
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiangiogenic platinum through glycan targeting†
Erica J.
Peterson
ab,
A. Gerard
Daniel
ab,
Samantha J.
Katner
ab,
Lisa
Bohlmann
c,
Chih-Wei
Chang
c,
Anna
Bezos
d,
Christopher R.
Parish
d,
Mark
von Itzstein
c,
Susan J.
Berners-Price
c and
Nicholas P.
Farrell
 *abc
*abc
aDepartment of Chemistry, Virginia Commonwealth University, Richmond VA 23284, Virginia, USA. E-mail: 5.npfarrell@vcu.edu
bThe Massey Cancer Center, Virginia Commonwealth University, Richmond 23294, Virginia, USA
cInstitute for Glycomics, Griffith University, Gold Coast Campus, Southport, Queensland 4222, Australia
dJohn Curtin School of Medical Research, The Australian National University, Canberra, Australia
First published on 12th August 2016
Abstract
Heparan sulfate is identified as a ligand receptor for polynuclear platinum anti-cancer agents through sulfate cluster binding. We present a new biological role for platinum and coordination compounds and a new target for metal-based drugs while presenting a new chemotype for heparanase and growth factor inhibition through modulation (metalloshielding) of their interactions. Masking of extracellular (ECM)-resident heparan sulfate (HS) through metalloshielding results in very effective inhibition of physiologically critical HS functions including enzyme (heparanase, HPSE) and protein growth factor recognition. The interaction of the highly cationic polynuclear platinum complexes (PPCs) with the highly sulfated pentasaccharide Fondaparinux (FPX, in this case as a model HS-like substrate) results in inhibition of its cleavage by the HS-related enzyme heparanase. Binding of the fibroblast growth factor FGF-2 to HS is also inhibited with consequences for downstream signalling events as measured by a reduction in accumulation of phospho-S6 ribosomal protein in human colon tumor HCT-116 cells. The end-point of inhibition of HPSE activity and growth factor growth factor signaling is the prevention of cell invasion and angiogenesis. Finally these events culminate in inhibition of HCT-116 cell invasion at sub-cytotoxic concentrations and the process of angiogenesis. A competition assay shows that Fondaparinux can sequester the 8+ TriplatinNC from bound DNA, emphasising the strength of PPC–HS interactions. Altering the profile of platinum agents from cytotoxic to anti-metastatic has profound implications for future directions in the development of platinum-based chemotherapeutics.
Introduction
Small molecule innovation is critical to development of new medicines. Within anti-cancer drug development the delineation of a target allows molecular approaches to modulation of target interactions thereby hopefully modifying the overall biological response. This has certainly been the case in the area of platinum-based anti-cancer agents where a rich fount of structure and reactivity relationships has been produced based on DNA as their ultimate cellular target. These chemical and biophysical aspects are further linked to understanding of cellular signalling effects and downstream responses, eventually leading to cytotoxicity. In parallel, the advances in our understanding of cancer disease biology provide novel targets for drug development. In this paper we show that the interactions of polynuclear platinum complexes with the cellular signalling molecule heparan sulfate can be conceptualized in a similar manner to DNA – analysing the structural features necessary for strong binding with consequences for inhibition of function in this case leading to a systematic anti-angiogenic, rather than cytotoxic, effect.An optimal goal for cancer treatment is to develop anti-angiogenic drugs that will prevent metastasis and by limiting the primary tumor to a relatively localized site, allow for more effective drug intervention at that site. Most steps in the angiogenic cascade including endothelial cell proliferation, migration and differentiation are understood.1 Central to many of these processes is the heparanase (HPSE)/heparan sulfate (HS) interaction. Soluble glycosaminoglycan (GAG)-containing proteoglycans (e.g., heparin-containing and HS-containing; HPGs and HSPGs respectively) are thought to form the basis of the ECM glycointeractome and are known to bind to a significant range of well characterized bioactive molecules including angiogenic growth factors, pro-angiogenic receptors and in some cases to angiogenesis inhibitors.1 Cleavage of ECM HS by the enzyme heparanase is directly linked to the processing of these bioactive molecules and ECM remodeling. As a consequence, heparanase modulates relevant tumor-related events including angiogenesis, cell invasion, metastasis and inflammation.2 Furthermore, heparanase is over-expressed in many tumors and there is a significant correlation between metastatic potential and heparanase activity.2 Proteoglycans and their associated enzymes are thus significant emerging drug targets of high biological relevance.1,2
The molecular level understanding of the myriad biological processes involved in angiogenesis suggests new targets for therapeutic intervention. General approaches to exploit the HPG/HSPG system to inhibit angiogenesis can be summarized as (i) inhibition of enzymatic cleavage of HS by heparanase and (ii) interference with glycan–protein interactions, by complementary approaches such as design of “decoy” compounds to bind and mask endothelial cell-surface HSPGs and/or sequestering of angiogenic growth factors, thereby preventing their interaction with the endothelial cells and (iii) chemical or enzymatic modification of HSPGs to forms that are less recognizable by angiogenic growth factors. With respect to drug development, there has been a heavy emphasis on design of oligosaccharide mimics to interfere with enzyme/protein binding. Relevant examples of enzyme-targeted inhibitors include PI-88 (Muparfostat), a yeast-derived mixture of highly sulfated, monophosphorylated mannose oligosaccharides,3 and, most recently, the “PG500” series such as PG-545 derived from PI-88.4 Strategies based on inhibition of HS–growth factor interactions (predominantly fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF) families) also include small molecules and HS mimetics such as chemically modified heparins with variable saccharide lengths and/or sulfation patterns.1,5 Designed synthesis of low-sulfate mimetics or HS-like molecules having chemically-induced sulfation gaps has also been proposed for inhibition of HS–FGF interactions.5 Inhibition of heparanase activity and/or growth factor binding are not mutually exclusive and mimetics such as PI-88, PG-545 and the oligomannurate JG3 may target both functions.3,4,6
Masking or removing the HSPGs to inhibit enzymatic cleavage and/or diminish growth factor recognition is an attractive approach to modulate HSPG function. The cationic highly arginine-rich protein protamine, for example, interacts with HSPGs and inhibits FGF-2-dependent angiogenesis in vitro and in vivo.7,8 We have previously shown that clinically relevant anticancer polynuclear platinum complexes (PPCs) with high positive charge (Fig. 1(a)) are very effective metalloshielding agents for sulfated oligosaccharides.9 Their 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts with an HS octasaccharide (DP8, containing 3 sulfate groups per disaccharide) were shown by Electrospray Ionisation Mass Spectrometry (ESI-MS) to protect against sulfate loss in the gas phase.9 The strong binding is sufficient to protect the highly sulfated pentasaccharide Fondaparinux (FPX) from enzymatic cleavage by bacterial heparinases.9,10 The molecular description of the PPC–HS metalloshielding (protection of an adducted polysaccharide against sulfate loss or recognition) was suggested to be through a “sulfate clamp” analogous to the phosphate clamp motif observed in the crystal structure of TriplatinNC bound to DNA11,12 (Fig. 1(b and c)). These canonical motifs bear high structural similarity to those suggested for arginine-mediated recognition of proteins, for example TBP (TATA Binding Protein), through “arginine forks” to the phosphate backbone of DNA13 and indeed to the molecular description of cellular internalization of arginine-containing peptides through binding of sulfate groups on cell-surface HSPGs14 (Fig. 1(d and e)). In the latter case, PPCs are competitive inhibitors of binding of TAMRA-R9 (a nonaarginine peptide coupled to the fluorescent label, 5-(6)-carboxytetramethylrhodamine) to HSPGs with consequent decrease in cellular internalization of the nonapeptide.15 By the use of wt CHO cells and mutant CHO-pgsD-677 (lacking HS), and CHO-pgsA (lacking HS/CS chondroitin sulfate) cells, the clear biological consequences of PPC–HS interaction correlated with reduced cellular accumulation of PPCs and related decrease in apoptosis and cytotoxicity.15 These results led to recognition of the HSPG-mediated cellular accumulation of PPCs, a pathway unique to the highly cationic compounds and not shared by neutral cisplatin or oxaliplatin.
:1 adducts with an HS octasaccharide (DP8, containing 3 sulfate groups per disaccharide) were shown by Electrospray Ionisation Mass Spectrometry (ESI-MS) to protect against sulfate loss in the gas phase.9 The strong binding is sufficient to protect the highly sulfated pentasaccharide Fondaparinux (FPX) from enzymatic cleavage by bacterial heparinases.9,10 The molecular description of the PPC–HS metalloshielding (protection of an adducted polysaccharide against sulfate loss or recognition) was suggested to be through a “sulfate clamp” analogous to the phosphate clamp motif observed in the crystal structure of TriplatinNC bound to DNA11,12 (Fig. 1(b and c)). These canonical motifs bear high structural similarity to those suggested for arginine-mediated recognition of proteins, for example TBP (TATA Binding Protein), through “arginine forks” to the phosphate backbone of DNA13 and indeed to the molecular description of cellular internalization of arginine-containing peptides through binding of sulfate groups on cell-surface HSPGs14 (Fig. 1(d and e)). In the latter case, PPCs are competitive inhibitors of binding of TAMRA-R9 (a nonaarginine peptide coupled to the fluorescent label, 5-(6)-carboxytetramethylrhodamine) to HSPGs with consequent decrease in cellular internalization of the nonapeptide.15 By the use of wt CHO cells and mutant CHO-pgsD-677 (lacking HS), and CHO-pgsA (lacking HS/CS chondroitin sulfate) cells, the clear biological consequences of PPC–HS interaction correlated with reduced cellular accumulation of PPCs and related decrease in apoptosis and cytotoxicity.15 These results led to recognition of the HSPG-mediated cellular accumulation of PPCs, a pathway unique to the highly cationic compounds and not shared by neutral cisplatin or oxaliplatin.
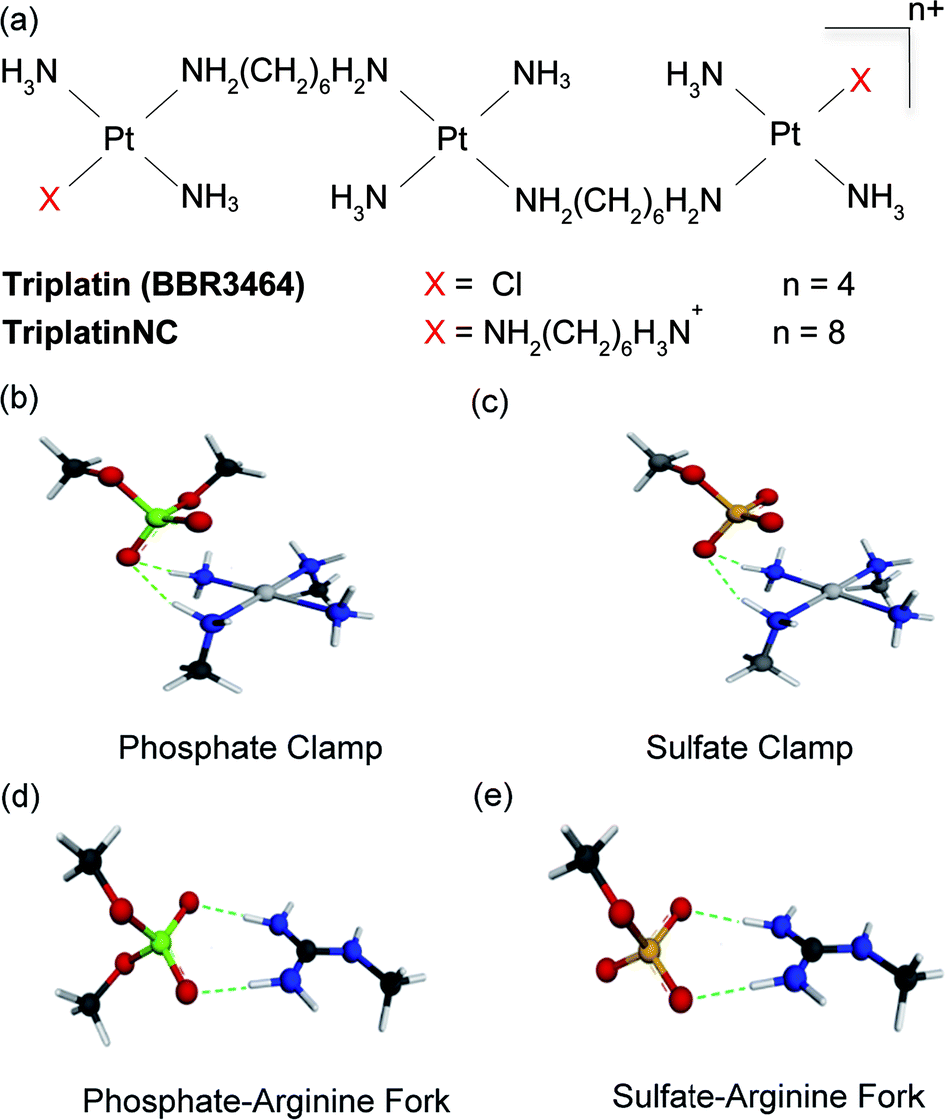 | ||
| Fig. 1 (a) Structures of platinum polynuclear complexes (PPCs). (b and c) Energy-minimized representations of one coordination unit trans-[Pt(NH3)2(NH2CH3)2]2+ of a PPC forming a phosphate and sulfate clamp, respectively, through hydrogen-bonding interactions. (d and e) Energy-minimized representations a Meguanidine moiety with hydrogen-bonding interactions to phosphate and sulfate, respectively, showing structural analogies of the arginine fork. The calculated charges on individual atoms and calculated bond distances are omitted for clarity but are consistent with the proposed structures. | ||
Given the correlation between HSPG-mediated cellular accumulation of PPCs and the PPC–HS interaction we have now asked the question – what are the functional consequences of such binding? In principle, metalloshielding could protect an adducted oligosaccharide from the action of glycan-degrading enzymes and/or protein recognition (analogous to inhibition of DNA–protein interactions through high-affinity DNA binding). Among PPCs, compounds capable of covalent interaction with biomolecules contain a Pt–Cl bond, as exemplified by Triplatin (Fig. 1(a)). Substitution-inert analogs capable of “non-covalent” binding to biomolecules through hydrogen-bonding and electrostatic interactions are obtained by substitution of the Pt–Cl bonds with NH3 or a “dangling” amine –H2N(CH2)nNH3+ (specifically TriplatinNC, n = 6 (Fig. 1(a))).15 In the case of glycans, formation of a Pt–sulfate bond may be preceded by a pre-association through the proposed sulfate clamp, a concept strictly analogous to the reactions observed in the kinetics of DNA binding by these compounds.16–18 In this paper we examine the effects of the two principal compounds, Triplatin and TriplatinNC, on limiting aspects of HS function. We show that the biological consequences of metalloshielding extend to modulation of HPSE activity and HS–fibroblast growth factor interactions leading to inhibition of cell invasion and angiogenesis. The PPC series represents a new multifunctional chemotype capable of systematic rational development as inhibitors of HS interactions while expanding the potential utility and description of platinum anti-cancer agents from cytotoxic to anti-angiogenic and anti-metastatic.
Results and discussion
Sulfate cluster anchoring as a model for PPC–HS interactions
In considering “non-covalent” ligand recognition we must examine how the interactions on extended systems such as DNA, heparin and HS differ from the simple phosphate and sulfate anions.10 The negative charge is more dispersed on a sulfate monoester because of delocalization involving three non-ester oxygen atoms compared to a phosphate diester with only two non-ester oxygen atoms. We first calculated the free energy of interaction (Eint) for isolated sulfate interactions and compared them with phosphate (see Fig. 1(b and c), (Table 1)). The trans-[Pt(NH3)2(NH2CH3)2]2+ compound was chosen because DNA–phosphate clamp interactions are always made by mutually cis-[Pt(NH3)(NH2R)] units in TriplatinNC.10 The interaction in water is not significantly favorable and only slightly stronger than the model methylguanidinium interaction (Table 1). The individual sulfate binding is also inherently weaker than that of phosphate.Density functional theory (DFT) along with molecular modeling (MM) and classical molecular dynamics (MD) have all been applied to understanding the structure and dynamics of heparin and GAG fragments.19,20 Sulfated GAGs, given the variability in the identity and conformation of the sugar and the number and position of sulfation, are highly complex systems. To reduce the complexity for molecular modeling and visualization, in this study we used the NMR-derived structure of heparin, which has also been used for DFT applications.19,20 Heparan sulfate is very closely related to heparin, in which the major repeating disaccharide consists of 2-O-sulfated iduronic acid and 6-O-sulfated, N-sulfated glucosamine, IdoA(2S)-GlcNS(6S). The structure of heparin is approximated by a ribbon with a cluster of sulfates and carboxylates on the edges and hydroxyl and sugar ring oxygens positioned on the surfaces between these negatively charged groups (Fig. 2(a)). We therefore applied DFT to elucidate the details of the sulfate masking under the influence of hydrogen bonding to various platinum tetram(m)ine coordination spheres. We first used the simple coordination unit [Pt(NH3)4]2+ on a GlcN(6S)-IdoA(2S)-GlcN(6S) trisaccharide heparin fragment, which constitutes one of the sulfate clusters, for validation (Fig. 2(b)). Now the sulfate binding by the platinum coordination spheres produces a more extended hydrogen-bonding network along the face of the cluster. Superimposition of the optimized structure of the association complex with the optimized structure of the free heparin trimer,19,20 shows no major conformational changes and the Eint is −53 kcal mol−1.
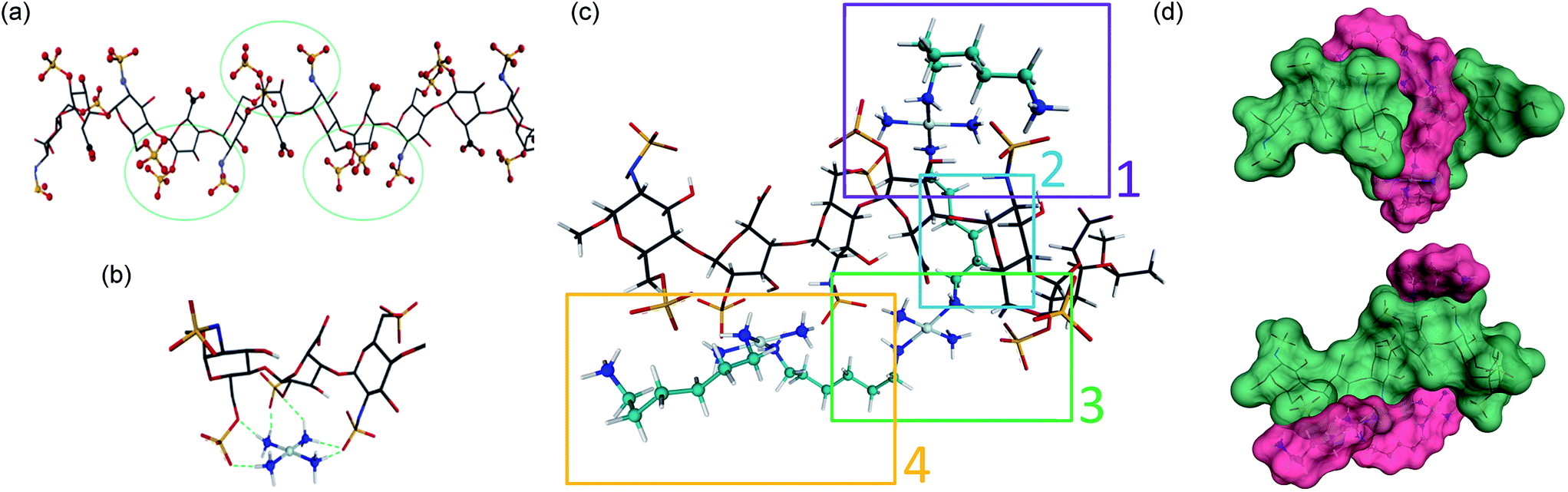 | ||
| Fig. 2 Sulfate cluster anchoring as a model for PPC–HS interactions. (a) Structure of the heparin dodecamer comprising dimers of IdoA(2S) and GlcNS(6S) where the iduronic acid residues are in the 2S0 conformation (1HPN). The green circles represent areas of negatively charged sulfate and carboxylate clustering. (b) The optimized structure of the trimer cluster [GlcNS(6S)-IdoA(2S)-GlcNS(6S)] modeled with the simple Pt–tetrammine [Pt(NH3)4]2+. (c) Optimized structure of TriplatinNC with a heparin hexamer [IdoA(2S)-GlcNS(6S)]3. Sections 1, 3 and 4 show regions of sulfate clamp interactions while Section 2 shows van der Waals contacts between sugar and diamine backbones. See Fig. S1† for detail of H-bonding interactions. (d) Surface maps (two views) showing the relationship of TriplatinNC (magenta) to the heparin hexamer (green). See methods for experimental details. | ||
The role of the linker and dangling amines in non-covalent interactions of PPCs with biomolecules, such as DNA and glycans, is of fundamental relevance for the design of molecules with more specific affinity for one biomolecule over the other. Using the trimer–Pt(tetrammine) interaction as a basis, the association of TriplatinNC with a heparin hexamer residue from the PDB 1HPN structure was modeled ((Fig. 2(c)), right to left [IdoA(2S)-GlcNS(6S)]3). The model was designed such that the carbon chain of one of the dangling amine groups spanned a hydrophobic groove in the middle of the helix formed by the hydrocarbon rings of the carbohydrates. This results in placing the other dangling amine at the edge of the helix, with the free –CH2NH3+ amine interacting with an N-sulfate of a central GlcNS(6S). The optimized structure now shows a number of regions of strong hydrogen-bonding contacts (regions 1, 3, 4) (Fig. 2(c)). The hydrocarbon chain along the groove establishes van der Waals contact with hydrogens from three of the sugar residues (region 2) (Fig. 2(c)). Moreover, the chain stretches all the way to the other side of the helix facilitating interaction of the second Pt center with two sulfates and a carboxylate residue on the other edge of the helix (region 3). From here, the second linker stretches along the edge to allow the third Pt center to interact with three sulfate groups from the last three sugar rings. Many areas of potential interaction are seen in the minimized structure. For clarity, the individual regions with potential interactions highlighted are shown in Fig. S1.† The interaction energy of the complex is −250 kcal mol−1, almost five times the energy released during the Pt–tetrammine:trisaccharide interaction (Fig. 2(b)) and significantly more than that of the isolated sulfate clamp. As visualized through the surface maps, the electrostatic interactions along with the hydrophobic interactions allow for a tight fit of the two molecules, (Fig. 2(d)). Metalloshielding in this delocalised manner should be especially effective in essentially reducing the availability of sulfate ions on the HS backbone with many functional consequences for HS template interactions.
Functional consequences of high-affinity glycan binding
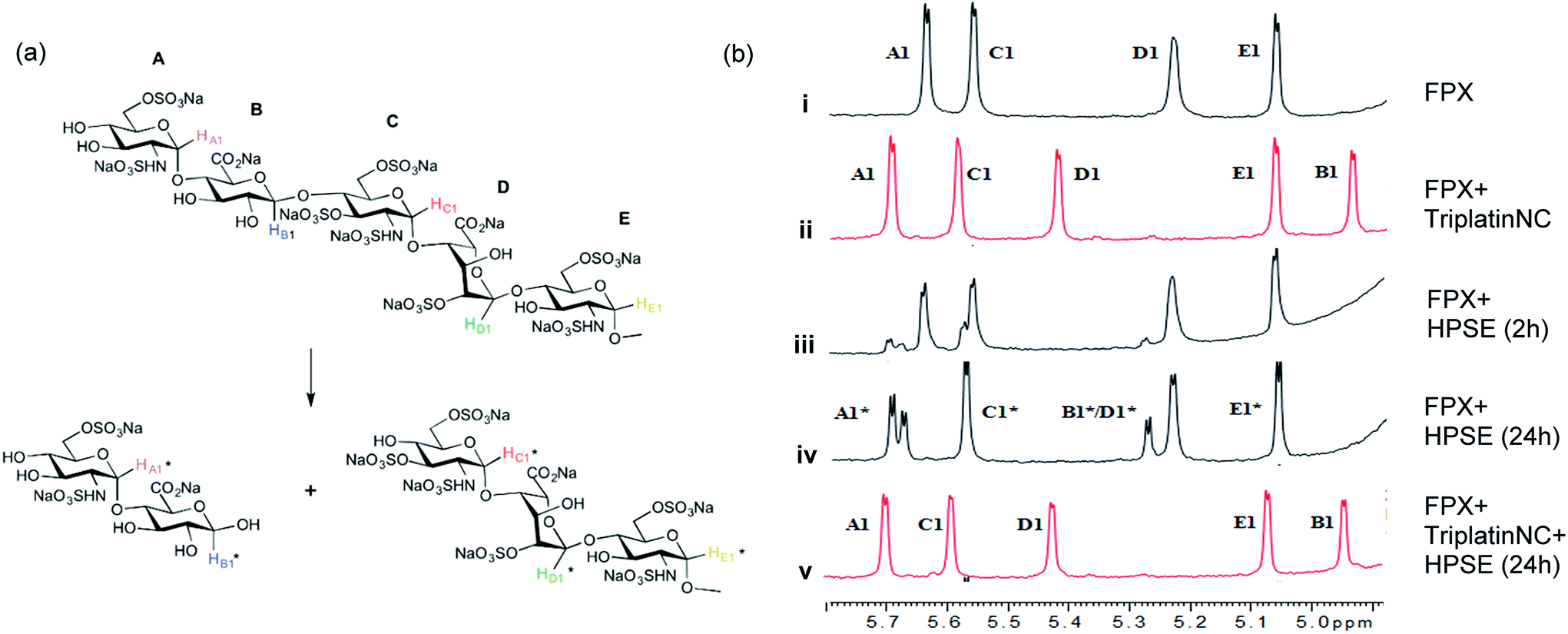 | ||
| Fig. 3 Blockade of heparanase activity. (a) Cleavage pattern of FPX by heparanase showing assignment of anomeric protons. Assignments followed published work.22,23,25 (b) Incubation of FPX with HPSE confirmed the cleavage of FPX, as expected (i → iii → iv, right). In the presence of one equivalent of TriplatinNC but in absence of enzyme, significant shifts are immediately seen in the anomeric signals of FPX (i → ii). Over the time course of 24 h with addition of enzyme (ii → iv), it is obvious that no further changes occur in the anomeric signals in particular with the absence of change around the A1/C1 signals clear evidence that there is complete inhibition of enzyme activity. | ||
The 1H NMR spectroscopy time course study of FPX incubated with HPSE (Fig. 3(b), i, iii and iv) confirmed the cleavage of FPX, as expected. Specifically, new signals arise at 5.65–5.7 ppm from the anomeric signals 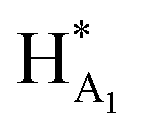 with concomitant decrease of the signal at 5.63 ppm (HA1). The signal for anomeric B1 is obscured by H2O in the absence of enzyme but upon cleavage undergoes an expected significant downfield shift (Fig. 3(b), iv). After 24 h, the FPX substrate is completely hydrolyzed, (Fig. 3(b), iv), in agreement with previous kinetic studies.22 In the presence of one equivalent of TriplatinNC and in the absence of the enzyme, a number of significant shifts are immediately seen in the anomeric signals of FPX (Fig. 3(b), ii). Firstly, the D1 signal undergoes a downfield shift of approx. 0.2 ppm while a similar downfield shift of B1 now makes the signal easily observable at 4.94 ppm. In the presence of the enzyme, over the time course of 24 h, it is evident that there is complete inhibition of enzyme activity as no obvious changes occur in the anomeric A1 proton signal (Fig. 3(b), v). A useful feature of this NMR assay is that the signals of the aliphatic carbon chains of the platinum complexes in the 1.50–3.00 ppm region are also easily observable with no overlap from the oligosaccharide protons. No major changes are apparent in the 1H NMR spectrum of TriplatinNC itself throughout the time course of the reaction.
with concomitant decrease of the signal at 5.63 ppm (HA1). The signal for anomeric B1 is obscured by H2O in the absence of enzyme but upon cleavage undergoes an expected significant downfield shift (Fig. 3(b), iv). After 24 h, the FPX substrate is completely hydrolyzed, (Fig. 3(b), iv), in agreement with previous kinetic studies.22 In the presence of one equivalent of TriplatinNC and in the absence of the enzyme, a number of significant shifts are immediately seen in the anomeric signals of FPX (Fig. 3(b), ii). Firstly, the D1 signal undergoes a downfield shift of approx. 0.2 ppm while a similar downfield shift of B1 now makes the signal easily observable at 4.94 ppm. In the presence of the enzyme, over the time course of 24 h, it is evident that there is complete inhibition of enzyme activity as no obvious changes occur in the anomeric A1 proton signal (Fig. 3(b), v). A useful feature of this NMR assay is that the signals of the aliphatic carbon chains of the platinum complexes in the 1.50–3.00 ppm region are also easily observable with no overlap from the oligosaccharide protons. No major changes are apparent in the 1H NMR spectrum of TriplatinNC itself throughout the time course of the reaction.
In the case of Triplatin, with the potential for Pt–Cl substitution reactions, a different situation arises when incubated with FPX and enzyme. Immediately upon mixing with FPX and in absence of enzyme, the spectral changes predominantly mirror those of the TriplatinNC–FPX interaction and, especially, the downfield shifts of B1 and D1 are indicative of pre-association, (Fig. S2(a)†). A number of peaks of very low intensity are also clearly visible in this region. No initial changes in this spectrum occur upon incubation with enzyme. After 1 h there is an increase in intensity of the minor peaks with concomitant decrease in intensity of the major peaks ascribed to pre-association. In contrast to TriplatinNC, new peaks are observed in the Pt–NH2CH2– region of the spectrum with the appearance of a slightly upfield multiplet growing in intensity over the 24 h of the reaction period (Fig. S2(b and c)†). Over the time course of 24 h, the anomeric region becomes quite complicated but very similar with or without enzyme. The overall results are also consistent with significant inhibition of FPX cleavage because of the lack of the new diagnostic 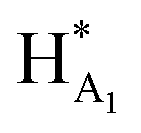 anomeric signals.
anomeric signals.
These NMR time course studies are therefore entirely consistent with pre-association of Triplatin on the oligosaccharide skeleton followed by bond formation through displacement of the Pt–Cl bond, presumably by sulfate and/or carboxylate groups on the edge of the FPX molecule. The spectral changes in the alkanediamine linkers of Triplatin mirror those observed upon covalent Pt–DNA bond formation.16,17 Both carboxylate and sulfate groups are weak ligands for dinuclear and trinuclear platinum compounds,16,26 but with strong pre-association can become viable coordinating sites.
The prototypical example of HS–protein interactions is the binding to fibroblast growth factors (FGFs), a family of (at least) 23 structurally related polypeptides involved in developmental and physiological processes including cell proliferation, differentiation, morphogenesis and angiogenesis.1,5 Binding of basic FGF (bFGF, also known as FGF-2) to cell surface HSPG is necessary for its recruitment of high affinity FGF receptors and for its mitogenic activity. In an initial step, these effects are exerted through binding to four highly related transmembrane tyrosine kinase receptors (FGFR1–FGFR4) resulting in FGF–FGFR dimerization1,27 (Fig. 4(a)). This results in trans-autophosphorylation of FGFRs at intracellular tyrosine residues and the activation of the Ras/mitogen-activated protein kinase and/or phosphoinositide 3-kinases (PI3K)/Akt signaling networks.28 In general, increased stimulation of receptor tyrosine kinases (RTKs) by growth factors is associated with the development and metastatic spread of cancerous cells.29
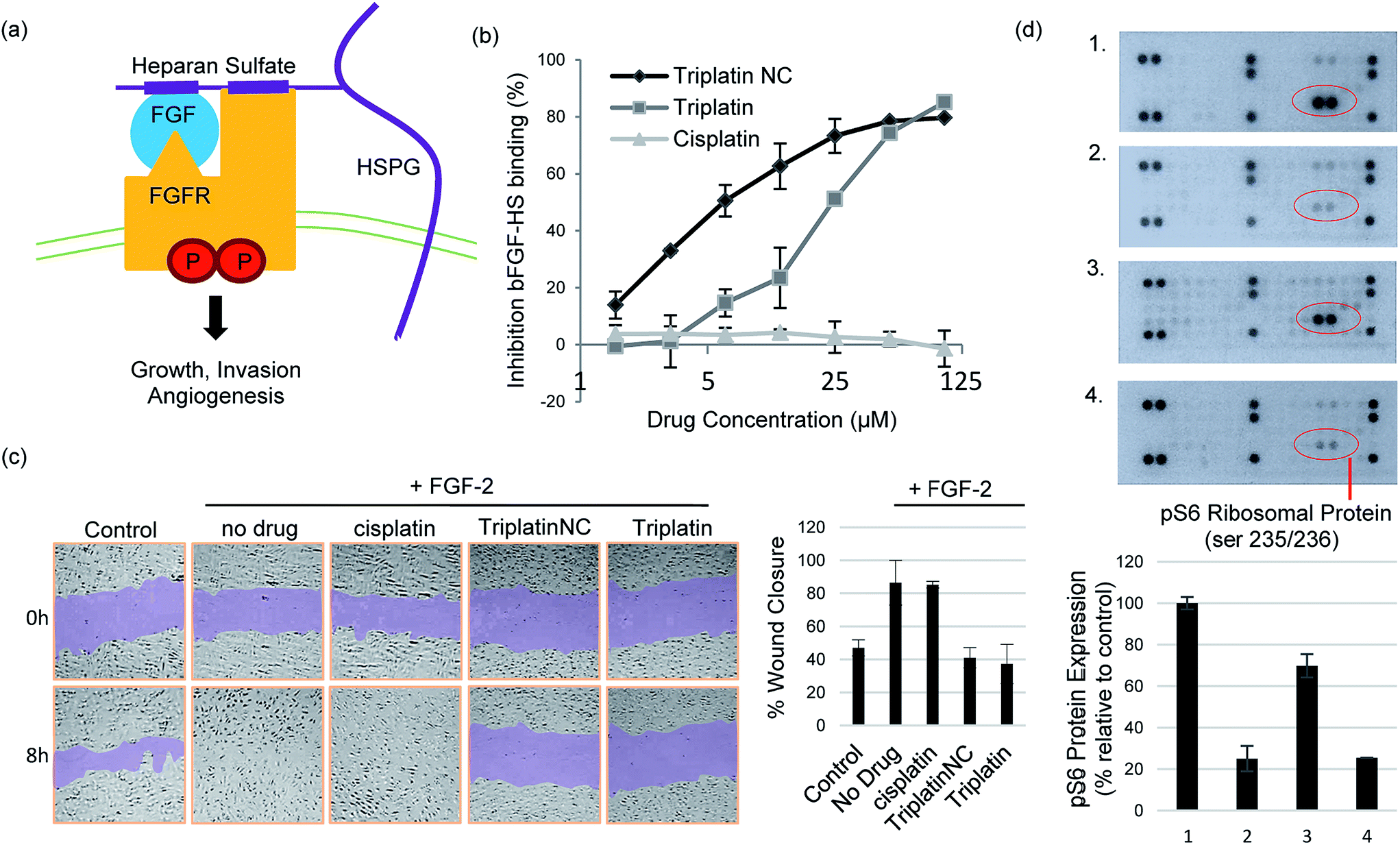 | ||
| Fig. 4 Inhibition of HS–protein interactions. (a) Growth signaling occurs when a growth factor (GF) binds and activates a cell surface receptor tyrosine kinase (FGFR). This interaction is facilitated by growth factor binding to heparan sulfate proteoglycans (HSPGs) forming stable high-affinity ternary complexes with FGFRs. (b) PPCs inhibit FGF-2 binding to heparan sulfate. (c) PPCs inhibit FGF-2-induced migration of human umbilical vein epithelial cells (HUVECs). Sub-cytotoxic concentrations of cisplatin, Triplatin or TriplatinNC (2 μM) and FGF-2 (10 ng mL−1) were added to the “wounded” cell monolayer. Cell migration was photographed at 8 h and compared to the 0 h control. (d) TriplatinNC inhibits FGF-2-induced accumulation of phospho-S6 ribosomal protein (pS6), measured by the PathScan RTK Signaling Antibody Array kit (Cell Signaling). (1) No treatment control (2) 24 h serum-starvation (3) 24 h serum-starvation + 10 min incubation with FGF-2 (100 ng mL−1) (4) 24 h serum-starvation + 5 min. TriplatinNC (20 μM) + 10 min incubation with FGF-2 (100 ng mL−1). | ||
Crystal structures of heparin-derived tetra and hexa-saccharides with FGF-2 show clear sulfate contacts with FGF-2 surfaces defined by asparagine, arginine, lysine and glutamine residues.30 We hypothesized that strong HS masking by PPCs would interfere with the interaction of FGF-2 with its receptors. A biotinylated-HS was incubated with varying concentrations of platinum complex prior to addition to a microtiter plate containing immobilized FGF-2. HS–FGF-2 binding was detected by a modified ELISA-type assay where binding of biotinylated HS to the growth factor is detected by an increase in absorbance at 450 nm (OD450).31 Triplatin and TriplatinNC directly inhibited FGF-2 binding to HS (Fig. 4(b)). Cisplatin is completely ineffective (Fig. 4(b)). Next, we asked whether PPCs would inhibit cellular migration in response to FGF-2 using the wound-healing assay, which measures directional migration of cells in a monolayer after a scratch or ‘wound’ is inflicted.6 Monolayers of confluent human umbilical vein epithelial cells (HUVECs) were scratched, exposing the growth surface. Within 8 h, FGF-2 (10 ng mL−1) induced the cells to migrate, closing the exposed area completely. Upon addition of 2 μM cisplatin, Triplatin, or TriplatinNC, only the PPCs inhibited growth factor induced migration (Fig. 4(c)). Because anti-proliferative and cytotoxic effects would also inhibit migration, the drug concentration used was determined to be well below the cytotoxic level for this short exposure time (Fig. S3†). Therefore, it was asked whether PPCs can block interactions of FGF-2 with its' receptor, FGFR1. Human colon carcinoma cells, HCT116, were chosen as an appropriate cell model as they overexpress FGFR1, and are susceptible to growth inhibition by a small molecule FGFR1 inhibitor.32,33 Although direct receptor activation was not detectable, TriplatinNC clearly reduced FGF-2-induced accumulation of phospho-S6 ribosomal protein (pS6) (Fig. 4(d)). pS6 is a primary initiator of protein synthesis that is activated in response to FGFR phosphorylation and PI3K/Akt/mTOR signaling. Therefore, a reduction in pS6 is indicative of PPC inhibition of FGF/FGFR signaling. Using the wound-healing assay, PPCs inhibited serum-induced migration of HCT116 s similarly to FGF-2-induced HUVECs (Fig. S4†).
We did not detect a significant increase in pFGFR in response to FGF-2 induction of HCT116 cells at the time points tested. This was somewhat expected as phosphorylation events at the receptor are highly transient, but a shorter induction time may allow for a more visible response. Downstream ERK1/2 phosphorylation was also examined under these conditions, but a decrease was not observed in serum-starved cells, nor an increase upon induction with FGF-2. This finding is consistent with a previous report showing no change in ERK1/2 activation in HCT116 (KRAS-mutated) cells with inhibition of pFGFR.32 FGF/FGFR signaling can be mediated by the Ras-Erk or PI3K-Akt-S6 pathways, both leading to changes in cell proliferation and mobility. In this case, pS6 provided the most dynamic response to FGF-2 induction and corresponding inhibition by PPCs.
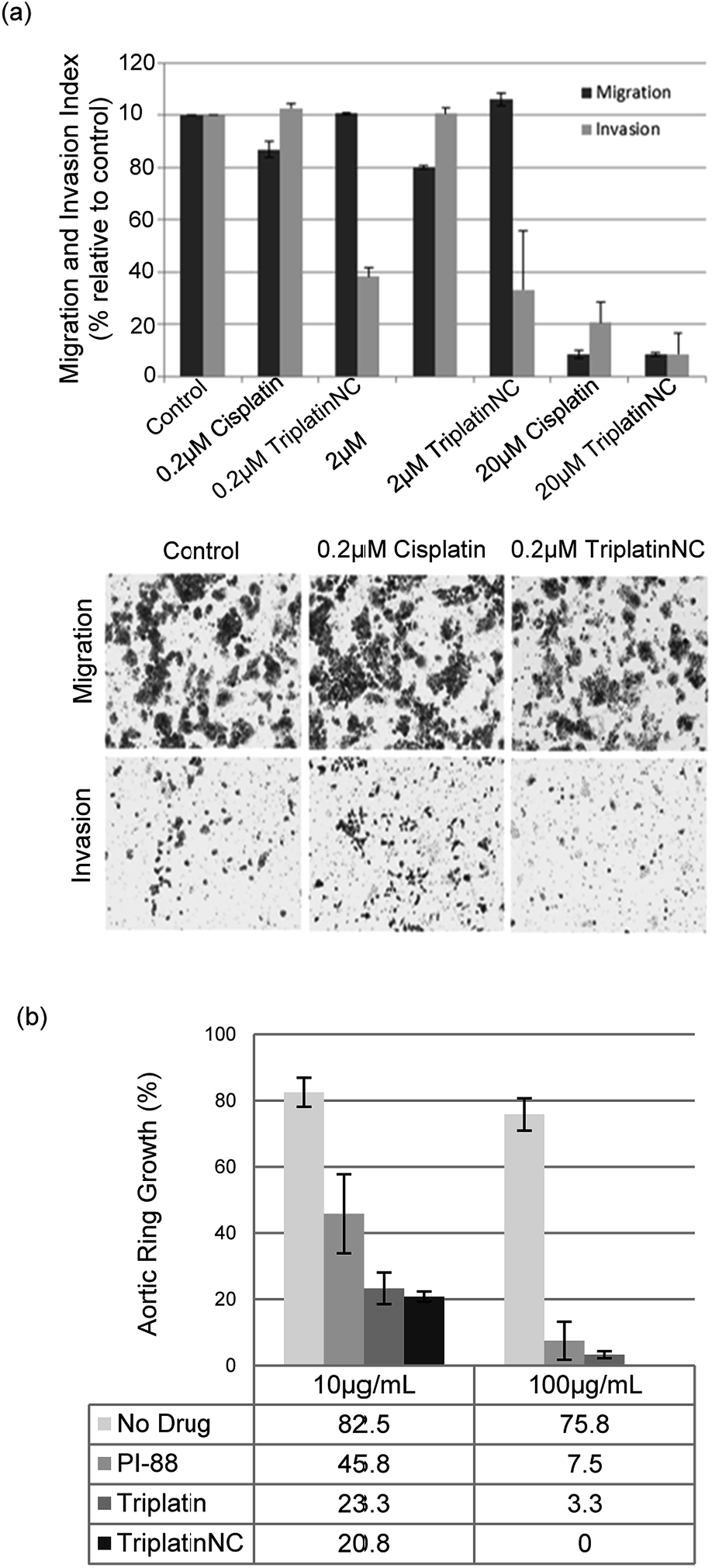 | ||
| Fig. 5 PPCs inhibit tumor cell invasion and angiogenesis. (a) Invasion of HCT116s through extracellular matrix is reduced after treatment with TriplatinNC (top). Representative images of the lower Boyden transwell chambers stained with crystal violet dye at 100× magnification (bottom). (b) PPCs inhibit the outgrowth of blood vessels from rat aortic ring. Aortic rings (1 mm) embedded and cultured in fibrin gel were stimulated with 20% serum and treated with or without BBR3464, TriplatinNC, or PI-88. The effects were observed on day 5 of the stimulation. Vessel growth is quantified as the percentage of the field (40×) around the vessel that is occupied by outgrowths. Typical new vessel growth in the control wells is 85% by day 5. The data shown is the mean percentage of outgrowth ± SD, n = 6.3 | ||
We finally assayed the ability of the two polynuclear compounds to inhibit angiogenesis in a rat aorta model and compared the results with the oligosaccharide mimic PI-88, which has undergone clinical trials as a heparanase inhibitor.3 The potency of anti-angiogenic activity is measured by the inhibition of new blood vessel growth sprouting from the original aortic ring.3 New vessel growth is quantified by estimation of growth as the percentage of the field around the vessel fragment that is occupied by vessel outgrowths (Fig. 5(b)). Typical new vessel growth in control wells is about 80–85% by day 5. TriplatinNC at 100 μg mL−1 completely inhibited new vessel growth on day 5 (there was no evidence of any growth even after 7 days) and was effective at 10 μg mL−1. Triplatin was also highly potent as an anti-angiogenic with very little new vessel growth. The inhibitory values at 10 μg mL−1 correspond to 8.07 and 6.06 μM for Triplatin and TriplatinNC respectively. The inhibition is remarkably efficient and comparable with that of PI-88.
To assess the strength of the non-covalent FPX–PPC interaction, novel competitive inhibition assays with DNA were developed. The displacement of intercalated ethidium bromide (EtBr) from DNA results in loss of fluorescence and has been widely used to measure platinum complex–DNA affinity. The assay was modified by titrating FPX into a TriplatinNC/DNA/EtBr reaction mixture, where the EtBr fluorescence is quenched due to strong TriplatinNC–DNA binding.40 The affinity of FPX for TriplatinNC is such that the complex is removed from the DNA, visualized by the reappearance of fluorescence as the intercalator again binds to the nucleic acid (Fig. 6(a)). Approximately equimolar concentrations of FPX relative to TriplatinNC are sufficient to restore the full fluorescence (full intercalation) of EtBr/DNA (Fig. 6(b)). While this experiment is purely biophysical, it demonstrates the high affinity of FPX for TriplatinNC. It is relevant to note that heparin itself is often cited as having the highest charge density of any biomolecule, higher even than polyanionic DNA. Further, we determined that sulfated residues contribute significantly to the affinity of the TriplatinNC–HS interaction using fluorescence polarization competition experiments (Fig. 6(c)). Unlike FPX, an unsulfated HS 5-mer (GlcA–GlcNAc–GlcA–GlcNAc–GlcA) did not efficiently compete for DNA bound TriplatinNC.
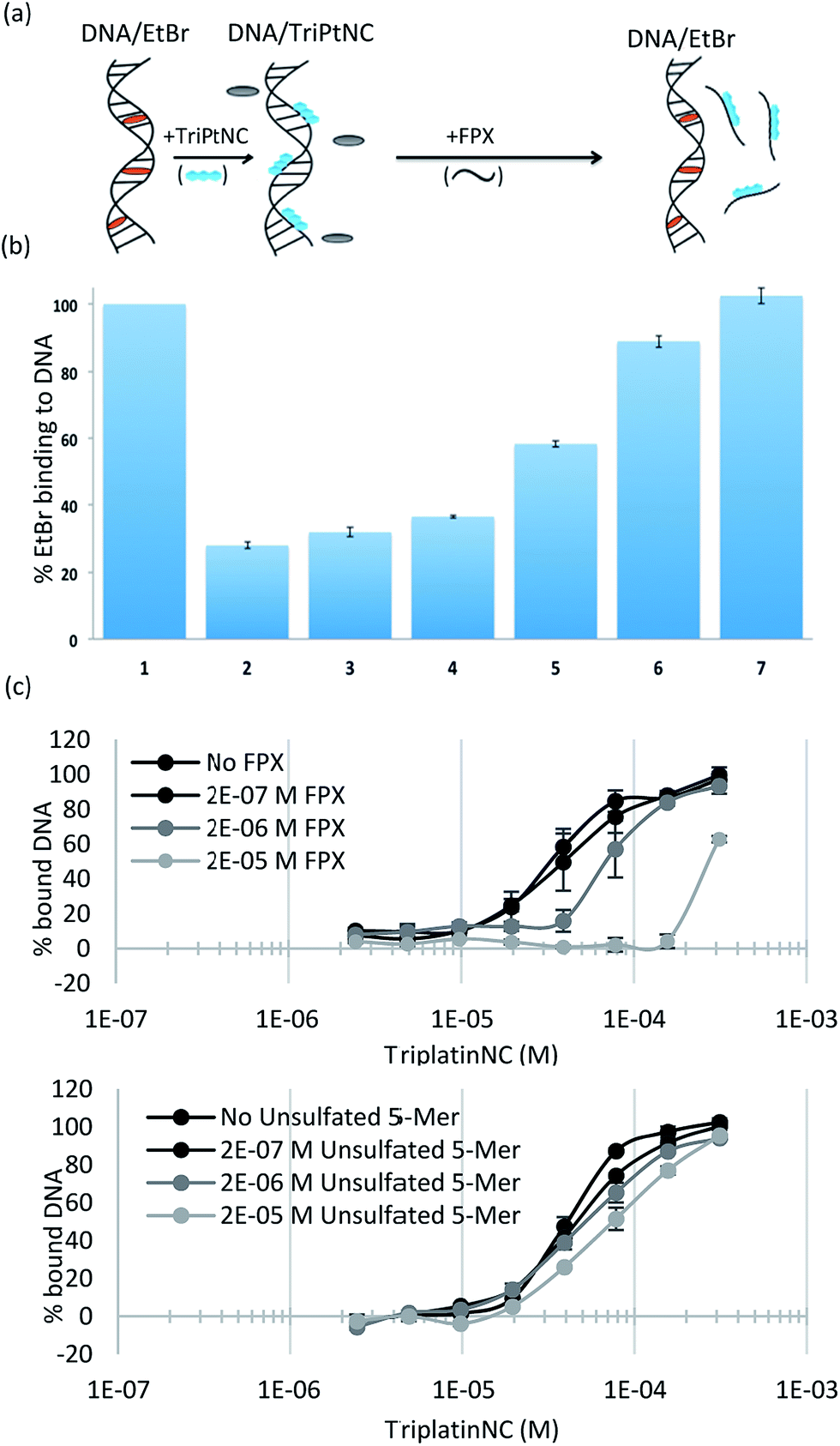 | ||
| Fig. 6 Sulfated HS competes with high affinity TriplatinNC–DNA binding. (a) Scheme of competitive inhibition experiment: the fluorescence of EtBr bound to DNA (DNA/EtBr), is reduced upon the addition of TriplatinNC but is regained upon addition of FPX (b) (1) EtBr/DNA only (2) EtBr/DNA/TriplatinNC (12.5 μM) (3–7). Fluorescence due to EtBr/DNA upon addition of increasing concentrations of FPX at 5, 7.5, 10, 12.5 and 15 μM respectively. EtBr bound to DNA (lane 1) was normalized to 100% with EtBr only as 0%. (c) Fluorescence polarization experiments showing comparison of TriplatinNC–DNA binding competition with sulfated 5-mer (FPX) (top) and unsulfated 5-mer (bottom). | ||
Conclusions
The sum of the data presented here show that PPCs represent a new, distinct and discrete chemotype for interference with HS/enzyme/protein interactions. In a systematic manner, we have shown that metalloshielding inhibits substrate cleavage by heparanase and prevents growth factor binding and growth factor receptor recruitment. Sulfate cluster anchoring of PPCs will shield the sulfates from recognition by charged protein residues preventing the exercise of the HS–enzyme/protein function. The cellular consequences are inhibition of invasion and angiogenesis. Diminishing available sulfate sites through metalloshielding is a uniform and general approach to reduce sulfate accessibility with possible consequences for regulation of HS sulfation and indeed may help define the existence of high-affinity regions within the heterogeneous biomolecule.1,41Both the covalently binding Triplatin and the substitution-inert TriplatinNC are effective at inhibiting heparanase cleavage of FPX and angiogenesis. The heparanase inhibition results confirm the previous data from the colorimetric assay using heparinase I, where inhibition was charge-dependent and comparison of a series of compounds also indicated a bond-forming contribution to the activity of Triplatin.9 The individual coordinating moieties on FPX and by extension the HS backbone – predominantly sulfate oxygen and possibly carboxylate – are inherently weaker ligands for covalent bond formation to Pt compared to those of DNA (N donor atoms of purines and pyrimidines) and proteins (N of histidine and S of cysteine and methionine residues). The aquation kinetics in 15 mM SO42− of a prototypical dinuclear compound structurally analogous to Triplatin, [{trans-PtCl(NH3)2}2(μ-NH2(CH2)6NH2)]2+, showed a very high kL for loss of sulfate suggesting that, when formed, the sulfate species will be substitution-labile.16 The strong non-covalent interactions assessed by DFT calculations (Fig. 2, Table 1) and observed in the NMR spectra of the initial interaction between Triplatin and FPX (Fig. S2†) may facilitate covalent Pt–heparan sulfate bond formation upon the sulfate cluster anchoring.
The identification of glycans as a viable target for therapeutic intervention raises the question of how we assess biomolecular targets of platinum in general. The advent of Triplatin (as BBR3464, see Fig. 1(a)) to human clinical trials altered the structure–activity paradigms for platinum anti-cancer agents but development was still predicated on the need for covalent Pt–DNA bond formation.10 The biological (including in vivo) activity of formally substitution-inert PPCs (e.g. TriplatinNC) is a further paradigm shift.35,42,43 With multiple points of contact, binding constants, Kapp, to calf thymus (ct) DNA are significantly higher than for canonical DNA binders such as minor groove binders, intercalators and even polyamines such as spermine.40,44 While phosphate and sulfate may be structurally similar there are significant chemical differences, especially when the anionic entities are incorporated into their respective biomolecules. The sulfate moieties are distinctly more conformationally flexible than the relatively constrained phosphodiester motif of the nucleotide, resulting in greater variability in neighboring sulfate–sulfate distances compared to the relatively fixed phosphate–phosphate distances in DNA/RNA. The “anchoring“ of the sulfate clusters on the HS backbone through the extended hydrogen-bonding framework and van der Waals interactions complements the modular nature of the PPCs to produce the strong non-covalent interactions observed.
The nature and extent of potentially strong platinum complex–heparin/HS interactions in general have not received as much attention as that of DNA and proteins but are required to give a full description of the biology of platinum complexes. Indeed, simple metal cations such as Zn2+ and Ca2+ affect conformation and activity in heparin derivatives.45 FGF–FGFR signaling is also modified by cation (Na+, Ca2+)-heparin polysaccharides.46,47 Our combined results further emphasize the relevance of the metalloglycomic concept – with the intrinsic ability to alter metal ion oxidation state, coordination number and geometry, and substitution lability of ligands to enhance shielding and the potential for specific conformational changes on the glycan template. The extent to which our observations may explain anti-angiogenic and vascular disturbing properties of recently reported platinum–metal complexes should be explored.48,49 Indeed our findings may be relevant to the biological function at the molecular level of the Ru-based NAMI-A, the prototypical anti-metastatic coordination compound, which has undergone clinical trials based on its anti-metastatic properties.50
Finally, recognition of HS as a target for PPCs also allows us to see the PPC chemotype as inherently possessing a dual-function. The activity demonstrated here is allied to DNA binding and nuclear effects such as bending, nuclear condensation and nucleolar targetting upon cellular entry.35,51 Just as Pt–DNA interactions are easily understood to affect downstream effects (protein recognition, signalling pathways) the work presented here clearly shows that Pt–HS interaction affects the downstream effects and signalling pathways of HS/heparanase.1,2,5 Indeed, it is provocative to consider that the effects described here may have been part of the early clinical findings of Triplatin but not appreciated at the time.10 The separate confirmation of significant anti-metastatic activity of Triplatin is supportive of this point and current efforts to confirm the biochemical mechanisms involved such as heparanase degradation of HS at a cellular level are ongoing.52 Cancer treatment may remain as an empirical combination of drugs that act on different mechanistic levels but are each designed for one specific target, in many cases, resulting in just incremental improvements in the outcome. Therapeutic strategies to develop inherently multifunctional agents hold promise for more effective combinations, overcoming the limitations such as resistance to single-targeted drugs. Development of new medicines and new chemical entities, which may simultaneously attack a range of targets, would represent a significant addition to the anti-cancer armamentarium. The recognition of a molecular pathway capable of systematic change enabling the transition from cytotoxic to anti-angiogenic holds major promise for a new era of platinum–metal chemotherapy.
Experimental
Synthesis and materials
The platinum complexes were prepared by published methods.42 Fondaparinux (GlcNS6S-GlcA-GlcNS3,6S-IdoA2S-GlcNS6S-OMe) and PI-88 (Muparfostat) were sourced as previously.3,23 The unsulfated 5-mer (GlcA–GlcNAc–GlcA–GlcNAc–GlcA) was purchased from Polysciences, Inc.Cell culture
Human umbilical vein endothelial cells (HUVECs) and Human Colon Tumor (HCT116s) were purchased from the American Type Tissue Collection. HUVECs were maintained in Vasculife Endothelial Medium containing 5% serum + growth factors (Lifeline Cell Technology). HCT116s were maintained in RPMI (Life Technologies) containing 10% serum (FBS, Serum Source International) and 1% penicillin–streptomycin solution (10000 U mL−1) from Gibco.
Molecular modeling and DFT calculations
All DFT computations were performed using the Gaussian 09 suite of programs.53 The dispersion corrected density functional M06L,54 which was shown to perform better for organometallic complexes, was used for these computations. The basis sets used are, 6-31g(3df,2pd) for calculations related to isolated sulfate and phosphate units and 6-31g(2d,2p) for all the other calculations involving larger entities, with diffuse functions only on anionic oxygen atoms in all cases. All systems were solvated using the IEFPCM solvation model with water as solvent. The interaction energies that are reported here include zero-point energy and basis set superposition error (BSSE) corrections for the larger systems and only zero-point energy correction for isolated sulfate and phosphate units interacting with Arg (Meguanidinium) or a single Pt center. The counterpoise method was implemented for BSSE correction.55 For the bigger systems the initial model was optimized using the semi-empirical method PM6, using MOPAC56 and the resulting geometry was used as the input for the DFT calculations. The heparin models were constructed based on the NMR structure, 1HPN.19 The structure is a dodecamer made of dimers formed by IdoA(2S) and GlcNS(6S). There were two models in the original PDB file, each corresponding to all the iduronic acid residues in either a 2S0 or 1C4 conformation. The structure resulting from the IdoA(2S) in a 2S0 conformation has sulfate groups well separated compared to the structure from the 1C4 conformation of IdoA(2S). Through an initial visual assessment of a crude model, the 2S0 form was found to be more suitable for Pt–tetrammine interactions. In order to validate this hypothesis, the trimer [GlcNS(6S)-IdoA(2S)-GlcNS(6S)] that forms the cluster was first modeled with Pt–tetrammine [Pt(NH3)4]2+. The interaction of TriplatinNC with a heparin hexamer was modeled by extension of this model.NMR HPSE inhibition assay
The assay followed previous reports.23 Briefly, FPX (0.5 mM) was pre-incubated with Pt inhibitor (0.5 mM TriplatinNC or Triplatin) and then was incubated with 1 μg human heparanase at 37 °C in 40 mM deuterated sodium acetate buffer, pH 5. 1H NMR spectroscopy was performed in 200 μL Shigemi tubes in an Avance 600 MHz Ultrashield NMR spectrometer (Bruker) with a 1H/13C/15N gradient cryoprobe system, data being collected and analysed with the TOPSPIN software (Bruker). The HDO-signal was suppressed using a 2 s rf-field during the relaxation delay.FGF-2 and HS binding assay
Varying concentrations of TriplatinNC, Triplatin, and cisplatin were incubated with human biotinylated heparan sulfate (Takara/Clontech) for 10 min at room temp. Each sample was transferred to the wells of a FGF-2 immobilized microtiter plate containing immobilised FGF-2 (Takara/Clontech) and incubated at 37 °C for 15 min. The mixtures were then removed, the wells washed, and the remaining biotinylated HS bound to FGF-2 was detected by the binding of an avidin–peroxidase conjugate. After addition of the peroxidase substrate, the absorbance was measured at 450 nm using a microplate reader. Data is reported as the average of two independent experiments ± SD.Wound-healing assay
5 × 105 HUVECs were seeded in 1 mL supplemented media (VascuLife Endothelial Cell Media, Lifeline Cell Technologies) in a 24-well plate. The cells were grown to confluence (∼3 days). A scratch was made on the monolayer using p10 pipette tips, removing the cells to expose the growth surface. The wells were washed 2× with PBS. The media was replaced with unsupplemented media containing 0.1% BSA for the control. For FGF-2-induction, 10 ng mL−1 recombinant FGF-2 (Cell Signaling) was added with or without drug to unsupplemented media containing 0.1% BSA. The extent of closure of the scratch at 8 h was compared to the control by light microscopy, digital imaging, and analysis by ImageJ software.Antibody array detection of FGF-2-induced signaling
For each condition, two 100 mm dishes were seeded with HCT116 cells (1 × 106) in 20 mL of RPMI media without serum and incubated at 37 °C for 24 h. HCT116s were (a) untreated (b) serum starved for 24 h or (c) serum starved for 24 h and treated with 100 ng mL−1 of recombinant FGF-2 (Cell Signaling) for 10 min or (d) serum starved for 24 h, treated with 20 μM TriplatinNC for 5 min, and then treated with 100 ng mL−1 FGF-2 for 10 min. The cell samples were harvested, lysed in the presence of protease and phosphatase inhibitors, and the amount of protein quantified using the Bradford protein assay. 150 μg of protein from each sample was incubated overnight at 4 °C on separate arrays from the PathScan RTK Signaling Antibody Array kit (Cell Signaling). The antibody array allows for simultaneous detection of 28 receptor tyrosine kinases and 11 downstream signaling nodes when phosphorylated at tyrosine or other residues. The chemiluminescent array images were captured following 3–5 s exposure times. The dot intensities were quantified by densitometry using ImageJ software (National Institutes of Health) and normalized with the intensity of internal positive controls. Densitometry data is reported as the average of two independent experiments ± SD.Matrigel invasion assay
Serum-starved HCT116 cells were seeded onto membranes coated with matrigel in the upper chamber of Boyden transwell chambers. The upper chamber contained serum-free medium with or without varying concentrations of platinum drug. The bottom chamber contained medium with 10% fetal bovine serum (FBS). To allow for invasion to occur, the transwell chambers were incubated at 37 °C for 48 h. The migration index was calculated as the number of cells migrating through membranes without matrigel (normalized to the untreated control). The invasion index is calculated as the number of cells invading through membranes coated with matrigel divided by the number of cells migrating through membranes without matrigel ×100 (normalized to the untreated control). The data shown are the mean number of cells that migrated or invaded (n = 4) in two separate experiments ± SD.Ex vivo rat aortic ring assay
An angiogenesis assay using rat aortic rings (1 mm vessel segments; 9 month-old Fischer rats) was performed as previously described.3,57 Briefly, aorta segments were embedded in a fibrin gel in 48-well plates (six replicates per treatment) and cultured in medium 199 containing 20% heat inactivated fetal calf serum (FCS) in the presence/absence of Triplatin (BBR3464) or TriplatinNC (10 and 100 μg mL−1). Vessel outgrowths were microscopically examined after 5 days culture for anti-angiogenic activity, growth being quantified manually as the percentage of the field (40×) around the vessel that is occupied by outgrowths, typically new vessel growth in the control wells being ∼85% by day 5. Triplatin and TriplatinNC were dissolved in water and diluted at least 1:100 in FCS supplemented medium M199. Control cultures received medium with an appropriate volume of water but without the test substance or with the anti-angiogenic control, PI-88 (Muparfostat), at 100 μg mL−1.
HS competition with PPC–DNA binding (EtBr reporter method)
Ethidium bromide (EtBr) at 5 mM in water was diluted in HEPES buffer (80 mM HEPES, 7.2 pH) and incubated with calf thymus (ct) DNA for 5 minutes at room temperature. An aliquot of 1 mM TriplatinNC in water was diluted in HEPES buffer, and incubated for 1 hour to reduce fluorescence to approximately 25%. The concentrations before addition of FPX were the following: EtBr 12.5 μM, ct DNA 10 μM, and TriplatinNC 12.5 μM. Finally, increasing concentrations of FPX from 5–164 μM final in buffer (data only shown for 5–15 μM) were incubated for an additional hour. All samples were read (after a total of 2 hours and 5 min) in a 96 well plate at 530/590 nm using a microplate reader (BioTek instruments). Samples were normalized to the controls (EtBr only as 0% and EtBr–DNA as 100%).HS competition with PPC–DNA binding (fluorescence polarization method)
50 μL binding reactions were carried out at room temperature in 96-well, low-binding, black plates (Greiner) using 100 nM to 25 μM of TriplatinNC in 1.25 mM NaCl, 0.125 mM HEPES pH 7.2, and 1 μM 3′ fluorescein-labeled hairpin DNA (GGGGCGACTGGTGAGTACGCCCC, MW 7705; Sigma). Readings were recorded immediately after mixing (for no competitor readings). Subsequently, 1 μL of 10 μM FPX (GlcNS6S-GlcA-GlcNS3,6S-IdoA2S-GlcNS6S-OMe, MW 1726.77) or unsulfated 5-mer (GlcA–GlcNAc–GlcA–GlcNAc–GlcA, MW 1073.87) was titrated into each reaction (final concentration of competitor is 2 × 10−6 M). Readings were recorded immediately. This was followed by 1 μL titrations of 100 μM competitor (for reactions containing 2 × 10−5 M) and 1 mM competitor (for reactions containing 2 × 10−4 M) with immediate readings after each addition. The data is reported as the average of 3 separate experiments ± SD.Acknowledgements
This work was supported by grants from NIH (RO1CA78754) to NPF and The Australian Research Council (DP150100308) to SBP, MvI and NPF and from the Australian National Health and Medical Research Council (455395) to CRP. Support from The Massey Cancer Center CA016059 is gratefully acknowledged. We thank Dr Yun Qu for assistance with NMR spectra. We thank Dr Daniel Lee for discussions on the competitive (DNA/FPX) inhibition assay. We sincerely thank Dr Robin Thomson for careful reading of the manuscript and suggestions.References
- P. Chiodelli, A. Bugatti, C. Urbinati and M. Rusnati, Molecules, 2015, 20, 6342–6388 CrossRef CAS PubMed.
- C. Pisano, I. Vlodavsky, N. Ilan and F. Zunino, Biochem. Pharmacol., 2014, 89, 12–19 CrossRef CAS PubMed.
- C. R. Parish, C. Freeman, K. J. Brown, D. J. Francis and W. B. Cowden, Cancer Res., 1999, 59, 3433–3441 CAS.
- K. Dredge, E. Hammond, P. Handley, T. Gonda, M. Smith, C. Vincent, R. Brandt, V. Ferro and I. Bytheway, Br. J. Cancer, 2011, 104, 635–642 CrossRef CAS PubMed.
- M. Guerrini, M. HricovIni and G. Torri, Curr. Pharm. Des., 2007, 13, 2045–2056 CrossRef CAS PubMed.
- H. Zhao, H. Liu, Y. Chen, X. Xin, J. Li, Y. Hou, Z. Zhang, X. Zhang, C. Xie, M. Geng and J. Ding, Cancer Res., 2006, 66, 8779–8787 CrossRef CAS PubMed.
- K. J. Brown and C. R. Parish, Biochemistry, 1994, 33, 13918–13927 CrossRef CAS PubMed.
- S. Taylor and J. Folkman, Nature, 1982, 297, 307 CrossRef CAS PubMed.
- J. B. Mangrum, B. J. Engelmann, E. J. Peterson, J. J. Ryan, S. J. Berners-Price and N. P. Farrell, Chem. Commun., 2014, 50, 4056–4058 RSC.
- N. P. Farrell, Chem. Soc. Rev., 2015, 44, 8773–8785 RSC.
- S. Komeda, T. Moulaei, K. K. Woods, M. Chikuma, N. P. Farrell and L. D. Williams, J. Am. Chem. Soc., 2006, 128, 16092–16103 CrossRef CAS PubMed.
- S. Komeda, T. Moulaei, M. Chikuma, A. Odani, R. Kipping, N. P. Farrell and L. D. Williams, Nucleic Acids Res., 2011, 39, 325–336 CrossRef CAS PubMed.
- Z. S. Juo, T. K. Chiu, P. M. Leiberman, I. Baikalov, A. J. Berk and R. E. Dickerson, J. Mol. Biol., 1996, 261, 239–254 CrossRef CAS PubMed.
- S. M. Fuchs and R. T. Raines, Cell. Mol. Life Sci., 2006, 63, 1819–1822 CrossRef CAS PubMed.
- H. Silva, F. Frézard, E. J. Peterson, P. Kabolizadeh, J. J. Ryan and N. P. Farrell, Mol. Pharmaceutics, 2012, 9, 1795–1802 CrossRef CAS PubMed.
- R. A. Ruhayel, B. Corry, C. Braun, D. S. Thomas, S. J. Berners-Price and N. P. Farrell, Inorg. Chem., 2010, 49, 10815–10819 CrossRef CAS PubMed.
- A. Hegmans, S. J. Berners-Price, M. S. Davies, D. S. Thomas, A. S. Humphreys and N. Farrell, J. Am. Chem. Soc., 2004, 126, 2166–2180 CrossRef CAS PubMed.
- Y. Qu, R. Kipping and N. P. Farrell, Dalton Trans., 2015, 44, 3563–3572 RSC.
- B. Mulloy and M. J. Forster, Glycobiology, 2000, 10, 1147–1156 CrossRef CAS PubMed.
- M. Hricovini, M. Guerrini, A. Bisio, G. Torri, M. Petitou and B. Casu, Biochem. J., 2001, 359, 265–272 CrossRef CAS PubMed.
- M. Petitou and C. A. van Boeckel, Angew. Chem., Int. Ed., 2004, 43, 3118–3133 CrossRef CAS PubMed.
- J. C. Wilson, A. E. Laloo, S. Singh and V. Ferro, Biochem. Biophys. Res. Commun., 2014, 443, 185–188 CrossRef CAS PubMed.
- L. Bohlmann, C. Chang, I. Beacham and M. von Itzstein, ChemBioChem, 2015, 16, 1205–1211 CrossRef CAS PubMed.
- E. Hammond, C. P. Li and V. Ferro, Anal. Biochem., 2010, 396, 112–116 CrossRef CAS PubMed.
- G. Torri, B. Casu, G. Gatti, M. Petitou, J. Choay, J. Jacquinet and P. Sinay, Biochem. Biophys. Res. Commun., 1985, 128, 134–140 CrossRef CAS PubMed.
- J. Zhang, D. S. Thomas, M. S. Davies, S. J. Berners-Price and N. Farrell, JBIC, J. Biol. Inorg. Chem., 2005, 10, 652–666 CrossRef CAS PubMed.
- W. Jorgen, H. Kaisa and M. H. Ellen, Biochem. J., 2011, 437, 199–213 CrossRef PubMed.
- J. H. Dey, F. Bianchi, J. Voshol, D. Bonenfant, E. J. Oakeley and N. E. Hynes, Cancer Res., 2010, 70, 4151–4162 CrossRef CAS PubMed.
- M. Koziczak, T. Holbro and N. E. Hynes, Oncogene, 2004, 23, 3501–3508 CrossRef CAS PubMed.
- S. Faham, R. E. Hileman, J. R. Fromm, R. J. Linhardt and D. C. Rees, Science, 1996, 271, 1116–1120 CAS.
- E. H. Knelson, J. C. Nee and G. C. Blobe, Trends Biochem. Sci., 2014, 39, 277–288 CrossRef CAS PubMed.
- F. Göke, A. Göke, A. Von Mässenhausen, A. Franzen, R. Sharma, R. Kirsten, D. Böhm, G. Kristiansen, A. Stenzinger, M. Wynes, F. R. Hirsch, W. Weichert, L. Heasley, R. Beuttner and S. Perner, Digestion, 2013, 88, 172–181 Search PubMed.
- M. Ayers, J. Fargnoli, A. Lewin, Q. Wu and J. S. Platero, Cancer Res., 2007, 67, 6899–6906 CrossRef CAS PubMed.
- M. D. Hulett, C. Freeman, B. J. Hamdorf, R. T. Baker, M. J. Harris and C. R. Parish, Nat. Med., 1999, 5, 803–809 CrossRef CAS PubMed.
- E. J. Peterson, V. R. Menon, L. Gatti, R. Kipping, D. Dewasinghe, P. Perego, L. F. Povirk and N. P. Farrell, Mol. Pharmaceutics, 2015, 12, 287–297 CrossRef CAS PubMed.
- J. Langdown, K. J. Belzar, W. J. Savory, T. P. Baglin and J. A. Huntington, J. Mol. Biol., 2009, 386, 1278–1289 CrossRef CAS PubMed.
- M. Hricovini, M. Guerrini, A. Bisio, G. Torri, M. Petitou and B. Casu, Biochem. J., 2001, 359, 265–272 CrossRef CAS PubMed.
- U. Desai, R. Swanson, S. C. Bock, I. Bjork and S. T. Olson, J. Biol. Chem., 2000, 275, 18976–18984 CrossRef CAS PubMed.
- V. Arocas, S. C. Bock, S. T. Olson and I. Björk, Biochemistry, 1999, 38, 10196–10204 CrossRef CAS PubMed.
- A. Prisecaru, Z. Molphy, R. G. Kipping, E. J. Peterson, Y. Qu, A. Kellett and N. P. Farrell, Nucleic Acids Res., 2014, 42, 13474–13487 CrossRef CAS PubMed.
- M. L. E. Galvis, J. Jia, X. Zhang, N. Jastrebova, D. Spillmann, E. Gottfridsson, T. H. van Kuppevelt, E. Zcharia, I. Vlodavsky and U. Lindahl, Nat. Chem. Biol., 2007, 3, 773–778 CrossRef PubMed.
- A. L. Harris, J. J. Ryan and N. Farrell, Mol. Pharmacol., 2005, 69, 666–672 CrossRef PubMed.
- B. T. Benedetti, E. J. Peterson, P. Kabolizadeh, A. Martínez, R. Kipping and N. P. Farrell, Mol. Pharmaceutics, 2011, 8, 940–948 CrossRef CAS PubMed.
- J. Malina, N. P. Farrell and V. Brabec, Angew. Chem., Int. Ed., 2014, 53, 12812–12816 CrossRef CAS PubMed.
- Y. Seo, M. R. Schenauer and J. A. Leary, Int. J. Mass Spectrom., 2011, 303, 191–198 CrossRef CAS PubMed.
- S. E. Guimond, T. R. Rudd, M. A. Skidmore, A. Ori, D. Gaudesi, C. Cosentino, M. Guerrini, R. Edge, D. Collison and E. McInnes, Biochemistry, 2009, 48, 4772–4779 CrossRef CAS PubMed.
- F. Zhang, X. Liang, J. M. Beaudet, Y. Lee and R. J. Linhardt, J. Biomed. Mater. Res., 2014, 1, 6000101 Search PubMed.
- J. Yellol, S. A. Pérez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak, A. Espinosa and J. Ruiz, J. Med. Chem., 2015, 58, 7310–7327 CrossRef CAS PubMed.
- J. K. Muenzner, T. Rehm, B. Biersack, A. Casini, I. A. M. De Graaf, P. Worawutputtapong, A. Noor, R. Kempe, V. Brabec, J. Kasparkova and R. Schobert, J. Med. Chem., 2015, 58, 6283–6292 CrossRef CAS PubMed.
- A. Bergamo and G. Sava, Chem. Soc. Rev., 2015, 44, 8818–8835 RSC.
- L. E. Wedlock, M. R. Kilburn, R. Liu, J. A. Shaw, S. J. Berners-Price and N. P. Farrell, Chem. Commun., 2013, 49, 6944–6946 RSC.
- E. Katsuta, S. Demasi, S. Katner, H. Aoki, E. Peterson, N. P. Farrell and K. Takabe, Proc. AACR, New Orleans, LA, 2016, Abstract #3064, .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. E. Petersson and H. Nakatsuji, Gaussian 09, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 1–18 Search PubMed.
- S. F. Boys and F. D. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. J. Stewart, Stewart Computational Chemistry, 2012 Search PubMed.
- K. J. Brown, S. F. Maynes, A. Bezos, D. J. Maguire, M. D. Ford and C. R. Parish, Lab. Invest., 1996, 75, 539–555 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed mapping of platinum complex–oligosaccharide interactions, NMR spectra, HUVEC cytotoxicity data and wound healing in HCT116 cells. See DOI: 10.1039/c6sc02515c |
| This journal is © The Royal Society of Chemistry 2017 |