
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heptamethylindenyl (Ind*) enables diastereoselective benzamidation of cyclopropenes via Rh(III)-catalyzed C–H activation†
Natthawat
Semakul
a,
Kelvin E.
Jackson
b,
Robert S.
Paton
 *b and
Tomislav
Rovis‡
*a
*b and
Tomislav
Rovis‡
*a
aDepartment of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA
bChemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: robert.paton@chem.ox.ac.uk
First published on 23rd September 2016
Abstract
The diastereoselective coupling of O-substituted arylhydroxamates and cyclopropenes mediated by Rh(III) catalysis was successfully developed. Through ligand development, the diastereoselectivity of this reaction was improved using a heptamethylindenyl (Ind*) ligand, which has been rationalized using quantum chemical calculations. In addition, the nature of the O-substituted ester of benzhydroxamic acid proved important for high diastereoselectivity. This transformation tolerates a variety of benzamides and cyclopropenes that furnish cyclopropa[c]dihydroisoquinolones with high diastereocontrol, which could then be easily transformed into synthetically useful building blocks for pharmaceuticals and bio-active molecules.
Introduction
Rh(III)-catalyzed C–H bond functionalization strategies have emerged as a powerful synthetic tool.1 The methodology allows for the functionalization of simple organic molecules and expedient synthesis of nitrogen-containing heterocycles from readily available precursors. Cyclopropenes constitute a class of building blocks with a special reactivity due to their high ring strain energy (54 kcal mol−1). In this context, a handful of reactions utilizing transition metals has been developed for the stereoselective functionalization of cyclopropenes.2 Under the aegis of Rh(III) catalysis, Wang and coworkers have shown that cyclopropenes participate in a Rh(III) catalyzed reaction with N-phenoxyacetamide to give 2H-chromenes (Fig. 1A, eqn (1)).3 Our group reported the Rh(III)-mediated coupling of O-pivaloyl benzhydroxamate 1a with 3,3-diester substituted cyclopropene 2a to afford 4-substituted isoquinolone 3a after ring opening of the three-membered ring (Fig. 1A, eqn (2)).4 During this study, when using the methyl 1-phenylcycloprop-2-ene-1-carboxylate 2b as a substrate, the Cp*Rh(III) catalyst gives the [4.1.0] bicyclic product 3b in low diastereoselectivity (1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, Fig. 1A, eqn (3)). We believe the lack of diastereoselectivity stems from the poor facial selectivity during coordination of the cyclopropene and the migratory insertion step of cyclopropene unit. We reasoned that creating anisotropy around the cyclopentadienyl ligand of the rhodium metal center could solve this selectivity issue.
:1 dr, Fig. 1A, eqn (3)). We believe the lack of diastereoselectivity stems from the poor facial selectivity during coordination of the cyclopropene and the migratory insertion step of cyclopropene unit. We reasoned that creating anisotropy around the cyclopentadienyl ligand of the rhodium metal center could solve this selectivity issue.
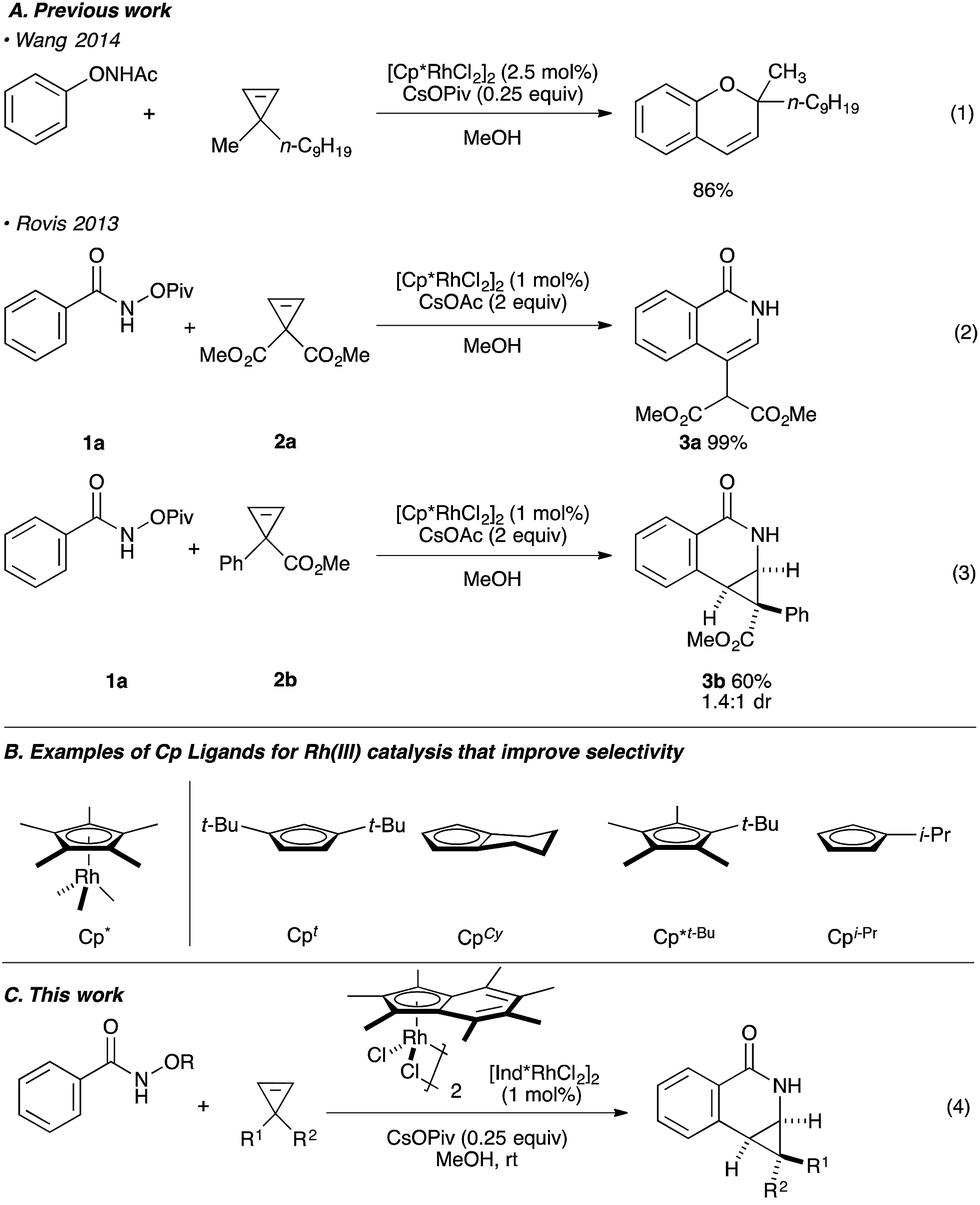 | ||
| Fig. 1 (A) The use of cyclopropenes in Rh(III) catalysis. (B) Examples of Cp ligands that improve selectivity. (C) This work. | ||
Our group6–12 and others5,13 have developed several Rh(III)-catalyzed transformations where the nature of the Cp ligand drastically impacts the reactivity5–7 and selectivity8–13 of the reaction (Fig. 1B). For example, the sterically bulky di-tert-butylcyclopentadienyl (Cpt) ligand has been shown to improve the regiochemistry of alkyne and alkene insertion events in the synthesis of pyridones,8 pyridines9 and dihydroisoquinolones.10 Interestingly, Cramer and coworkers found a divergent regioselective synthesis of 3- and 4-substituted dihydroisoquinolone from O-Boc arylhydroxamate and styrene when using cyclohexane-fused cyclopentadienyl (CpCy) and pentamethylcyclopentadienyl (Cp*) ligands.13 Recently, our group disclosed a cyclopropanation reaction with the coupling of N-enoxyphthalimides and alkenes. Monoisopropylcyclopentadienyl (CpiPr) outperforms the more common Cp* ligand, furnishing the trans-cyclopropane in high diastereoselectivity.11 Alternatively, a divergent carboamination path was identified when using a hindered tert-butyltetramethylcyclopentadienyl (Cp*t-Bu) ligand delivering the acyclic adduct with high chemoselectivity.12 Motivated by these results, we believed ligand design could provide a solution to the inherent selectivity issues encountered for the coupling of benzamide and 3,3-disubstituted cyclopropenes (Fig. 1C, eqn (4)).
Results and discussion
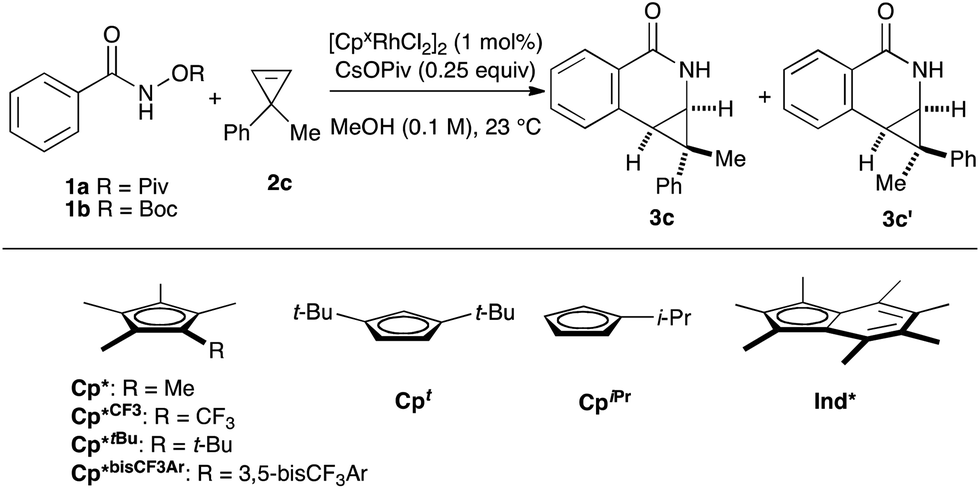
We began our investigation by employing O-pivaloyl benzhydroxamate ester 1a and cyclopropene 2c as model substrates for the optimization of the catalytic process (Table 1). [Cp*RhCl2]2 provides the desired product in a moderate yield and diastereoselectivity (5.8:1 dr, entry 1). The relative stereochemistry of the major diastereoisomer of 3c was confirmed by NOESY (see ESI†). By modulating the steric and electronic properties of the Cp ligand, we have shown that the diastereoselectivity of the reaction is considerably affected. Sterically hindered di-tert-butylcyclopentadienyl8–10 (Cpt) and the electron-poor trifluoromethyl tetramethylcyclopentadienyl7 (Cp*CF3) ligands give only modest diastereoselectivity (entries 2 and 3). The monoisopropylcyclopentadienyl ligand11 (CpiPr) gave the desired product in a good yield albeit with no diastereocontrol (entry 4). 3,5-Bis(trifluoromethyl)aryl tetramethylcyclopentadienyl6 (Cp*bisCF3Ar) provides the desired product in good yield with slightly improved diastereoselectivity (7.0:1 dr, entry 5). Good level of diastereocontrol (8.8:1 dr) is achieved when tert-butyl tetramethylcyclopentadienyl12 (Cp*tBu) ligand was employed (entry 6). Gratifyingly, heptamethylindenyl ligand14 (Ind*) provides high reactivity and diastereoselectivity with 90% yield and 15.2:1 dr (entry 7). To demonstrate the scalability of the transformation, the reaction was performed in 2 mmol scale of substrate 1a, which gives the expected product with comparable yield and selectivity. The catalyst loading can be lowered to 0.5 mol% [Ind*RhCl2]2 without affecting reactivity (entry 8). We then examined the nature of the directing group. It was found that using O-Boc benzhydroxamate ester 1b as a substrate gave excellent diastereoselectivity (>20:1 dr) but with slightly lower yield (entry 9), presumably due to a competitive Lossen rearrangement under the basic conditions.15
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Cpx | Yieldb | dr (3c:3c′)b |
| a Reaction conditions: 1a or 1b (0.1 mmol), 2c (0.11 mmol), Rh catalyst (1 mol%), CsOPiv (0.25 equiv.) in MeOH (0.1 M) at 23 °C for 18 h. b The yield and diastereoselectivity were measured from the 1H-NMR analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yield. d Catalyst loading of 0.5 mol% on 1 mmol scale. | ||||
| 1 | 1a | Cp* | 63 | 5.8:1 |
| 2 | 1a | Cpt | 82 | 5.0:1 |
| 3 | 1a | Cp*CF3 | 75 | 5.3:1 |
| 4 | 1a | CpiPr | 73 | 1.1:1 |
| 5 | 1a | Cp*bisCF3Ar | 80 | 7.0:1 |
| 6 | 1a | Cp*tBu | 64 | 8.8:1 |
| 7 | 1a | Ind* | 90c | 15.2:1 |
| 8d | 1a | Ind* | 85c | 15:1 |
| 9 | 1b | Ind* | 69c | >20:1 |
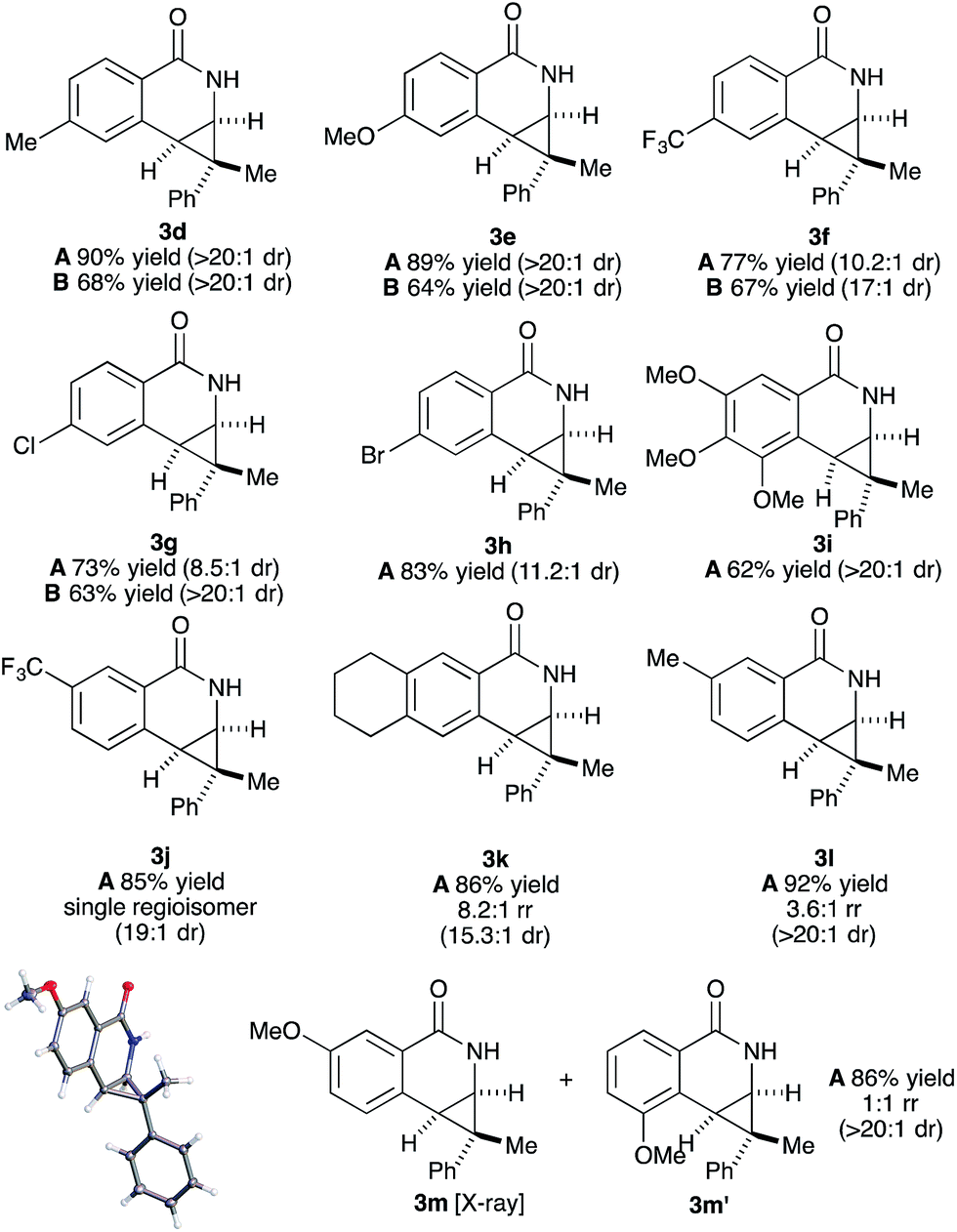
Both benzamide directing groups, O-Piv 1a (condition A) and O-Boc 1b (condition B), were used for studying the scope of the transformation (Table 2). Substituents at the para position of the benzamide are tolerated in the reaction (Table 2, 3d–3h). The O-Piv directing group with an electron rich para-methoxy substituent (OMe) gave excellent diastereoselectivity (>20:1 dr, product 3e) compared to electron deficient substituents (∼10:1 dr, products 3f, 3g and 3h). The electron-rich benzamide derived from gallic acid furnishes the desired product with good yield and excellent diastereoselectivity (>20:1 dr, product 3i). The O-Boc directing group gives the products in good to excellent diastereoselectivity (3d–3g). Of interest are halogen substituents at the para positions (Cl and Br) which provide a functional group handle for further chemical modification. The ortho-methyl arylbenzhydroxamate substrate retards the transformation presumably due to steric hindrance. Substituents at the meta position on the arylhydroxamates can potentially deliver two regioisomeric products arising from the selectivity of C–H activation. meta-Trifluoromethyl arylhydroxamate exclusively provides the 6-substituted product (3j) in good yield and diastereoselectivity. Tetrahydronaphthalene-derived arylhydroxamate underwent the transformation with good regioselective C–H activation at less hindered position (8.6:1 ratio) to give the desired product (3k) in good yield and high diastereoselectivity. However, meta-methyl arylhydroxamate gave ∼3.6:1 regioisomeric ratio of C–H activation in good yield and diastereoselectivity (3l). meta-Methoxy arylhydroxamate provided 1:1 mixture of regioisomeric products (3m and 3m′) in good diastereocontrol, presumably a consequence of a combination of steric effects and kinetic acidity issues. In addition, X-ray structure of 3m ambiguously confirmed the relative stereochemistry of trans-diastereomer.
|
|
|---|
| a Conditions: 1a (for A) or 1b (for B) (0.1 mmol), 2c (0.11 mmol), Rh catalyst (1 mol%), CsOPiv (0.25 equiv.) in MeOH (0.1 M) at 23 °C for 18 h. b Isolated yield of the major diastereomer after silica gel column chromatography. c Diastereoselectivity was measured by 1H-NMR spectra of the unpurified material. |
|
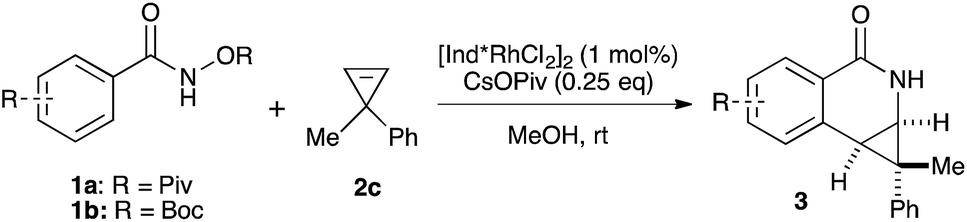
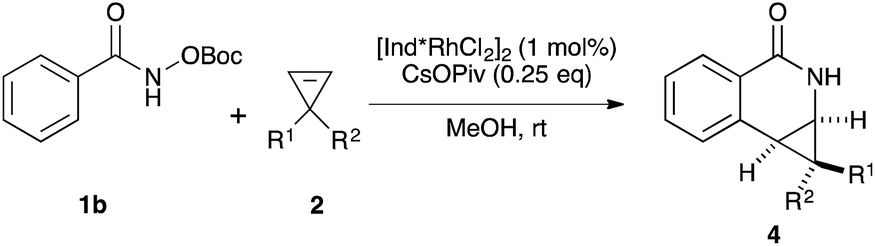
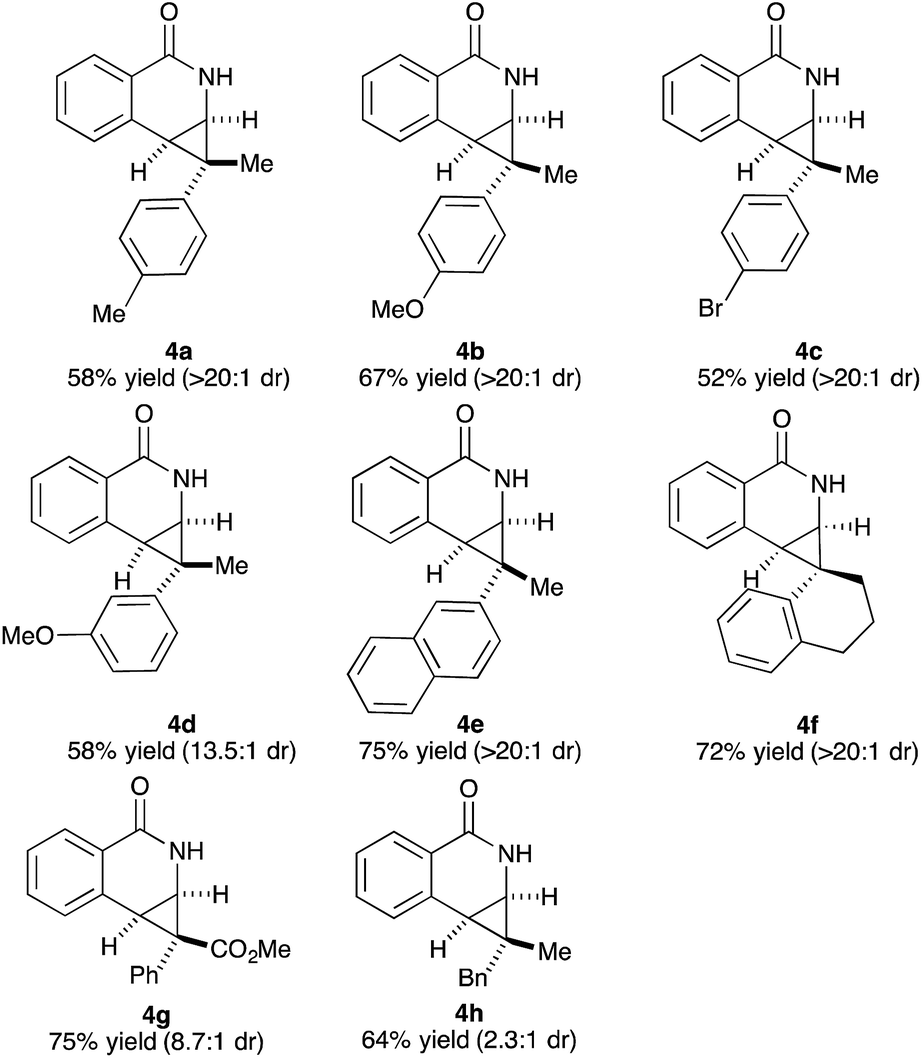
Variations of the cyclopropene coupling partner were explored for the transformation using the O-Boc benzhydroxamate 1b. Cyclopropenes bearing substituents at the para position gave the desired products in moderate yields and excellent diastereoselectivity regardless of the electronic nature of substituents (Table 3, 4a, 4b and 4c). Cyclopropene with a meta-methoxy group undergoes the transformation with slightly lower diastereoselectivity relative to the para-methoxy group (Table 3, 4d). A naphthalene-substituted cyclopropene 2e and a spiro-tetralin containing substrate 2f each furnish the desired products 4e and 4f in good yield and excellent diastereoselectivity. In our previous studies,4 we found that methyl 1-phenylcycloprop-2-ene-1-carboxylate 2b reacts with benzamide 1b and gives the desired product with low diastereoselectivity (1.4:1 dr) using [Cp*RhCl2]2 as the precatalyst. With the [Ind*RhCl2]2 ligand, we were pleased to find that cyclopropene 2b afforded the dihydroisoquinolone 4g with improved diastereoselectivity (8.7:1 dr). The relative stereochemistry of the major diastereomer of 4g was confirmed by NOESY (see ESI†). The observed major diastereomer can be rationalized by the size of the substituents on the cyclopropane ring. Thus, the phenyl group is larger than the carboxylate ester (A-values for Ph- and –CO2Me are 3.0 and 1.3, respectively) leading to higher diastereoselectivity observed in these reactions. The amidoarylation with benzyl substituted cyclopropene affords the desired product 4h in good yield but with lower diastereoselectivity. This observation can be explained by the steric differences of phenyl vs. benzyl groups (A-values for Ph and Bn are 3.0 and 1.75, respectively). 2,3,3-Trisubstituted cyclopropenes did not participate in the Rh(III)-catalyzed coupling with benzamides.
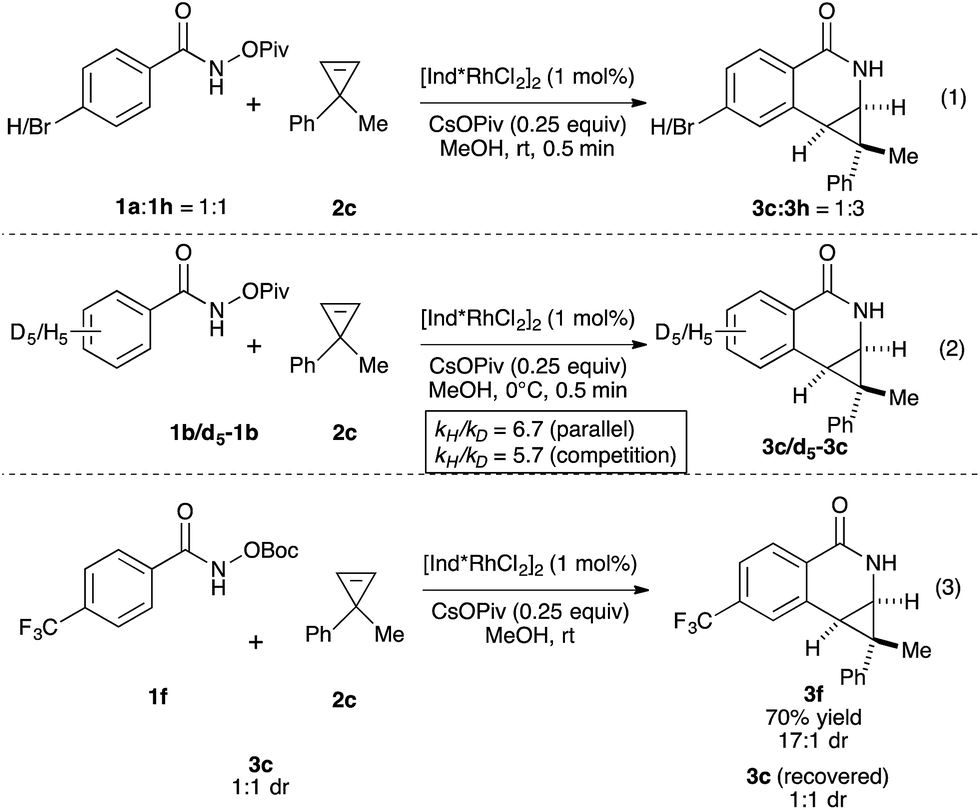
We then investigated the mechanism of the transformation. The reversibility of C–H activation was first examined. Trace deuterium incorporation (<5%) was observed when the reaction was run in CD3OD, suggesting the C–H activation is largely irreversible (see ESI†). The competitive reaction between p-bromobenzamide (1h) and unsubstituted benzamide (1a) was conducted to probe the electronic preference of reaction (Scheme 1, eqn (1)). The product formation favors an electron deficient substrate in a 3:1 ratio. Kinetic isotope studies revealed KIE values of 6.7 and 5.7 for the parallel and competition experiments, respectively (Scheme 1, eqn (2)). These studies together suggest that the C–H activation occurs via concerted metallation-deprotonation (CMD) mechanism and is the turnover-limiting step, as seen in several previous examples of C–H activation with Rh(III).16,17 To determine if epimerization of the product occurs under the reaction conditions, we independently prepared product 3c (1:1 dr) and resubjected it to the reaction conditions of benzamide 1f and cyclopropene 2c. After full conversion to 3f (70% yield, 17:1 dr), we did not observe any change of the dr of 3c, indicating the products are not epimerized under the reaction conditions.
 | ||
| Scheme 1 Mechanistic experiments. | ||
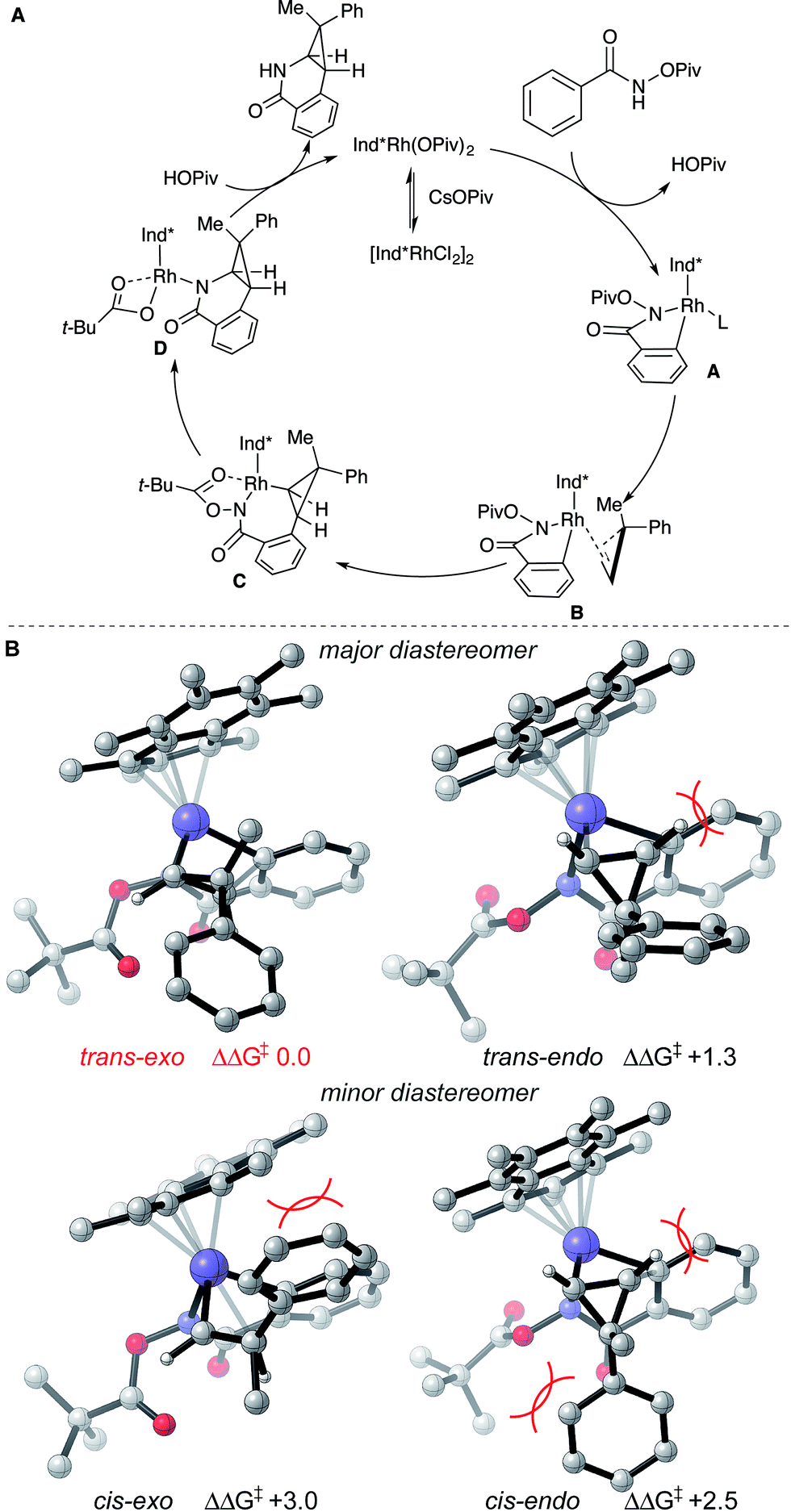
Based on literature precedent17 and our mechanistic studies, the mechanism of the transformation is proposed in Scheme 2A. The Ind*Rh(OPiv)2 species is generated in situ by an anion exchange of [Ind*RhCl2]2 and CsOPiv. The amide directed C–H activation occurs via a CMD mechanism to give the five-membered rhodacycle intermediate A, which then coordinates the cyclopropene giving intermediate B.
 | ||
| Scheme 2 (A) Proposed reaction mechanism and (B) stereochemical model for diastereoselectivity. Gibbs energies in kcal mol−1. | ||
To understand diastereoselectivity and the effect of the Ind* ligand we performed density functional theory (DFT) calculations.18 Transition structures (TSs) were optimized at the TPSS/def2-TZVP level of theory, which was the most accurate of several functionals tested (see ESI†), for the reaction of benzamide 1a with cyclopropene 2c using Cp* and Ind* ligands. Firstly, we confirmed that the product diastereoselectivity arises from the facial selectivity of the coordination of the cyclopropene and subsequent migratory insertion step (Scheme 2B). Our calculations indicate a facile migratory insertion step (barriers of 8.0 and 9.7 kcal mol−1) which is substantially exergonic, so that the barriers in the reverse direction are prohibitively large (25.7 kcal mol−1) given the reaction conditions. We predict this step will occur irreversibly,19 thus determining the diastereoselectivity. With both Cp* and Ind* ligands, we found that the insertion step can proceed via four distinct TSs. For both diastereofaces of the cyclopropene, two conformers exist in which the cyclpropenyl gem-disubstituted carbon can be oriented towards (endo) or away (exo) from the benzamide. In terms of the nomenclature adopted the trans-diastereomer is the major product experimentally. All four possibilities are shown for the Ind* ligand in Scheme 2B (with Cp* structures in the ESI†).
The most favorable TS (trans–exo) agrees with the observed sense of diastereoselectivity and the computations also quantitatively reproduce the increase in selectivity of Ind* vs. Cp* ligands (cis–trans ΔΔG‡ increasing from 1.8 to 2.5 kcal mol−1; note that the favored cis TS changes from cis–exo for Cp* to cis–endo for Ind*). In the favored TS the cyclopropene substrate is oriented with the methyl group towards the ligand. The alternative approach (trans–endo) is less favorable, suffering from a more severe H…H clash (2.16 Å) about the incipient C–C bond. The Piv group is also oriented towards the ligand in this less favorable TS. The minor diastereomer results from trying to orient the larger phenyl group toward the Ind* ligand (cis–exo TS) or toward the substrate and directing group (cis–endo TS), causing unfavorable steric interactions. These structures show the synergistic effect of steric interactions involving both the Ind* ligand and directing group on the facial selectivity. Migratory insertion of cyclopropene gives intermediate C. Reductive elimination (C–N bond formation) occurs to generate a Rh(I) species.19 The saturated coordination of acyl directing group to Rh(III) of intermediate C is important for the reductive elimination step since O-methyl benzhydroxamate is not reactive for the transformation (see ESI†).
The prevalence of nitrogen-containing heterocycles in pharmaceuticals led us to investigate the derivatization of the dihydroisoquinolones bearing [4.1.0] bicycles.20 For example, the chloro- and O-triflate substituted dihydroisoquinolines, which are versatile functional group handles for further cross-coupling reactions could be easily prepared from the dihydroisoquinolone products in good yields, allowing for easy incorporation of these bicycles into pharmaceuticals or bio-active molecules (Scheme 3).
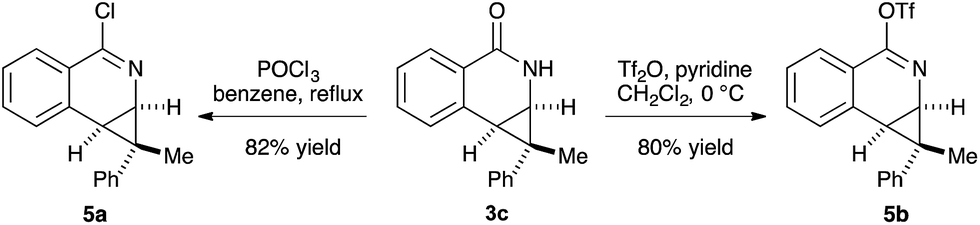 | ||
| Scheme 3 Derivatizations of product. | ||
Conclusions
In summary, we have developed a heptamethylindenyl (Ind*) ligand that enables high diastereoselectivity for cyclopropene insertion in the Rh(III)-catalyzed synthesis of cyclopropa[c]dihydroisoquinolone. The steric interaction of the ligand on rhodium and the ester substitution of O-substituted benzhydroxamate work cooperatively to improve the diastereoselectivity of cyclopropene insertion. Mechanistically, the C–H activation proceeds via a concerted metallation-deprotonation pathway and is the turnover-limiting step. This methodology is useful for the rapid synthesis of nitrogen-containing heterocycles with a [4.1.0] motif and their derivatives.Acknowledgements
We thank the NIGMS for generous support of this research (TR – GM80442), and Johnson Matthey for a generous loan of Rh salts. We thank Dr Tiffany Piou for preparing a number of the ligands examined in this study and Dr Brian Newell for assistance with X-ray crystallography. NS thanks Royal Thai Government for a DPST graduate scholarship. RSP acknowledges the use of the EPSRC UK National Service for Computational Chemistry Software (CHEM773) and funding from SCG.Notes and references
- (a) For the review on Rh(III) catalyzed C–H bond functionalization, see: D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (b) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (c) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31–41 CAS; (d) G. Y. Song, F. Wang and X. W. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (e) G. Y. Song and X. W. Li, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef CAS PubMed; (f) C. Zhu, R. Wang and J. R. Falck, Chem.–Asian. J., 2012, 7, 1502–1514 CrossRef CAS PubMed.
- For the review: (a) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed; Selected recent works: (b) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2002, 124, 11566–11567 CrossRef CAS PubMed; (c) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2003, 125, 7198–7199 CrossRef CAS PubMed; (d) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2004, 126, 3688–3689 CrossRef CAS PubMed; (e) D. H. T. Phan, K. G. M. Kou and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16354–16355 CrossRef CAS PubMed; (f) B. Tian, Q. Liu, X. F. Tong, P. Tian and G. Q. Lin, Org. Chem. Front., 2014, 1, 1116–1122 RSC; (g) A. Parra, L. Amenos, M. Guisan-Ceinos, A. Lopez, J. L. G. Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833–15836 CrossRef CAS PubMed; (h) D. S. Muller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414–15417 CrossRef CAS PubMed.
- H. Zhang, K. Wang, B. Wang, H. Yi, F. D. Hu, C. K. Li, Y. Zhang and J. B. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234–13238 CrossRef CAS PubMed.
- T. K. Hyster and T. Rovis, Synlett, 2013, 24, 1842–1844 CrossRef CAS PubMed.
- (a) M. Fukui, Y. Hoshino, T. Satoh, M. Miura and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1638–1644 CrossRef CAS; (b) Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577–1585 CrossRef CAS; (c) Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2011, 50, 10917–10921 CrossRef CAS PubMed.
- T. A. Davis, C. Q. Wang and T. Rovis, Synlett, 2015, 26, 1520–1524 CrossRef CAS PubMed.
- (a) F. Romanov-Michailidis, K. F. Sedillo, J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2015, 137, 8892–8895 CrossRef CAS PubMed; (b) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735–2738 CrossRef CAS PubMed.
- T. K. Hyster and T. Rovis, Chem. Sci., 2011, 2, 1606–1610 RSC.
- T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846–11848 RSC.
- T. K. Hyster, D. M. Dalton and T. Rovis, Chem. Sci., 2015, 6, 254–258 RSC.
- T. Piou and T. Rovis, J. Am. Chem. Soc., 2014, 136, 11292–11295 CrossRef CAS PubMed.
- T. Piou and T. Rovis, Nature, 2015, 527, 86–90 CrossRef CAS PubMed.
- M. D. Wodrich, B. H. Ye, J. F. Gonthier, C. Corminboeuf and N. Cramer, Chem.–Eur. J., 2014, 20, 15409–15418 CrossRef CAS PubMed.
- A. K. Kakkar, G. Stringer, N. J. Taylor and T. B. Marder, Can. J. Chem., 1995, 73, 981–988 CrossRef CAS.
- N. J. Webb, S. P. Marsden and S. A. Raw, Org. Lett., 2014, 16, 4718–4721 CrossRef CAS PubMed.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- (a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS PubMed; (b) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed; (c) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed; (d) W. Guo and Y. Z. Xia, J. Org. Chem., 2015, 80, 8113–8121 CrossRef CAS PubMed.
- M. J. Frisch, et al., Calculations were performed with Gaussian 09 rev. D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed. Computational details and references are given in full in the ESI.†.
- This is consistent with several DFT studies published on Rh(III) catalyzed reactions. See: (a) L. Xu, Q. Zhu, G. Huang, B. Cheng and Y. J. Xia, Org. Chem., 2012, 77, 3017–3024 CrossRef CAS PubMed; (b) W. Guo and Y. Z. Xia, J. Org. Chem., 2015, 80, 8113–8121 CrossRef CAS PubMed; (c) T. Zhou, W. Guo and Y. Xia, Chem.–Eur. J., 2015, 21, 9209–9218 CrossRef CAS PubMed; (d) S. R. Neufeldt, G. Jimeńez-Oseś, J. R. Huckins, O. R. Thiel and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 9843–9854 CrossRef CAS PubMed; (e) Y.-F. Yang, K. N. Houk and Y.-D. Wu, J. Am. Chem. Soc., 2016, 138, 6861–6868 CrossRef CAS PubMed.
- (a) A. Cromarty, K. E. Haque and G. R. Proctor, J. Chem. Soc. C, 1971, 3536–3540 RSC; (b) I. Lantos, D. Bhattacharjee and D. S. Eggleston, J. Org. Chem., 1986, 51, 4147–4150 CrossRef CAS; (c) C. D. Perchonock, I. Lantos, J. A. Finkelstein and K. G. Holden, J. Org. Chem., 1980, 45, 1950–1953 CrossRef CAS; (d) J. Pedroni, T. Saget, P. A. Donets and N. Cramer, Chem. Sci., 2015, 6, 5164–5171 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterization. CCDC 1472771. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02587k |
| ‡ Current Address: Department of Chemistry, Columbia University, New York, NY 10027, USA, Email: E-mail: tr2504@columbia.edu |
| This journal is © The Royal Society of Chemistry 2017 |