatic and non-enzymatic reactions. The role of the crucial active site residues during GTP/GDP hydrolysis in hGBP1 was probed and the nature of the transition state (TS) and the mechanisms of GTP/GDP hydrolysis in hGBP1 were unveiled. Going beyond most of the previous approaches that usually focused on preselected reaction pathways, freezing degrees of freedom, and considered only the selective parts of the enzyme, our study incorporates finite temperature effects and dynamics of the fully solvated protein while simulating the reaction. In this way our study has the advantage of mimicking the experimental system and its biochemical processes as closely as possible. With this we were able to obtain a detailed picture of the catalytic action of the hGBP1 enzyme, which can then be extended and utilized to help unravel the biological function of other large GTPases on a molecular level.
1 Methods
In a nutshell, force field molecular dynamics simulations were carried out to setup and thermalize the catalytically active dimerized form of hGBP1 using the GROMACS simulation package.25 The OPLS all-atom force field26 was used to describe the protein and the substrate, whereas the water molecules were treated using the TIP3P model. These simulations were then followed by fully dynamical ab initio QM/MM molecular dynamics simulations,24 which were performed using the CP2k program package27,28 without imposing any con- or restraints. The underlying BLYP density functional has been validated based on MP2 reference calculations along the minimum free energy path of the rate-determining step (see Fig. ESI 1†). Electronic structure analysis of representative configurations has also been carried consistently out using the same methods.27,28 Finally, extended Lagrangian ab initio QM/MM metadynamics24 was used in order to compute all shown free energy surfaces at 300 K. Comprehensive details and references are provided in the ESI Section 1.†2 Results
2.1 First step: GTP → GDP conversion
In order to simulate the GTP hydrolysis reaction in hGBP1 we have sampled the free energy landscape in a reaction subspace spanned by two very flexible generalized coordinates. The reaction mechanism, including the associated activation free energies, is given by the minimum free energy path within that subspace and the reaction coordinate is approximated in terms of these so-called collective variables (CVs); see ESI Section 1† for background and details. The first CV was chosen to probe the bond-breaking and bond-forming events (being the coordination number difference C[Pγ–OLG] − C[ONu–Pγ] involving the leaving group and the attacking Nu), whereas the second CV was selected to probe deprotonation of the catalytic water nucleophile (in terms of the coordination of ONu to both of its protons, C[ONu–HNu]). We emphasize that the CVs utilized here do not specify a particular location for proton transfer to take place, therefore, they are not biased in preselecting the base candidate as generally seen with most of the mechanisms already studied.The hydrolysis reaction comprises nucleophilic attack by a closeby water molecule Nu on Pγ, which occurs simultaneously with the cleavage of the Pγ–OLG bond. This process converts structure 1P into 3P as analyzed in Fig. 1 in terms of the free energy landscape (a) spanned by the aforementioned two CVs, the free energy profile (b) along the minimum free energy path, the resulting reaction mechanism (e), and representative configuration snapshots nP. In the TS structure 2P, the Pγ–OLG bond was completely broken (d[Pγ–OLG] = 3.5 Å), and the Nu was repositioned with respect to Pγ by Thr75 in order to form a bond with it (d[Pγ–ONu] = 1.8 Å). Nu remains an intact H2O molecule at the TS, i.e. nucleophilic attack is not via OH−, however, one of its protons was already slightly displaced towards the Oγ,Ser73 site (see snapshot 2P in Fig. 1). Ser73 is able to activate the Nu water molecule due to the presence of the vicinal Glu99, which, via the bridging water molecule, serves to polarize the Ser73 for activating the Nu. The stabilization of the TS is achieved mainly by the interaction of the metaphosphate with the positively charged side chains of Lys51 and Arg48, the backbone NH group of Thr75 and Gly100, as well as the substrate-bound Mg2+ (see Fig. ESI 2†). These interactions are important as to stabilizing the detaching metaphosphate and preventing reformation of the bond between Pγ and OLG. Moreover, it can be seen that the nucleophilic water is stabilized/oriented suitably in the TS by interacting with Ser73 and with the backbone carbonyl group of Thr75. Hence, these interactions contribute significantly to enzymatic catalysis in hGBP1 via TS stabilization. Progressing from this TS being greatly stabilized by the specific enzyme environment toward the product 3P, the ONu–Pγ bond is found to form completely. In addition to this, the proton from the Nu was fully transferred in 3P to Oγ,Ser73, assisted by another proton transfer of the original hydroxyl proton Hγ,Ser73 from Oγ,Ser73 to Oε,Glu99via the WAT, see electron transfer arrows in 1P of Fig. 1(e). Thus, WAT is found to serve as a proton shuttle from Nu via Ser73 to Glu99 through a local hydrogen bond network. Moreover, only minimal changes were observed in the interaction of GTP with the surrounding active site residues while moving from reactant to the TS (see Table 3 in ESI†). The rate-limiting free energy barrier of the whole process from 1Pvia2P to 3P (see Fig. 1(b)) was found to be 18 kcal mol−1 and, strikingly, correlates well to the one estimated from the experimentally calculated rate constant.22
Although the proton ended up on Glu99, leading to HPO42− as a product, it is quite feasible that this proton can undergo a further transfer to form H2PO4−, thereby reinstating the deprotonated form of Glu99, making it ready to catalyze the next cleavage step. This was proposed based on the fact that Glu99 interacts with HPO42− in structure 3P through a hydrogen bond network formed by Ser73 and WAT (see snapshot 3P in Fig. 1). Moreover, these interactions remained stable in a simulation carried out starting with structure 3P. Hence, to verify this possibility, another simulation was performed in order to generate the free energy landscape for that process starting with the equilibrated structure of 3P. The following generalized coordinates were chosen for this purpose: the coordination number of Hγ,Ser73 to the oxygen of γ-phosphate that forms a hydrogen bond with Hγ,Ser73 and the coordination number difference C[OWAT–HWAT] − C[OWAT–Hε,Glu99]. The first CV was selected to probe the proton transfer from Ser73 to the γ-phosphate oxygen, whereas the second CV was utilized to sample the proton transfer from WAT to Ser73 and from Glu99 to WAT. With these CV selections, we were able to simulate the free energy landscape for proton transfer from Ser73 to the γ-oxygen thus leading from 3Pvia4P to the product 5P (see Fig. 1(c) and (d)). The proton transfer from Ser73 to γ-oxygen took place in a concerted fashion with the proton transfer from WAT to Ser73 and from Glu99 to WAT. Again, WAT is the crucial proton shuttle that allows for this long-distance de/reprotonation process. The free energy barrier for the whole process is 9 kcal mol−1, which is considerably less than the lower limit of the reverse barrier (3P → 1P) obtained for the previous process in Fig. 1(b). This implies that subsequent to the formation of HPO42−, the proton eventually transfers from Glu99 to HPO42−, resulting in H2PO4− as the final product and restoring Glu99 to its deprotonated state.
As previously mentioned, recent studies on an EF-Tu favored reaction pathway correspond to direct transfer of a proton from the water Nu to a phosphate oxygen.29,30 In order to examine this direct SAC scenario in hGBP1, we performed an additional simulation (see ESI Section 5†) in which the reaction subspace that spans the free energy landscape was optimal to simulate the direct proton transfer from Nu to GTP. However, the proton transfer to the γ-phosphate oxygen took place, once again, indirectly via a sequence of proton relays involving Ser73, Glu99, and WAT, which are all strongly engaged in hydrogen bonding. Overall, these data further support our finding that GTP hydrolysis in hGBP1 proceeds with the help of a complex two-step proton-shuttling mechanism from the Nu to GTP via Ser73, Glu99, and WAT.
In order to explore additional pathways to the proton relay through Ser73 we carried out a further simulation starting again with 1P, but where the proton transfer from the water Nu to Ser73 was blocked thus forcing the proton to look for alternative pathways (achieved by imposing a soft repulsive potential at distance 1.60 Å between HNu and Oγ,Ser73). This can be viewed as mimicking a “mild” mutation of Ser73 understood in the sense of fully inhibiting its participation in the proton transfer relay while neither altering the steric demand nor changing the electrostatic properties of residue #73. This is in stark contrast to experimental mutations where both steric and electrostatic properties are necessarily changed upon exchanging an individual amino acid in a protein. Therefore, our computational approach can not be directly compared with the respective experimental mutagenesis study19 in terms of mutation-induced changes of kinetics and activation free energetics; the mutation of serine to alanine certainly results in significant differences and thus altered hydrogen bonding at the active site, which in turn can create alternative proton translocation pathways with altered activity in the mutant. Thus, our “mild” mutation of Ser73 is an exquisite probe of the involvement of this particular residue in the reaction mechanism without perturbing the environment at all, whereas elucidating the hydrolysis mechanism in the presence of Ala73 is not part of the present study. Within this mind-set in conjunction with employing the same reaction subspace as before in Fig. 1, it was found that the hydrolysis of GTP did not take place up to a free energy of at least 41 kcal mol−1 above the reactant minimum 1P (see Fig. ESI 6†), which effectively fully blocks the preferred proton relay mechanism. This finding is in accord with the acknowledged importance of Ser73 according to experiment19 and specifically indicates that GTP hydrolysis in hGBP1 will follow the proton relay pathway consisting of a hydrogen bonding network formed by Nu, Ser73, WAT, Glu99, and GTP. The other suggested pathways, including the one corresponding to direct proton transfer to the substrate GTP (direct SAC), are all consistent with high energy barriers and, hence, can safely be excluded for hGBP1.
2.2 Second step: GDP → GMP conversion
As hGBP1 is capable of hydrolyzing GDP further to GMP, we also looked into the reaction mechanism of this particular step. In this context, it is important to note that, in the GDP-bound state, the ribose plane of GDP slides by about 2.5 Å towards the catalytic center by positioning the α-phosphate of GDP at the same position as the β-phosphate of GTP was formerly placed.19 Thus, α- and β-phosphates of GDP occupy the same positions as those of the β- and γ-phosphates of GTP, which is assured in our simulations by starting from the proper X-ray structure as detailed in the ESI (see ESI Section 1.1†). In order to investigate GDP hydrolysis within that setup, the reaction free energy landscape was determined within the subspace spanned by the following generalized coordinates: the coordination number differences C[Pβ–OLG] − C[ONu–Pβ] and C[ONu–HNu,Hζ,Lys51] − C[Nζ,Lys51 − Hζ,Lys51]. The first CV, as described before, is a requisite for the bond-breaking and bond-forming events, whereas the second CV is chosen to probe the deprotonation of the catalytic water; see ESI Section 9† for further details. With this setup we were successful in simulating also the second hydrolysis step from GDP to GMP in hGBP1.The free energy barrier for GDP-hydrolysis was determined to be 22 kcal mol−1 as can be seen in Fig. 2. The reactant minimum on the free energy landscape is represented by an intact GDP molecule, which is coordinated to the Mg2+ ion by one of its α- and one of its β-oxygens (see 6P in Fig. 2). The TS structure obtained during the reaction characterizes the breaking of the Pβ–OLG bond and the repositioning of ONu in the vicinity of Pβ, ready for nucleophilic attack. Moreover, in line with the GTP hydrolysis reaction, the Nu attains a catalytically competent position in the TS by forming a hydrogen bond with Thr75, see 7P. Eventually, the Pβ–ONu bond is formed and the proton from the Nu is transferred to Glu99 via a proton relay utilizing two bridging water molecules, which both correspond to crystallographic water molecules (see reaction scheme in Fig. 2). The proton was not passed on to HPO42−, leaving it as the final product.
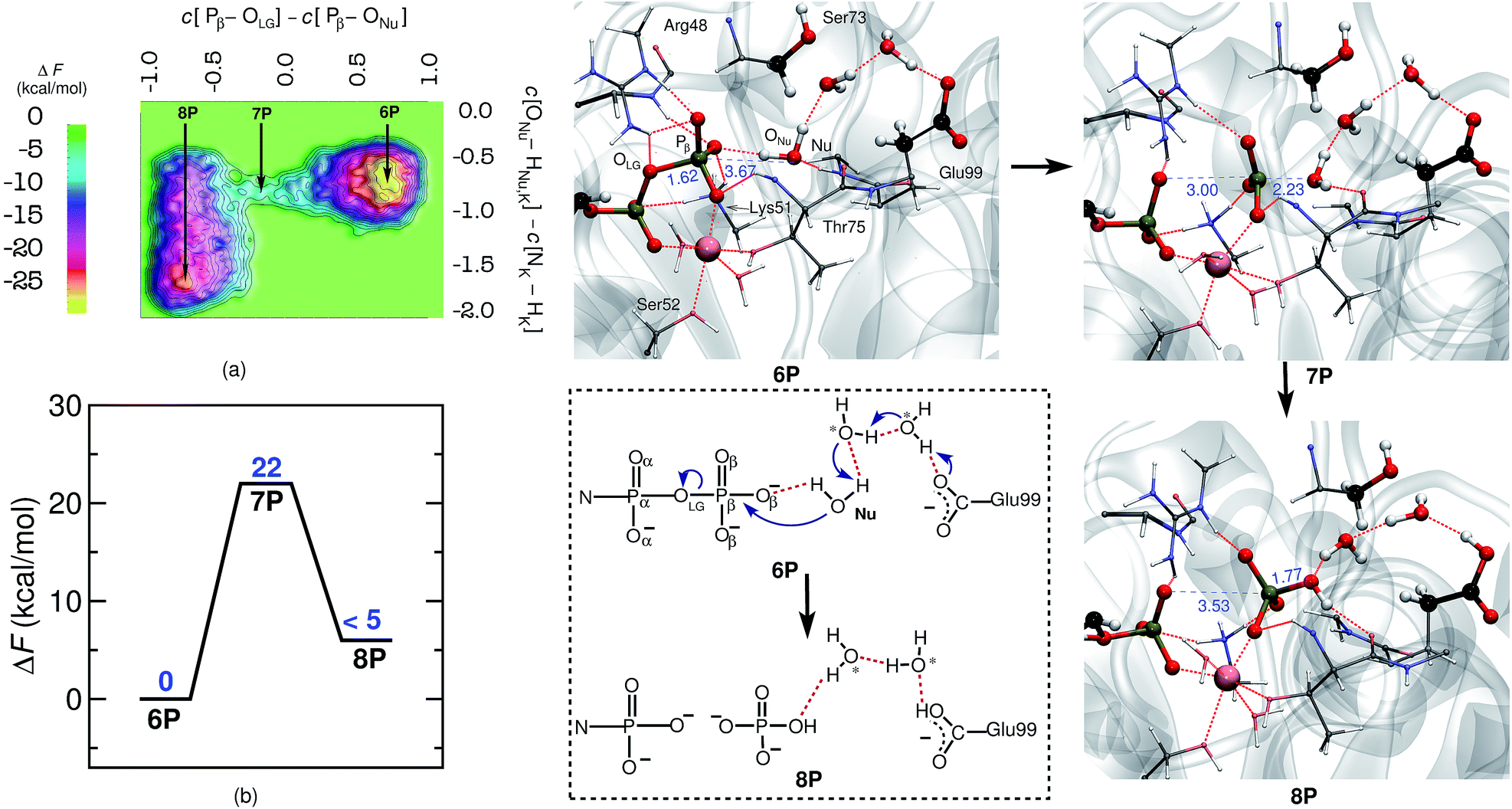 | ||
| Fig. 2 Mechanism of GDP to GMP hydrolysis by hGBP1. Free energy surface, (a), and corresponding free energy profile, (b) obtained for GDP hydrolysis by hGBP1. Representative snapshots of the reactant (GDP) and product (GMP) minima 6P and 8P as well as the intervening TS structure 7P are depicted using representative configuration snapshots; the P⋯O distances for leaving group and nucleophile being relevant to this hydrolysis step are provided (blue numbers) in these snapshots. The mechanism of GDP hydrolysis by hGBP1 according to these results is summarized schematically in the dotted box. The water molecules, other than the nucleophilic water, participating in the reaction are marked with asterisk. | ||
2.3 Non-enzymatic reference scenario: MeTP hydrolysis in bulk water
Last but not least, a molecular level understanding of the mechanism of phosphate hydrolysis in a bulk water environment is desired in order to enable a one-to-one comparison between the enzymatic reaction and a suitable non-enzymatic reference scenario. In the present case, we use a MeTP molecule together with a suitably coordinated Mg2+ cation in aqueous solution (see ESI Section 2† for methods and details). Note that Mg2+ is usually placed between the γ- and β-phosphates also in the reference system when studying GTP or ATP hydrolysis in order to establish a most direct comparison with the enzymatic reaction but in the absence of the protein environment. Free energy sampling was performed in analogy to hGBP1 to generate the non-enzymatic reaction where the following generalized coordinates were considered: the coordination number difference C[Pγ–OLG] − C[ONu–Pγ] and the coordination number of ONu to all water protons, C[ONu–HWater]. The free energy barrier was calculated to be 33 kcal mol−1, as can be seen from Fig. 3(a) and (b), which is consistent with results obtained from previous computational studies on GTP/ATP/MeTP hydrolysis in bulk water.29,31–34 The reactant minimum on the free energy landscape underlying the reference reaction, 1R, is represented by an intact MeTP, which is coordinated to the Mg2+ ion by one of its γ- and one of its β-oxygens being both negatively charged, see Fig. 3. The rest of the hexacoordinated shell of the magnesium ion is made up by four water molecules. The saddle point on the free energy surface, 2R, represents (i) the breaking of the Pγ–OLG bond, (ii) repositioning of the catalytic water molecule in the vicinity of Pγ, and (iii) repositioning of two water molecules in order to enable a proton transfer from Nu to a γ-oxygen. Eventually, the bond between ONu and Pγ was formed, accompanied by the proton transfer from Nu to a γ-oxygen via a proton relay through two water molecules, resulting in H2PO4− as the final product, 3R.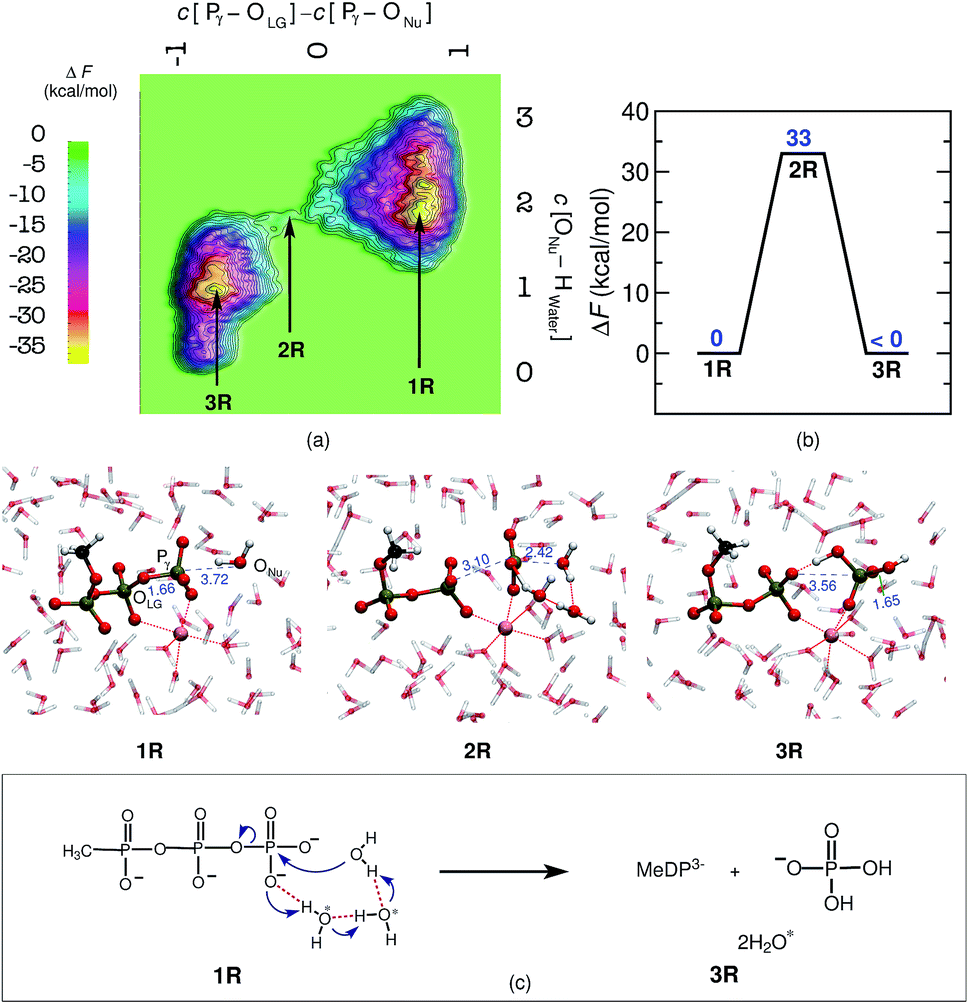 | ||
| Fig. 3 Mechanism of MeTP to MeDP hydrolysis in aqueous bulk solution. Free energy surface, (a), and corresponding free energy profile, (b), obtained for MeTP hydrolysis in bulk water. The reactant minima, 1R and 3R (R signifies the reference system), and the intervening TS structure, 2R, are depicted using representative configuration snapshots the P⋯O distances for leaving group and attacking water molecule (nucleophile) are provided (blue numbers) in these snapshots. The mechanism of MeTP hydrolysis in bulk water obtained from this study is shown schematically in panel (c). The water molecules, other than the nucleophilic water, participating in the reaction are marked with asterisk. | ||
3 Discussion
Before entering the extensive discussion and interpretation of our data, including their relation to both other enzymes and previous work, we provide an outline of what follows in order to guide the reader. Based on our computational results, we first analyze the reaction mechanism of GTP hydrolysis in hGBP1 being the first step. Given that, the mechanistic comparison of GTP hydrolysis in the enzyme can be made relative to the same process in bulk aqueous solution, which serves as the usual non-enzymatic reference system. For the enzymatic reaction, both the nature of the nucleophilic activation and the reaction pathway are characterized in detail. Subsequently, the crucial role of important active site residues of hGBP1 during the GTP hydrolysis step is discussed. Furthermore, a possible rationale for the significant increase in the catalytic activity via the enzyme in comparison to its aqueous solution counterpart is elaborated. Last but not least, we discuss the mechanistic details of the subsequent GDP hydrolysis step by hGBP1 thus leading to the GMP final state.The preferred two-step reaction mechanism of GTP to GDP hydrolysis by hGBP1 obtained from our study is depicted in Fig. 1(e). The rate-determining first step of this hydrolysis reaction comprises the dissociation of the γ-phosphate from GTP, which occurs in a clearly concerted fashion with bond formation between the Nu and the γ-phosphate. This is accompanied by proton transfer from Nu to Ser73, which took place in conjunction with the movement of a proton from Ser73 to Glu99 via a bridging water molecule. In a second step, a proton transfer from Ser73 to HPO42−, resulting in H2PO4−, is observed and Ser73 gains its proton back from Glu99 again via the bridging water molecule. Overall, the GTP hydrolysis according to this two-step indirect SAC process follows a DNAN + AHDxh mechanism in the IUPAC nomenclature, see ref. 35 and 36 for definitions. The net free energy barrier of GTP hydrolysis obtained from our study correlates very well with the one estimated from the experimental rate,22 hence lending confidence to our results.
To decipher the GTP hydrolysis in a non-enzymatic system, the hydrolysis of the methyl triphosphate molecule, MeTP, was studied in bulk water. The hydrolysis in this reference system was found to follow a DNANDxhAH mechanism. Interestingly, the identical mechanism has been revealed earlier for concerted one-step catalysis in hGBP1, whereas the energetically preferred mechanism proceeds via a stable intermediate and thus a two-step proton relay process, DNAN + AHDxh. The free energy barrier for MeTP hydrolysis in our reference bulk water solution of 33 kcal mol−1 is consistent with previous theoretical results for GTP/ATP/MeTP hydrolysis in bulk water.29,31–34 This barrier only exceeds slightly that of the one-step process in the enzyme (26 kcal mol−1), whereas the rate-determining barrier of the two-step indirect SAC reaction is significantly lower (18 kcal mol−1) in hGBP1.
Identification of the nature of nucleophile activation and characterization of the reaction mechanism are the most controversial topics concerning GTP hydrolysis. We have shown here that GTP ultimately acts as the proton acceptor in GTP hydrolysis by hGBP1, suggesting a SAC mechanism. However, the obtained pathway does not correspond to a direct proton transfer from Nu to an Oγ but comprises a series of proton transfers between Nu, Ser73, WAT, Glu99, and a γ-oxygen. Nevertheless, it indicates that the GTP substrate is important for the actual mechanism of cleavage as it restores Glu99's former deprotonated state in readiness for the next reaction step. Interestingly, the independent simulation that was performed to simulate the direct proton transfer from Nu to γ-oxygen also results in the same indirect SAC pathway involving proton transfer from the Nu to γ-oxygen via a proton relay through Ser73, WAT, and Glu99 and, therefore, rules out direct SAC. Additionally, it was found that blocking the indirect SAC via Ser73 results in an enormously high free energy barrier for the hydrolysis, hence corroborating our findings. Indeed, Ser73, which is introduced into the active site of hGBP1 only during dimerisation, is found to interact with the nucleophilic water in the X-ray structure,19 thus is expected to play a key role in water Nu deprotonation. The significance of Ser73 and Glu99 in GTP hydrolysis by hGBP1 according to the two-step indirect SAC mechanism is fully consistent with experimental observations,19,23 which showed that mutation of these residues resulted in a notable decrease in the catalytic rate/intrinsic activity of GTP hydrolysis. Concerning ATPases, the catalytic role of Glu99 and Ser73 as a proton relay group during the phosphoryl transfer reaction has already been well established by previous studies.37–42 What is unique in hGBP1 is that Glu99 and Ser73 must act concertedly in both, proton abstraction from Nu and protonation of HPO42−.
The indirect SAC mechanism of GTP hydrolysis in hGBP1 as obtained from our study is somewhat similar to the indirect SAC mechanism proposed for the extensively studied Ras GTPases, in which however only Gln61 is the responsible mediator of catalytic proton transfer from Nu to GTP,11,12,43,44 whereas recent studies on EF-Tu clearly supported instead a direct SAC mechanism in that case.10,30 We believe that these mechanistic differences stem from distinctly different active site architectures of the three enzymes. In enzymes like hGBP1 or Ras, where the catalytic water is connected to γ-phosphate via an active site residue and/or water molecules (such as only Gln61 in the case of Ras whereas Ser73, WAT and Glu99 are involved in hGBP1), indirect SAC seems to be preferred over the direct SAC mechanism. In EF-Tu, in stark contrast, the presence of a protonated His10,30 adjacent to the nucleophilic water cannot support an indirect SAC but, instead, drives the reaction to follow a direct SAC mechanism. Overall, in comparison to other GTPases such as Ras or EF-Tu, the active site of hGBP1 possesses a much more complex extended hydrogen bonded network composed of Nu, Ser73, WAT, and Glu99 (see 1P in Fig. 1). This network rearranges after nucleophilic attack of the Nu on GTP, thereby connecting the Glu99, which carries the Nu proton, to the HPO42− that is formed by the attack of Nu on the PγO3− (see 3P in Fig. 1). These dynamic rearrangements of the hydrogen bonding network in hGBP1 ultimately connect the Nu proton to the Oγ of GTP, hence rendering the proton relay mechanism favorable for hGBP1. The subsequent GDP hydrolysis further confirms a proton relay mechanism, where two bridging crystal water molecules, connecting Nu to Glu99 in terms of a water wire without Ser73, participate in proton shuttling. The same sort of proton relay mechanism can also be extended to other GTPases like MnmE (PDB ID 2GJ8,452GJ9 (ref. 45)), FeoB (3SS8 (ref. 46)) and also to the ATPases like myosin (1VOM,472JJ9,481MND37etc.), Eg5 (3HQD49etc.) and F1-ATPase (2JDI50etc.); all of which maintain a well-organized hydrogen bonding network that connects the nucleophilic water to a catalytic Glu via one or more water molecule(s).
Interestingly, it has been shown recently for Rho GTPase that the specific γ-oxygen that directly coordinates the Mg2+ cation, denoted herein as O1γ, possesses the highest electron density in the TS, suggesting that O1γ is the strongest base during the proton transfer step.51 Nevertheless, no direct interaction has been found between the nucleophilic water proton and O1γ in that TS structure, which would be the geometric prerequisite to accept a proton. Instead, the catalytic Gln of Rho is in a perfect position to translocate the proton from water to O3γ within that TS structure, akin to what we find in hGBP1 to occur via Ser73 according to the TS structures 2P and 4P thus leading from 1P to 5P (see Fig. 1). The same involvement of O3γ as the proton acceptor has also been found in Ras.11,12,43,44,52 In line with these previous proposals, our study also supports O3γ to act as the proton acceptor for GTP hydrolysis in hGBP1 whereas O1γ is a spectator since it stays firmly coordinated to Mg2+ (see Fig. 1).
The nature of the TS structure 2P obtained during the GTP hydrolysis can be interpreted by various observables, such as bond distances between Pγ–OLG and Pγ–ONu, the corresponding bond orders, or the change in atomic charges on crucial atoms along the reaction pathway. The average distance between the attacking water oxygen to the Pγ of GTP and its average distance to the bridging oxygen for the reactant and TS structures along the GTP hydrolysis reaction pathway are compiled in Table 2 in ESI.† It can be seen that, at the TS, the Pγ–OLG bond distance is elongated up to 3.5 Å, whereas the Pγ–ONu bond distance is decreased to 1.8 Å. Since bond formation is nearly synchronous with bond cleavage, this is clearly suggestive of a concerted pathway, however the TS is found to be geometrically more similar to the product structure, possibly suggesting a significant dissociative component. In order to examine the reaction coordinate and thus the nature of the TS in more detail, we make use of More O'Ferrall–Jencks (MOFJ) analysis,53 which correlates the evolution of bond cleavage and bond formation in terms of bond distances, thus Pγ–OLG and Pγ–ONu, respectively, in the present case. The four corners of the resulting graph represent the reactant, product, and two limiting intermediate structures corresponding to the metaphosphate ion and the pentavalent phosphorane for GTP hydrolysis (see Fig. 4(b)). The correlation obtained from the simulated trajectory clearly shows that GTP hydrolysis in hGBP1 neither follows an associative nor dissociative mechanism but a concerted pathway, where the TS, however, is found to be shifted very much towards the region of the dissociated product state (see Fig. 4(a)). Note that MOFJ analysis is carried out in a quite restricted coordinate space compared to that spanned by the more flexible generalized coordinates used to determine the free energy landscape and thus the TS according to Fig. 1(a). Based on this analysis, we conclude that GTP hydrolysis in hGBP1 follows a so-called “concerted-dissociative” pathway2 in its rate-limiting step. The dissociative character of the concerted reaction pathway is extracted even more clearly from the trajectory when the MOFJ analysis is carried out in terms of bond orders (see Fig. ESI 9†) instead of bond differences. A similar concerted mechanism with TS possessing a considerable amount of dissociative character was also proposed for Ras based on an infrared spectroscopy study.54 Finally, electronic structure-based charge transfer analysis of the three key atoms i.e. Pγ, OLG and ONu further confirms the existence of a concerted pathway in hGBP1 (for more details, see ESI Section 7†).
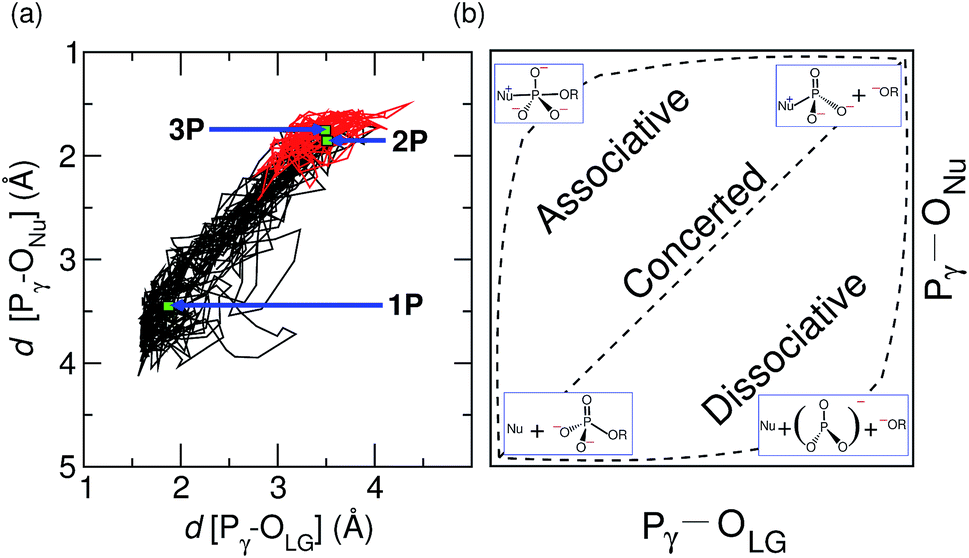 | ||
| Fig. 4 Two-dimensional analysis of the concertedness of the reaction mechanism of GTP to GDP hydrolysis by hGBP1. More O'Ferrall–Jencks analysis of the reaction coordinate based on the generated trajectory corresponding to GTP hydrolysis by the enzyme, see panel (a). The location of the TS structure, 2P, as well as the corresponding reactant and product structures, 1P and 3P, in hGBP1 are marked in the diagram. The trajectory piece after the deprotonation of Nu is highlighted in red. Panel (b) illustrates the idealized associative, concerted, and dissociative reaction pathways (where the respective TS is midway between reactant and product) and the corresponding schematic molecular structures for GTP to GDP hydrolysis within this reaction coordinate diagram; here R symbolizes GDP. | ||
Mutational studies were successful in indicating the crucial role played by many active site residues during GTP hydrolysis by hGBP1,19,23 however, the precise roles of these residues during the course of reaction have not been elucidated. The missing information is contributed by our dynamical simulations that span the entire catalyzed hydrolysis process. Our study shows that Arg48 interacts with the γ-phosphate oxygen through the amine group of the guanidinium side chain during the hydrolysis. Also, Arg48 interacts with OLGvia an amide NH and this interaction becomes stronger as the system approaches the TS (see Table 3 in ESI†). These interactions are vital in order to prevent the bond between Pγ and OLG from reforming and, thus, contribute significantly to the catalysis. Lys51 is engaged in hydrogen bonds with both β- and γ-oxygens throughout the reaction, thus playing an important role in positioning the substrate and in stabilizing the negative charge on the oxygen atoms of GTP during the reaction. Ser52 interacts with a β-oxygen and Mg2+ and these interactions remain intact during the hydrolysis. Thr75, which is highly conserved in GTP-binding proteins, coordinates the Mg2+ ion with its hydroxyl group and also a γ-oxygen with its backbone NH. More importantly, Thr75 forms a hydrogen bond with the Nu only in the TS, thus, aligning it with the Pγ–OLG bond for in-line attack at the phosphorus. The drastic reduction in GTPase activity of the T75A mutant23 further supports our findings. Gly100 also interacts with the Nu through its backbone NH, helping to keep the Nu in close proximity to the γ-phosphate in the reactant state. Since many of these residues are highly conserved in most GTPases, the disclosed roles of these residues in hGBP1 can be generalized to other classes of GTPases.
Enzymes catalyze chemical reactions by reducing the activation energy of the reaction, often by forming a pre-organized active site structure complementary to the TS structure.55 Our study shows nearly a two-fold reduction in the free energy barrier when transferring GTP hydrolysis from a bulk water environment to an enzymatic biomatrix. Interestingly, only minimal changes in the hydrogen bonding patterns are seen while moving from reactant to TS during GTP hydrolysis in hGBP1 (see Table 3 in ESI†). This reflects that the hydrogen bonding interactions in the active site of hGBP1 are already partially oriented toward the developing TS charges and, hence, are likely to contribute to large rate accelerations relative to the reaction in bulk water. Thus, the active site of the enzyme is a kind of blueprint of the TS, forcing the substrate to take on a conformation that facilitates the cleavage reaction and, as a consequence, lowering the activation barrier.
The hydrolysis of GDP to GMP was also studied in order to gain a molecular level understanding of this unique second step in hGBP1. GDP hydrolysis also exhibits the simultaneous breaking and forming of the Pβ–OLG and Pβ–ONu bonds, respectively, that occur concurrently with the proton transfer from Nu to Glu99 via a proton relay through two bridging crystal water molecules. The proton ends up on the Glu99 residue resulting in HPO2−4 as the final product. The role of Glu99 as a proton acceptor is in accordance with the mutagenesis study,23 where mutation of this residue resulted in a significant reduction in the GDP hydrolysis rate. Also, we found that Ser73 does not participate directly in GDP hydrolysis as opposed to the key role it plays in GTP cleavage. Therefore, the low intrinsic GDPase activity and loss of cooperativity observed in the experimental study of the S73A mutation19 is explained here based on the observation that, in the wild-type enzyme, Lys76 interacts with Ser73, leaving Glu99 free to be part of the proton relay pathway. In the S73A mutant, however, the Ser73 residue is no longer present, leaving Lys76 free to form a salt bridge with Glu99 and, consequently, blocking the proton relay mechanism (see ESI Section 10 in ESI†). On an additional note, the TS structure obtained from our study fits excellently with the GMP-AlF4− bound crystal structure (see Fig. ESI 8†), which mimics the TS of GDP hydrolysis, once again substantiating our findings.
4 Conclusion
We have shown in this work that hGBP1 catalyzes GTP hydrolysis through a complex proton relay pathway with an extended and dynamical hydrogen bonding network involving Nu, Ser73, WAT, Glu99, and GTP. The architecture of the active site of hGBP1 is optimally designed to facilitate this proton shuttling. GTP was found to be the ultimate proton acceptor, suggesting an indirect SAC mechanism for hGBP1, which is in keeping with the mechanism proposed for the Ras GTPase. The subsequent GDP hydrolysis in hGBP1 was also found to follow a proton relay mechanism in which Glu99 activates the nucleophilic water utilizing two intervening water molecules in a wire-like arrangement without involving Ser73. Overall, we have shown that GTP hydrolysis in hGBP1 follows a concerted-dissociative pathway in the first rate-determining step, followed in a second step by subsequent proton transfer from Glu99 to the GTP, restoring the deprotonated state of Glu99 for another cleavage reaction. A two-fold reduction in the activation free energy barrier was observed when comparing the hydrolysis of GTP in hGBP1 with non-enzymatic hydrolysis of the methyl triphosphate molecule MeTP in bulk water, which we chose as our reference system. The fact that the hydrogen bond interactions present in the active site of hGBP1 are partially oriented toward the developing transition state charges is the main reason behind the more efficient enzymatic hydrolysis compared to the reference system. The TS structure of GDP hydrolysis and free energy barriers obtained from our studies are in excellent agreement with the available experimental results, thus further corroborating our findings. Overall, our study provides a detailed molecular-level understanding of both GTP and GDP hydrolysis in hGBP1. We can expect similar proton relay mechanisms to take place in other classes of GTPases/ATPases depending on how well the hydrogen bonding network is organized within the active site of these enzymes. We believe that the molecular insights gained from our study on GTP/GDP hydrolysis can also help to understand the hitherto unknown mechanisms of the inhibition of MMP-1 expression and HCV replication by hGBP1.Acknowledgements
It gives us great pleasure to thank Christian Herrmann for insightful discussions and Theodros Zelleke for technical assistance. This work was partially supported by Deutsche Forschungsgemeinschaft via MA 1547/15 and is also part of the Cluster of Excellence RESOLV (EXC 1069). The simulations were carried out using resources from HPC – RESOLV, HPC@ZEMOS, BOVILAB@RUB, as well as RV – NRW.References
- A. T. P. Carvalho, K. Szeler, K. Vavitsas, J. Åqvist and S. C. L. Kamerlin, Arch. Biochem. Biophys., 2015, 582, 80–90 CrossRef CAS PubMed.
- S. C. L. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1–132 CrossRef CAS PubMed.
- J. K. Lassila, J. G. Zalatan and D. Herschlag, Annu. Rev. Biochem., 2011, 80, 669–702 CrossRef CAS PubMed.
- E. F. Pai, U. Krengel, G. A. Petsko, R. S. Goody, W. Kabash and A. Wittinghofer, EMBO J., 1990, 9, 2351–2359 CAS.
- H. R. Bourne, D. A. Sanders and F. McCormick, Nature, 1990, 348, 125–132 CrossRef CAS PubMed.
- A. Wittinghofer and I. R. Vetter, Annu. Rev. Biochem., 2011, 80, 943–971 CrossRef CAS PubMed.
- I. R. Vetter and A. Wittinghofer, Science, 2001, 294, 1299–1304 CrossRef CAS PubMed.
- A. Wittinghofer, Chem. Biol., 1998, 379, 933–937 CAS.
- H. R. Bourne, D. A. Sanders and F. McCormick, Nature, 1991, 349, 117–127 CrossRef CAS PubMed.
- G. Wallin, S. C. L. Kamerlin and J. Åqvist, Nat. Commun., 2013, 4, 1733 CrossRef PubMed.
- R. B. Prasad, N. V. Plotnikov, J. Lameira and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20509–20514 CrossRef PubMed.
- M. G. Khrenova, B. L. Grigorenko, A. B. Kolomeisky and A. V. Nemukhin, J. Phys. Chem. B, 2015, 119, 12838–12845 CrossRef CAS PubMed.
- C. Lubeseder-Martellato, E. Guenzi, A. Jörg, K. Töpolt, E. Naschberger, E. Kremmer, C. Zietz, E. Tschachler, P. Hutzler, M. Schwemmle, K. Matzen, T. Grimm, B. Ensoli and M. Stürzl, Am. J. Pathol., 2002, 161, 1749–1759 CrossRef CAS PubMed.
- S. Martens and J. Howard, Annu. Rev. Cell Dev. Biol., 2006, 22, 559–589 CrossRef CAS PubMed.
- B. H. Kim, A. R. Shenoy, P. Kumar, R. Das, S. Tiwari and J. D. MacMicking, Science, 2011, 332, 717–721 CrossRef CAS PubMed.
- B. Prakash, G. J. K. Praefcke, L. Renault, A. Wittinghofer and C. Herrmann, Nature, 2000, 403, 567–571 CrossRef CAS PubMed.
- B. Prakash, L. Renault, G. J. K. Praefcke, C. Herrmann and A. Wittinghofer, EMBO J., 2000, 19, 4555–4564 CrossRef CAS PubMed.
- M. Schwemmle and P. Staeheli, J. Biol. Chem., 1994, 269, 11299–11305 CAS.
- A. Ghosh, G. J. K. Praefcke, L. Renault, A. Wittinghofer and C. Herrmann, Nature, 2006, 440, 101–104 CrossRef CAS PubMed.
- E. Guenzi, K. Töpolt, C. Lubeseder-Martellato, A. Jörg, E. Naschberger, R. Benelli, A. Albini and M. Stürzl, EMBO J., 2003, 22, 3772–3782 CrossRef CAS PubMed.
- Y. Itsui, N. Sakamoto, S. Kakinuma, M. Nakagawa, Y. Sekine-Osajima, M. Tasaka-Fujita, Y. Nishimura-Sakurai, G. Suda, Y. Karakama, K. Mishima, M. Yamamoto, T. Watanabe, M. Ueyama, Y. Funaoka, S. Azuma and M. Watanabe, Hepatology, 2009, 50, 1727–1737 CrossRef CAS PubMed.
- S. Kunzelmann, G. J. K. Praefcke and C. Herrmann, J. Biol. Chem., 2006, 281, 28627–28635 CrossRef CAS PubMed.
- G. J. K. Praefcke, S. Kloep, U. Benscheid, H. Lilie, B. Prakash and C. Herrmann, J. Mol. Biol., 2004, 344, 257–269 CrossRef CAS PubMed.
- D. Marx and J. Hutter, Ab initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, 2009 Search PubMed.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–487 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- M. Klähn, E. Rosta and A. Warshel, J. Am. Chem. Soc., 2006, 128, 15310–15323 CrossRef PubMed.
- A. Adamczyk and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9827–9832 CrossRef CAS PubMed.
- J. Akola and R. O. Jones, J. Phys. Chem. B, 2003, 107, 11774–11783 CrossRef CAS.
- R. Glaves, G. Mathias and D. Marx, J. Am. Chem. Soc., 2012, 134, 6995–7000 CrossRef CAS PubMed.
- C. B. Harrison and K. Schulten, J. Chem. Theory Comput., 2012, 8, 2328–2335 CrossRef CAS PubMed.
- C. Wang, W. Huang and J.-L. Liao, J. Phys. Chem. B, 2015, 119, 3720–3726 CrossRef CAS PubMed.
- R. D. Guthrie, Pure Appl. Chem., 1989, 61, 23–56 CrossRef CAS.
- R. D. Guthrie and W. P. Jencks, Acc. Chem. Res., 1989, 22, 343–349 CrossRef CAS.
- A. J. Fisher, C. A. Smith, J. B. Thoden, R. Smith, K. Sutoh, H. M. Holden and I. Rayment, Biochemistry, 1995, 34, 8960–8972 CrossRef CAS PubMed.
- G. Li and Q. Cui, J. Phys. Chem. B, 2004, 108, 3342–3357 CrossRef CAS.
- B. L. Grigorenko, A. V. Rogov, I. A. Topol, S. K. Burt, H. M. Martinez and A. V. Nemukhin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7057–7061 CrossRef CAS PubMed.
- B. L. Grigorenko, I. A. Kaliman and A. V. Nemukhin, J. Mol. Graphics Modell., 2011, 31, 1–4 CrossRef CAS PubMed.
- M. J. McGrath, I.-F. W. Kuo, S. Hayashi and S. Takada, J. Am. Chem. Soc., 2013, 135, 8908–8919 CrossRef CAS PubMed.
- F. A. Kiani and S. Fischer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E2947–E2956 CrossRef CAS PubMed.
- B. L. Grigorenko, A. V. Nemukhin, I. A. Topol, R. E. Cachau and S. K. Burt, Proteins: Struct., Funct., Bioinf., 2005, 60, 495–503 CrossRef CAS PubMed.
- B. L. Grigorenko, A. V. Nemukhin, M. S. Shadrina, I. A. Topol and S. K. Burt, Proteins: Struct., Funct., Bioinf., 2006, 66, 456–466 CrossRef PubMed.
- A. Wittinghofer, Trends Biochem. Sci., 2006, 31, 20–23 CrossRef CAS PubMed.
- M. R. Ash, M. J. Maher, J. M. Guss and M. Jormakka, PLoS One, 2011, 6, e23355 CAS.
- C. A. Smith and I. Rayment, Biochemistry, 1996, 35, 5404–5417 CrossRef CAS PubMed.
- R. Fedorov, M. Böhl, G. Tsiavaliaris, F. K. Hartmann, M. H. Taft, P. Baruch, B. Brenner, R. Martin, H. J. Knölker, H. O. Gutzeit and D. J. Manstein, Nat. Struct. Mol. Biol., 2009, 16, 80–88 CAS.
- C. L. Parke, E. J. Wojcik, S. Kim and D. K. Worthylake, J. Biol. Chem., 2010, 285, 5859–5867 CrossRef CAS PubMed.
- R. I. Menz, J. E. Walker and A. G. W. Leslie, Cell, 2001, 106, 331–341 CrossRef CAS PubMed.
- Y. Jin, R. W. Molt, J. P. Waltho, N. G. L. Richards and G. M. Blackburn, Angew. Chem., Int. Ed., 2016, 55, 3318–3322 CrossRef CAS PubMed.
- V. A. Mironov, M. G. Khrenova, L. A. Lychko and A. V. Nemukhin, Proteins: Struct., Funct., Bioinf., 2015, 83, 1046–1053 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, 2006 Search PubMed.
- X. Du, H. Frei and S.-H. Kim, J. Biol. Chem., 2000, 275, 8492–8500 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02045c |
| ‡ Present address: Lehrstuhl für Physikalische Chemie II, Ruhr-Universität Bochum, 44780 Bochum, Germany. |
| This journal is © The Royal Society of Chemistry 2017 |