
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general strategy for colloidal stable ultrasmall amorphous mineral clusters in organic solvents†
Shengtong
Sun‡
,
Denis
Gebauer
and
Helmut
Cölfen
*
Department of Chemistry, Physical Chemistry, University of Konstanz, Universitätsstrasse 10, Box 714, D-78457 Konstanz, Germany. E-mail: helmut.coelfen@uni-konstanz.de
First published on 13th October 2016
Abstract
While nature exerts precise control over the size and chemical composition of minerals, this is still a challenging task for artificial syntheses. Despite its significance, until now, there are still no reports on colloidal mineral nanoparticles in the subnanometer range. Here we developed a general gas diffusion strategy using 10,12-pentacosadiynoic acid as a ligand and ethanol as a solvent to fabricate stable amorphous mineral clusters with a core size of less than 2 nm. First discovered for CaCO3, the method was successfully extended to produce monolayer protected clusters of MgCO3, SrCO3, Eu2(CO3)3, Tb2(CO3)3, Ce2(CO3)3, Cax(PO4)y, CaC2O4 and their hybrid minerals, CaxMgy(CO3)z and Cax(CO3)y(PO4)z. All the mineral clusters can be well dispersed in organic solvents like toluene, and are stable for a long period without further crystallization. Our work paves a way for the artificial synthesis of colloidal mineral clusters, which may have various uses in both fundamental research and industry.
Introduction
Natural minerals, abiotic or biotic, possess a diversity of functions due to their complex morphologies, chemical compositions, crystalline forms as well as hierarchical structures. During formation, all minerals go through a nanophase stage, which, in most cases, is transitional.1 Stopping the nanophase evolution of minerals on the nanoscale size range can produce mineral nanoparticles. Owing to the variation of atomic and electronic structures with size, mineral nanoparticles behave distinctly from their macroscopic counterparts, often with superior physical and chemical properties. In the case of biominerals, living organisms exert precise control over the mineral size, shape and structure, and it is still challenging to synthetically gain a similar control.2 Various methods such as biomimetic synthesis,3 microemulsion,4,5 hydrothermal reaction,6 postsynthesis,7 and polymer-templating8 have been reported to fabricate colloidal mineral nanoparticles with different shapes and sizes. However, none of the previous efforts have downsized mineral particles to the subnanometer level or even clusters, due to poor control over inherent severe ionic association, although clusters with sizes less than 2 nm, in particular monolayer-protected clusters, have shown more advantages than larger particles.9 For example, in the case of noble metals like Au or Ag, the alkanethiolate or alkanecarboxylate clusters are stable in air and behave like “molecules”, i.e., they can be repeatedly isolated from, and redissolved in common organic solvents without irreversible aggregation or decomposition.10On the other hand, in recent years, arising evidence suggests the wide existence of stable pre-nucleation or primary clusters as dissolved precursors during mineral formation, which are involved in a new concept of “non-classical” nucleation/crystallization.11–13 Pre-nucleation clusters have been found either experimentally or by modelling the nucleation of calcium carbonate (CaCO3),11,14 calcium phosphate,15,16 magnetite,17 silica,18 amino acids,19 quantum dots,20,21 YVO4,22 NaClO3,23 1,3,5-tris(4-bromophenyl)benzene,24 and many more. Analytical ultracentrifugation (AUC) measurements confirmed the presence of pre-nucleation clusters even in salts of simple monovalent ions like NaCl and LiI.19 Although pre-nucleation clusters were found to be highly dynamic for CaCO3,25 they provide a direct way to produce colloidal mineral clusters by stopping the nucleation/growth process at the very early stage by stabilization.26 In our previous papers, we showed that a gas diffusion method using a unique amphipathic ligand, 10,12-pentacosadiynoic acid (PCDA, structure shown in Scheme 1), as a ligand and ethanol as a solvent, can produce well-defined monolayer protected CaCO3 clusters.27,28 The amorphous CaCO3 core with a size of ∼1.4 nm contains only 7 CaCO3 units which are as few as that approximately contained in the primitive unit cell of calcite, and in that a proto-calcite short-range order can even be identified but with a relatively high degree of disorder. Herein, we further explored the applicability of this method to other minerals, and we found that this method can be a general strategy to prepare stable mineral (carbonate, phosphate, and oxalate) clusters as well as their hybrid clusters.
 | ||
| Scheme 1 Chemical structure of PCDA. | ||
Experimental
Synthesis of mineral clusters
For carbonate-based mineral clusters, the procedure is similar to the method for CaCO3 clusters.27 In brief, 10 mM cation salt (MgCl2·6H2O, SrBr2·6H2O, EuCl3·6H2O, TbCl3·6H2O, or CeCl3) and 4 mM 10,12-pentacosadiynoic acid (PCDA) were dissolved in 40 mL of ethanol in a vial. Note that PCDA dissolved in ethanol must be filtrated with a 0.45 μm PTFE filter to remove oligomeric impurities. The vial was then sealed with parafilm and three pinholes were punctured allowing for gas diffusion. Three glass bottles filled with 10 mL of crushed (NH4)2CO3 powder and covered with parafilm, punctured with three pinholes, were put together with the reaction vial in a desiccator. After three days of diffusion, the precipitate found at the bottom of the vial was collected by centrifugation (9000 rpm, 5 min) and then dissolved in a small amount of toluene with the assistance of ultrasonication, which resulted in a transparent solution. The product was further precipitated by adding a large volume of ethanol. This process was repeated at least six times to remove all free PCDA and any possible salt impurities. The final solid was dissolved in toluene for storage.For the Cax(PO4)y cluster, 10 mM CaCl2, 4 mM PCDA and 6 mM polyphosphoric acid (PPA, concentration based on repeat unit) were dissolved in 40 mL of ethanol. The reaction vial was put in a desiccator together with another bottle filled with 2 mL of NH4OH aqueous solution (25%). Both the reaction vial and the bottle with NH4OH were covered with parafilm with three punctured pinholes followed by gas diffusion in a desiccator. After three days of diffusion, the washing process was the same as described above for the carbonate-based minerals.
For CaC2O4 clusters, PPA was replaced by 4 mM dimethyl oxalate. The rest of the method is the same as for the Cax(PO4)y cluster.
For CaxMgy(CO3)z clusters, 5 mM CaCl2, 5 mM MgCl2 and 4 mM PCDA were mixed in ethanol. Gas diffusion occurred from the decomposition of (NH4)2CO3.
For Cax(CO3)y(PO4)z clusters, 10 mM CaCl2, 4 mM PCDA and 3 mM PPA were dissolved in ethanol. Gas diffusion occurred from the decomposition of (NH4)2CO3.
Characterization
TEM images and selected area electron diffraction (SAED) patterns were acquired on a Zeiss Libra 120 microscope operating at 120 kV. Energy dispersive X-ray (EDX) spectra were acquired on a JEOL JEM-2200FS high-resolution microscope operating at 200 kV. TEM samples were prepared from the cluster solution in toluene (∼0.5 mg mL−1) on conventional TEM grids (carbon-coated copper grids, 400 mesh, supplied by Quantifoil GmbH). X-ray powder diffraction (XRD) data were acquired on a PANalytical X'pert PRO diffractometer utilizing Cu Kα radiation. Dynamic light scattering (DLS) was performed on a Malvern ZS90 at a detection angle of 90°. 1H NMR spectra of mineral clusters were recorded using CDCl3 as the solvent on a Bruker AV (500 MHz) spectrometer. UV-vis spectra of mineral clusters and PCDA in hexane were measured on a UV-3150 photospectrometer (Shimadzu, Japan). Attenuated-total-reflection-infrared spectroscopy (ATR-FTIR) was conducted on a Perkin Elmer device (Spectrum 100 FTIR, equipped with a diamond ATR crystal). Analytical ultracentrifugation (AUC) was carried out on a Beckman-Coulter XL-I using Rayleigh interference optics and running the sample in self-build Ti double-sector cells at 25 °C. Thermal gravimetric analysis (TGA) was performed on a Netzsch STA 449 F3 by heating from 50 to 900 °C with a heating rate of 10 K min−1 under air flow. The matrix-assisted laser desorption/ionization time-of-flight mass (MALDI-TOF MS) spectrum was recorded on a Bruker Microflex MALDI-TOF with α-cyano-4-hydroxycinnamic acid (HCCA) as the matrix and toluene as the solvent. The radial integration of SAED patterns was calculated utilizing software packages, ImageJ 1.50i and JEMS-SAAS. Simulation of XRD curves for nanocrystalline mineral phases (standard CIF files from the ICSD database) was performed using the Reflex Module of Materials Studio 7.0. The crystallite size was set to 1.5 × 1.5 × 1.5 nm3.Results and discussion
For clarity, all the mineral clusters that were successfully synthesized by the gas diffusion method are compiled in Table 1. All the synthesized clusters exhibit an average size between 2.7 and 6.6 nm. To synthesize carbonate-based mineral clusters, typically, 10 mM cation salt (CaCl2, MgCl2, SrBr2, EuCl3, TbCl3, or CeCl3) and 4 mM PCDA were dissolved in ethanol, followed by gas diffusion from (NH4)2CO3 decomposition for three days. These cation salts were chosen because they show good solubility in ethanol. Released CO2 serves as a carbonate source and NH3 renders the solution basic. For calcium phosphate and calcium oxalate, polyphosphoric acid (PPA) and dimethyl oxalate were added into the solution as the sources of phosphate and oxalate from hydrolysis reactions controlled by ammonia diffusion. After thorough washing with repeated precipitation in ethanol and redissolution in toluene to remove any possible impurities, the resultant clusters can be well dispersed in organic solvents like toluene, and are stable without any changes for a long period of time (at least one year, see Fig. S2, ESI† for the XRD profiles of several clusters after long-term storage). Only the solution of Ce2(CO3)3 clusters is light-yellow, while all the others are transparent.| Mineral | Cation source | Anion source | Diffusion by | Sizea/nm |
|---|---|---|---|---|
| a The size distribution was determined from TEM images by manually counting about 300 particles. Enlarged TEM images of the clusters can be found in Fig. S1, ESI. b The data for CaCO3 clusters were reported in our previous paper,27 and are presented here only for comparison. c PPA: polyphosphoric acid. | ||||
| MgCO3 | MgCl2·6H2O | CO2 | (NH4)2CO3 | 6.6 ± 2.4 |
| CaCO3b | CaCl2 | CO2 | (NH4)2CO3 | 4.9 ± 1.7 |
| SrCO3 | SrBr2·6H2O | CO2 | (NH4)2CO3 | 4.4 ± 1.5 |
| Eu2(CO3)3 | EuCl3·6H2O | CO2 | (NH4)2CO3 | 3.6 ± 1.1 |
| Tb2(CO3)3 | TbCl3·6H2O | CO2 | (NH4)2CO3 | 4.0 ± 1.0 |
| Ce2(CO3)3 | CeCl3 | CO2 | (NH4)2CO3 | 2.9 ± 0.9 |
| Cax(PO4)y | CaCl2 | PPAc | NH4OH (25%) | 5.3 ± 2.0 |
| CaC2O4 | CaCl2 | Dimethyl oxalate | NH4OH (25%) | 2.7 ± 0.9 |
| CaxMgy(CO3)z | CaCl2, MgCl2·6H2O | CO2 | (NH4)2CO3 | 3.9 ± 1.7 |
| Cax(CO3)y(PO4)z | CaCl2 | CO2, PPA | NH4OH (25%) | 3.7 ± 1.3 |
The key roles of ligand structure and feed ratio have been discussed before in the case of CaCO3 clusters.28 Ethanol is also essential for the formation of mineral clusters, while methanol and isopropanol did not work well (data not shown). Using ethanol as the solvent not only dissolves PCDA, and metal cations (M+) and anions (A−), but also promotes the formation of stable amorphous precursors. Ethanol is known to be able to stabilize amorphous phases of minerals like CaCO3,29,30 Cax(PO4)y,31 and CaC2O4.32 The ammonia diffusion renders the solution basic, and thus the carbonate, phosphate or oxalate minerals can precipitate. Once mineral primary clusters formed in ethanol, PCDA molecules protected the clusters from further aggregation into larger particles, resulting in well-defined monolayer protected mineral clusters that become insoluble in ethanol and precipitate in the reaction vessel. A schematic mechanism for the mineral cluster formation is presented in Fig. 1. In fact, our method is very similar to the single-phase Brust–Schiffrin method,33 which is normally used to synthesize various monolayer protected metal clusters such as Ag34 and Cu.35 Our results further extend the applicability of the Brust–Schiffrin method to minerals.
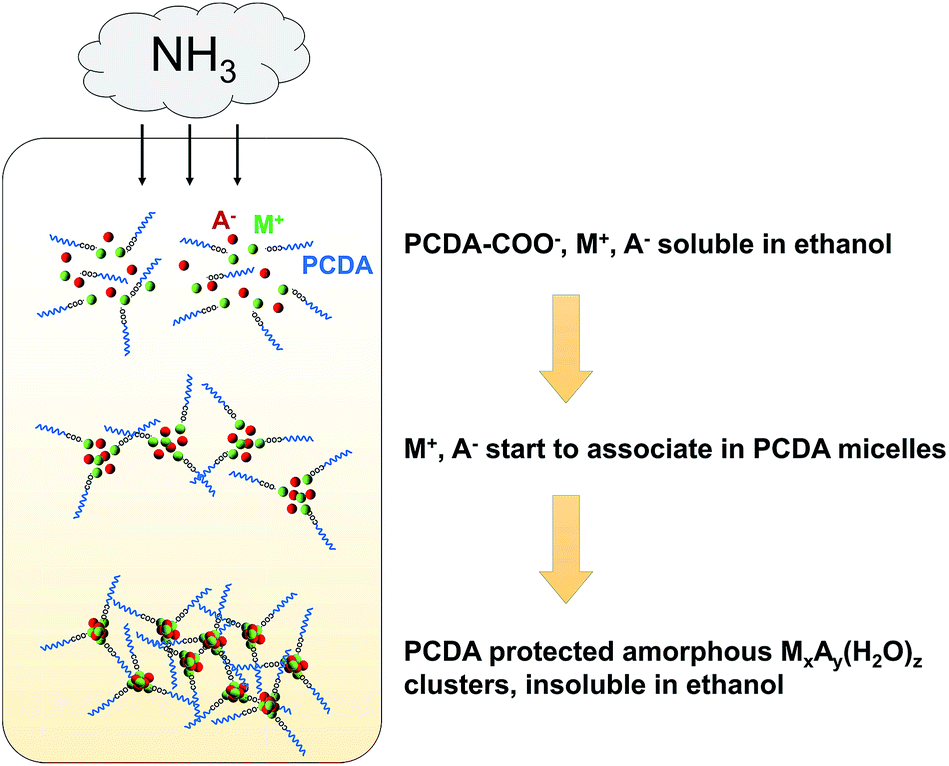 | ||
| Fig. 1 Possible mechanism for the gas diffusion method to synthesize PCDA monolayer protected mineral clusters in ethanol. | ||
Recently we also reported a solvothermal method to prepare PCDA protected CaCO3 clusters using the thermal decomposition of NaHCO3 as a CO2 source.36 However, the solvothermal method can only be applied to carbonate-based mineral clusters and the yield is relatively low due to the partial loss of products in the filtration process to remove Na2CO3 solid impurities. The here reported gas diffusion method is more general, and also provides access to phosphate and oxalate-based mineral clusters. Also, the yield of clusters by the gas diffusion method is very high (mostly >95% by weighing the final products), which is superior to the solvothermal method.
TEM images of all carbonate-based mineral (MgCO3, SrCO3, Eu2(CO3)3, Tb2(CO3)3, Ce2(CO3)3) clusters are shown in Fig. 2 (see enlarged TEM images in Fig. S1, ESI†). Among them, Mg and Sr are alkaline earth metals, while Eu, Tb and Ce are rare-earth lanthanides, which are often used as dopants of Ca-based minerals due to their similar atomic size to Ca.6,37 Taking MgCO3 and Eu2(CO3)3 clusters for example, as in the case of CaCO3,28 the similarity between the UV-vis absorption of the mineral clusters and PCDA in hexane (Fig. S3, ESI†) shows that the PCDA chains are binding on the mineral core. The 1H NMR spectra of MgCO3 and Eu2(CO3)3 clusters in d-chloroform (Fig. S4, ESI†) exhibit a resonance peak broadening effect, and a large shift of Ha (to Ha′) corresponding to the methylene group adjacent to COOH, indicating chelation between Mg2+/Eu3+ and PCDA as well as the core–shell structure of the clusters.
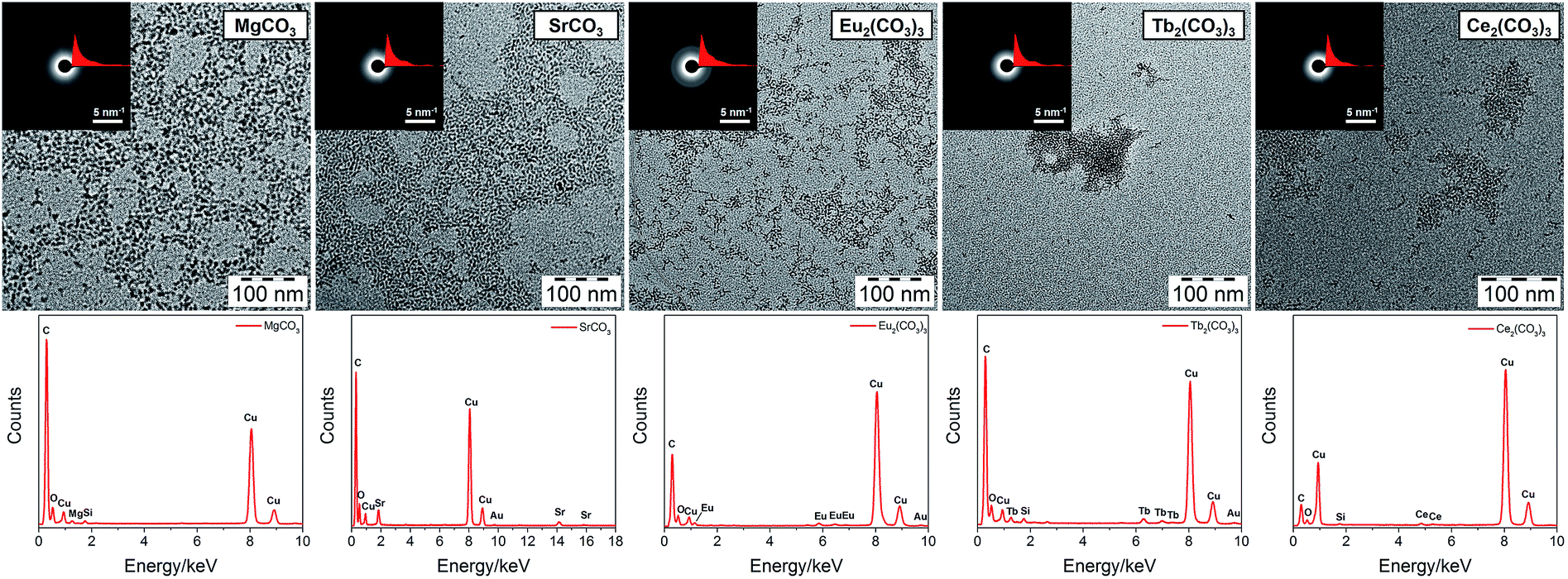 | ||
| Fig. 2 TEM images (top) and corresponding energy-dispersive X-ray (EDX) spectra (bottom) of PCDA monolayer protected carbonate-based mineral (MgCO3, SrCO3, Eu2(CO3), Tb2(CO3)3, Ce2(CO3)3) clusters. The insets in the TEM images are selected area electron diffraction (SAED) patterns and corresponding radial integration curves obtained from a round area with a diameter of 200 nm. In the EDX spectra, Cu and some C come from the TEM grid; Si and Au come from the detector. | ||
PCDA can stabilize these clusters in solution with different sizes (2.9–6.6 nm), which is further confirmed by dynamic light scattering (DLS) measurements (Fig. S5, ESI†). Considering PCDA has a chain length of 1.5–3 nm, depending on its conformation, the mineral cores of all these clusters should have sizes of less than 2 nm. Most of the clusters are isolated but some aggregation can also be observed, which can be interpreted to correspond to the interdigitation of hydrophobic PCDA chains. However, Tb2(CO3)3 is an exception with more aggregated particles than isolated ones. The reason for this is still unknown. Selected area electron diffraction (SAED) patterns reveal that these clusters are all amorphous, as in the case of CaCO3.27 The differences of SAED radial integration curves show that the diffraction does not arise from the supporting amorphous carbon film. From energy-dispersive X-ray spectroscopy (EDX) analyses (Fig. 1), the presence of Mg, Sr, Eu, Tb, and Ce is apparent in the respective clusters, proving the successful incorporation of mineral cores inside the PCDA chains. The successful synthesis of mineral clusters is also confirmed by ATR-FTIR spectra of the dried clusters (Fig. S6, ESI†). The O–H stretching band of structural water around 3300 cm−1, C–H stretching bands from PCDA (3000–2780 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of COO− from PCDA (1573–1530 cm−1) and two characteristic stretching bands for CO32− (ν2, 870–850 cm−1; ν3, 1410–1400 cm−1) can be observed, indicating that the deprotonated PCDA binds to the mineral core by chelation. As in the case of CaCO3, all the mineral clusters have disordered and hydrated cores.28 The exact hydration and ionic association status is not yet known and needs to be clarified further. Additionally, judging from the size, it is interesting to find that a larger atomic size (Sr > Ca > Mg) and a higher ionic charge (trivalent for Eu3+, Tb3+, Ce3+, divalent for Mg2+, Ca2+, Sr2+) lead to a smaller cluster size, perhaps due to a higher affinity to the carboxylate group in the deprotonated PCDA chains.
O stretching band of COO− from PCDA (1573–1530 cm−1) and two characteristic stretching bands for CO32− (ν2, 870–850 cm−1; ν3, 1410–1400 cm−1) can be observed, indicating that the deprotonated PCDA binds to the mineral core by chelation. As in the case of CaCO3, all the mineral clusters have disordered and hydrated cores.28 The exact hydration and ionic association status is not yet known and needs to be clarified further. Additionally, judging from the size, it is interesting to find that a larger atomic size (Sr > Ca > Mg) and a higher ionic charge (trivalent for Eu3+, Tb3+, Ce3+, divalent for Mg2+, Ca2+, Sr2+) lead to a smaller cluster size, perhaps due to a higher affinity to the carboxylate group in the deprotonated PCDA chains.
CaCO3, Cax(PO4)y and CaC2O4 are important biominerals abundant in animals and plants. For example, Cax(PO4)y in the form of hydroxyapatite is the main component of bone and teeth,38 and CaC2O4 is generated within membrane chambers of specialized cells in higher plants.39 Again, our method showed good control over the size of Cax(PO4)y and CaC2O4, as shown in Fig. 3 (enlarged TEM images are shown in Fig. S1, ESI†). It is noted that for the preparation of dispersible Cax(PO4)y clusters, the hydrolysis of PPA was employed to produce phosphate ions. From the SAED patterns, the two clusters are also amorphous and the signals of Ca and P can be detected by EDX. Similarly, UV-vis and 1H NMR spectra of Cax(PO4)y clusters (Fig. S3 and 4, ESI†) confirm their core–shell structure. DLS and AUC measurements (Fig. S5 and S7, ESI†) both reveal the rather small size of Cax(PO4)y clusters in solution. IR spectra of Cax(PO4)y and CaC2O4 clusters (Fig. S6, ESI†) show the C–H and CO stretching bands from PCDA and the O–H stretching band from structural water. A few characteristic bands for Cax(PO4)y and CaC2O4 can also be observed.
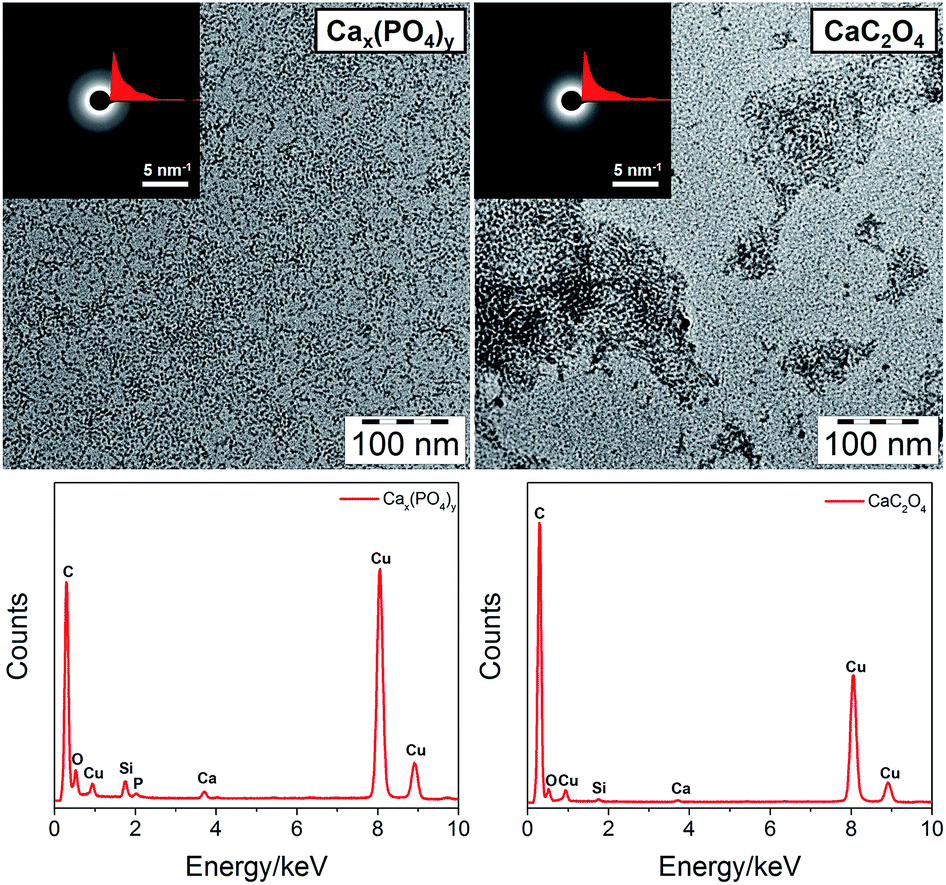 | ||
| Fig. 3 TEM images (top, insets are SAED patterns and radial integration curves) and corresponding EDX spectra (bottom) of PCDA monolayer protected Cax(PO4)y and CaC2O4 clusters. | ||
Due to the significant importance of Cax(PO4)y as well as its relatively complex structure involving both PO43− and HPO42−, we performed further thermogravimetric analysis (TGA) and matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS) measurements to elucidate its structure, as shown in Fig. 4. In the TGA, as temperature increases, the removal of structural water and PCDA decomposition take place successively. The remaining material at 900 °C may be represented by the formula Ca1.5x+2y(PO4)x(P2O7)y, or present a mixture of Ca3(PO4)2 and Ca2P2O7. Ca2P2O7 forms upon decomposition of CaHPO4. To know the exact chemical composition of the Cax(PO4)y cluster, MALDI-TOF MS was recorded, which allows for the assessment of the proportion of the different species. The largest detected species with a probable formula of (CaHPO4)8(H2O)3(PCDA)3 agrees well with the TGA results and should represent the actual formula of the cluster. It is also noted that the Cax(PO4)y core has a stoichiometry similar to amorphous dicalcium phosphate produced in ethanol media.40 This conversely reveals that PCDA stops the nucleation/growth of calcium phosphate in ethanol at a very early stage.
 | ||
| Fig. 4 (a) TGA curve (under air flow) and (b) MALDI-TOF MS of PCDA monolayer protected Cax(PO4)y cluster. For identification, corresponding assignments for the main peaks in MALDI-TOF MS are labelled. | ||
Natural minerals often have a hybrid structure. For example, Mg2+ is an important modifier of CaCO3 morphology and growth, and changes in the Mg/Ca ratio of sea water may have governed the polymorphic transitions of carbonate-based minerals during the geologic past.41 Natural hydroxyapatite in animal bone contains about 4–7 wt% carbonate.42 Therefore, to test the ability of our method to stabilize hybrid mineral clusters, we mixed CaCl2 and MgCl2 for CaxMgy(CO3)z clusters and used both (NH4)2CO3 and PPA as the anion sources for Cax(CO3)y(PO4)z clusters. TEM images (Fig. 5, enlarged images in Fig. S1, ESI†) indicate that PCDA can indeed stabilize these two hybrid clusters, which are also amorphous as revealed by SAED patterns. EDX spectra prove the co-existence of Ca and Mg in the CaxMgy(CO3)z cluster and the presence of Ca and P in the Cax(CO3)y(PO4)z cluster. IR spectra (Fig. S6, ESI†) can further confirm their hybrid structure judging from the combined bands from different components. Additionally, a shift of ν2(CO32−) for CaxMgy(CO3)z is observed for two peaks at 878 and 860 cm−1, while for MgCO3 the peaks are located at 852 cm−1, for CaCO3 at 860 cm−1 and for Cax(CO3)y(PO4)z at 864 cm−1. This reveals that the hybrid cores are not simply mixtures of two minerals but formed by mutual ionic substitution.
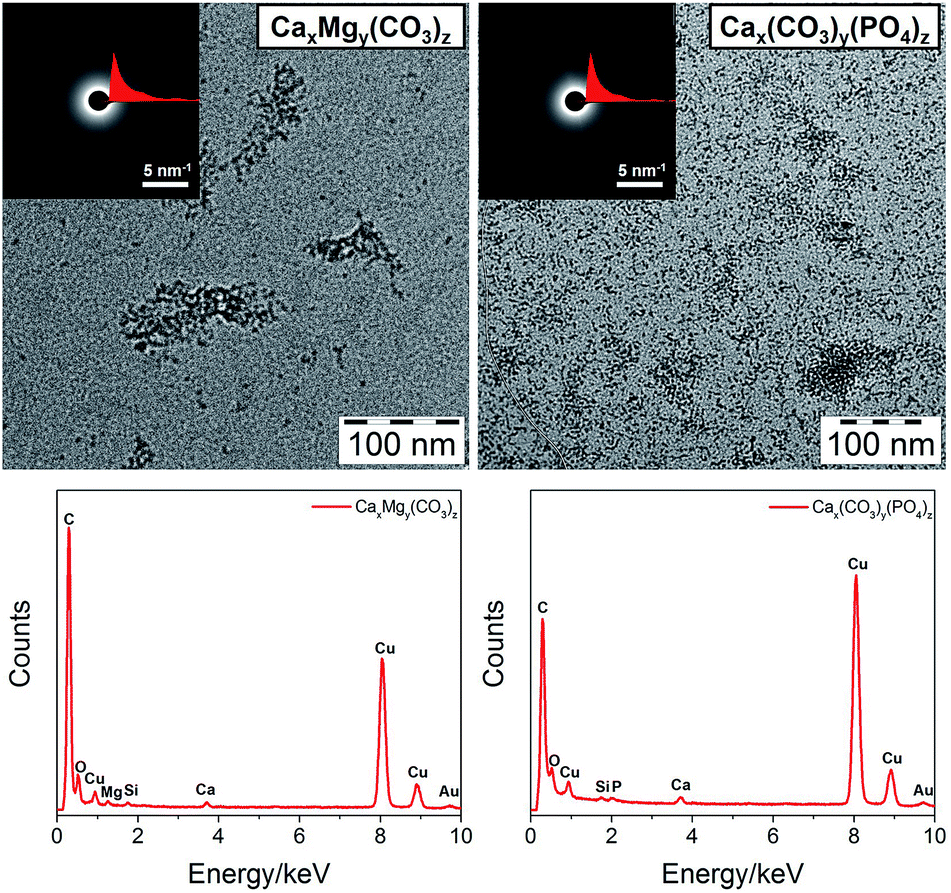 | ||
| Fig. 5 TEM images (top, insets are SAED patterns and radial integration curves) and corresponding EDX spectra (bottom) of PCDA monolayer protected CaxMgy(CO3)z and Cax(CO3)y(PO4)z clusters. | ||
To further investigate the internal structures of the mineral clusters, we compared the radial integration plots of SAED patterns with the simulated XRD curves of their corresponding crystalline phases, as shown in Fig. S8, ESI.† The size of crystalline phases was set to 1.5 × 1.5 × 1.5 nm3, and thus broad peaks are obtained due to the Bragg peak broadening effect from size reduction. Interestingly, the SAED radial integration curves of the mineral clusters exhibit several peaks that are in similar positions to the simulated XRD curves of corresponding nanocrystalline phases. This reveals that even in such small mineral entities, less than 2 nm, short-range order may already exist like in the case of CaCO3 clusters as we reported recently.28 Nevertheless, this still needs to be confirmed by more advanced techniques such as solid-state NMR and X-ray absorption spectroscopy (XAS).
Conclusions
In this paper, we developed a general gas diffusion strategy using PCDA as a ligand and ethanol as a solvent to fabricate stable amorphous mineral clusters with a core size of less than 2 nm. First discovered for CaCO3, the method was successfully extended to produce monolayer protected clusters of MgCO3, SrCO3, Eu2(CO3)3, Tb2(CO3)3, Ce2(CO3)3, Cax(PO4)y, CaC2O4 and their hybrid minerals, CaxMgy(CO3)z and Cax(CO3)y(PO4)z. All the mineral clusters show core–shell structures similar to the CaCO3 cluster we reported before,27,28 but with different sizes (2.7–6.6 nm) and compositions. The chemical formula of the Cax(PO4)y cluster was further confirmed to be (CaHPO4)8(H2O)3(PCDA)3. Although we cannot directly link the here presented monolayer protected mineral clusters to pre-nucleation clusters,13 the successful stabilization of such small mineral entities implies that mineral clusters as small as pre-nucleation clusters indeed exist. Our work paves a way for the artificial synthesis of colloidal mineral clusters, which may have various uses in both fundamental research and industry (e.g. as filler nanomaterials or nanocarriers).Acknowledgements
S. T. S. gratefully acknowledges the financial support from the Alexander von Humboldt Foundation and the National Science Foundation of China (NSFC; No. 21604024). D. G. is a Research Fellow of Zukunftskolleg of the University of Konstanz, and is supported by the Fonds der Chemischen Industrie. The authors thank Rose Rosenberg for AUC measurements and calculations and Zhouyue Lei of Fudan University (China) for experimental help.Notes and references
- M. F. Hochella, S. K. Lower, P. A. Maurice, R. L. Penn, N. Sahai, D. L. Sparks and B. S. Twining, Science, 2008, 319, 1631–1635 CrossRef CAS PubMed.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- Y. Boyjoo, V. K. Pareek and J. Liu, J. Mater. Chem. A, 2014, 2, 14270–14288 CAS.
- M. Li, H. Schnablegger and S. Mann, Nature, 1999, 402, 393–395 CrossRef CAS.
- D. H. M. Buchold and C. Feldmann, Adv. Funct. Mater., 2008, 18, 1002–1011 CrossRef CAS.
- J. F. Hui, Q. Y. Yu, Y. Long, Z. C. Zhang, Y. Yang, P. P. Wang, B. Xu and X. Wang, Chem.–Eur. J., 2012, 18, 13702–13711 CrossRef CAS PubMed.
- A. F. Demirors and A. Imhof, Chem. Mater., 2009, 21, 3002–3007 CrossRef CAS.
- M. Sasidharan and K. Nakashima, Acc. Chem. Res., 2014, 47, 157–167 CrossRef CAS PubMed.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 1999, 33, 27–36 CrossRef.
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- D. Gebauer and H. Cölfen, Nano Today, 2011, 6, 564–584 CrossRef CAS.
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergström and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC.
- E. M. Pouget, P. H. H. Bomans, J. Goos, P. M. Frederik, G. de With and N. Sommerdijk, Science, 2009, 323, 1455–1458 CrossRef CAS PubMed.
- A. Dey, P. H. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. de With and N. Sommerdijk, Nat. Mater., 2010, 9, 1010–1014 CrossRef CAS PubMed.
- F. Nudelman, K. Pieterse, A. George, P. H. Bomans, H. Friedrich, L. J. Brylka, P. A. Hilbers, G. de With and N. A. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- C. C. M. C. Carcouët, M. W. P. van de Put, B. Mezari, P. C. M. M. Magusin, J. Laven, P. H. H. Bomans, H. Friedrich, A. C. C. Esteves, N. A. J. M. Sommerdijk, R. A. T. M. van Benthem and G. de With, Nano Lett., 2014, 14, 1433–1438 CrossRef PubMed.
- M. Kellermeier, R. Rosenberg, A. Moise, U. Anders, M. Przybylski and H. Cölfen, Faraday Discuss., 2012, 159, 23–45 RSC.
- A. Schiener, A. Magerl, A. Krach, S. Seifert, H. G. Steinrueck, J. Zagorac, D. Zahn and R. Weihrich, Nanoscale, 2015, 7, 11328–11333 RSC.
- D. C. Gary, M. W. Terban, S. J. L. Billinge and B. M. Cossairt, Chem. Mater., 2015, 27, 1432–1441 CrossRef CAS.
- B. Fleury, M.-A. Neouze, J.-M. Guigner, N. Menguy, O. Spalla, T. Gacoin and D. Carriere, ACS Nano, 2014, 8, 2602–2608 CrossRef CAS PubMed.
- Y. Kimura, H. Niinomi, K. Tsukamoto and J. M. García-Ruiz, J. Am. Chem. Soc., 2014, 136, 1762–1765 CrossRef CAS PubMed.
- M. Salvalaglio, F. Giberti and M. Parrinello, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 132–136 CrossRef CAS PubMed.
- R. Demichelis, P. Raiteri, J. D. Gale, D. Quigley and D. Gebauer, Nat. Commun., 2011, 2, 590 CrossRef PubMed.
- M. Kellermeier, D. Gebauer, E. Melero-Garcia, M. Drechsler, Y. Talmon, L. Kienle, H. Cölfen, J. M. Garcia-Ruiz and W. Kunz, Adv. Funct. Mater., 2012, 22, 4301–4311 CrossRef CAS.
- S. Sun, D. Gebauer and H. Cölfen, 2016, unpublished results.
- S. Sun, D. M. Chevrier, P. Zhang, D. Gebauer and H. Cölfen, Angew. Chem., Int. Ed., 2016, 55, 12206–12209 CrossRef CAS PubMed.
- H. S. Lee, T. H. Ha and K. Kim, Mater. Chem. Phys., 2005, 93, 376–382 CrossRef CAS.
- S.-F. Chen, H. Cölfen, M. Antonietti and S.-H. Yu, Chem. Commun., 2013, 49, 9564–9566 RSC.
- Y. Kojima, A. Shiraishi, K. Ishii, T. Yasue and Y. Arai, Phosphorus Res. Bull., 1993, 3, 79–84 CrossRef CAS.
- M. Hajir, R. Graf and W. Tremel, Chem. Commun., 2014, 50, 6534–6536 RSC.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- S. Murthy, T. P. Bigioni, Z. L. Wang, J. T. Khoury and R. L. Whetten, Mater. Lett., 1997, 30, 321–325 CrossRef CAS.
- T. P. Ang, T. S. A. Wee and W. S. Chin, J. Phys. Chem. B, 2004, 108, 11001–11010 CrossRef CAS.
- S. Sun, D. Gebauer and H. Cölfen, Chem. Commun., 2016, 52, 7036–7038 RSC.
- F. Chen, P. Huang, Y.-J. Zhu, J. Wu and D.-X. Cui, Biomaterials, 2012, 33, 6447–6455 CrossRef CAS PubMed.
- G. He, T. Dahl, A. Veis and A. George, Nat. Mater., 2003, 2, 552–558 CrossRef CAS PubMed.
- J. Ihli, Y.-W. Wang, B. Cantaert, Y.-Y. Kim, D. C. Green, P. H. H. Bomans, N. A. J. M. Sommerdijk and F. C. Meldrum, Chem. Mater., 2015, 27, 3999–4007 CrossRef CAS.
- P. Layrolle and A. Lebugle, Chem. Mater., 1994, 6, 1996–2004 CrossRef CAS.
- K. J. Davis, P. M. Dove and J. J. De Yoreo, Science, 2000, 290, 1134–1137 CrossRef CAS PubMed.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02333a |
| ‡ Current address: School of Chemical Engineering, State Key Laboratory of Chemical Engineering, Shanghai Key Laboratory of Multiphase Materials Chemical Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, P. R. China. |
| This journal is © The Royal Society of Chemistry 2017 |