
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zirconocene catalyzed diastereoselective carbometalation of cyclobutenes†
Sudipta Raha
Roy
,
Hendrik
Eijsberg
,
Jeffrey
Bruffaerts
and
Ilan
Marek
*
The Mallat Family Laboratory of Organic Chemistry, Schulich Faculty of Chemistry and Lise Meitner-Minerva Center for Computational Quantum Chemistry, Technion-Israel Institute of Technology, Technion City, Haifa 32000, Israel. E-mail: chilanm@tx.technion.ac.il; Fax: +972-4-829-37-09; Tel: +972-4-829-37-09
First published on 30th August 2016
Abstract
The regio- and diastereoselective zirconocene-catalyzed carbomagnesiation of cyclobutenes is herein reported to afford configurationally stable cyclobutylmagnesium species that could subsequently react with a large variety of electrophiles to give polysubstituted cyclobutane species as a single diastereoisomer.
Introduction
In the repertoire of strategies using organometallic species that could lead to the efficient formation of two carbon–carbon bonds per chemical step, the carbometalation reaction to an unsaturated C–C bond represents a powerful strategy. The carbometalation reaction, defined as “the addition of a carbon–metal bond of an organometallic across a carbon–carbon unsaturated system leading to a new organometallic species that can be further functionalized” – is one of the most powerful approaches that have been used extensively to perform the 1,2-bis-alkylation of alkynes.1 In this context, organocopper,2 as well as zirconocene-catalyzed methylalumination,3 occupies a significant place due to its high stereoselectivity, typically controlled by the nature of the substituents on the triple bond (Scheme 1, path a for an example of carbocupration). Besides forming stereodefined polysubstituted double bonds, the carbometalation reaction of alkynes has recently been considered as a new stereodefined chemical handle to prepare reactive intermediates for the subsequent creation of more complex molecular structures possessing sp3-configurated stereocenters including quaternary carbon stereocenters (Scheme 1, path b).4 1,2-Disubstituted alkyl chains possessing sp3 stereocenters could theoretically also be obtained through the carbometalation of appropriate alkenes (Scheme 1, path c).5 However, these transformations are much more challenging than the carbometalation reactions of alkynes, since the carbometalated product is usually of similar reactivity to the starting organometallic species and an oligomerization reaction typically occurs.6 Moreover, when the reaction is performed on the α,β-disubstituted double bond, several issues, such as: (1) regio- and stereoselectivity of the addition; (2) configurational stability of the resulting sp3 organometallic species; and (3) diastereoselectivity of the reaction with electrophiles; are of major concern.7 Finally, the enantioselectivity of the addition of a carbon nucleophile across an unactivated double bond still represents a very challenging problem despite the fact that it would acquire significant utility as a method for the creation of asymmetric vicinal carbon centers (Scheme 1, path c).8 Due to the inherent difficulty to achieve an efficient carbometalation reaction across unactivated alkenes, most of the studies have focused on strained double bonds. As such, the copper-mediated carbometalation reaction of cyclopropenyl derivatives9 has been investigated in detail to provide a new route to enantio- and diastereoenriched configurationally stable cyclopropyl metal species (Scheme 1, path d).10 However, all attempts to extend the concept of carbometalation to less-strained compounds such as cyclobutenes failed, most probably due to the lower energy release during the addition step.11 None of the copper-catalyzed carbomagnesiation, copper-catalyzed carbozincation, carbocupration with organocopper or organocuprate reactions in Et2O or THF could lead to the desired addition of the organometallic species across a double bond embedded in a 4-membered ring (Scheme 1, path e).12 Only Tortosa and coworkers recently reported the highly enantioselective desymmetrization of meso-cyclobutene through the copper-catalyzed borylation reaction.13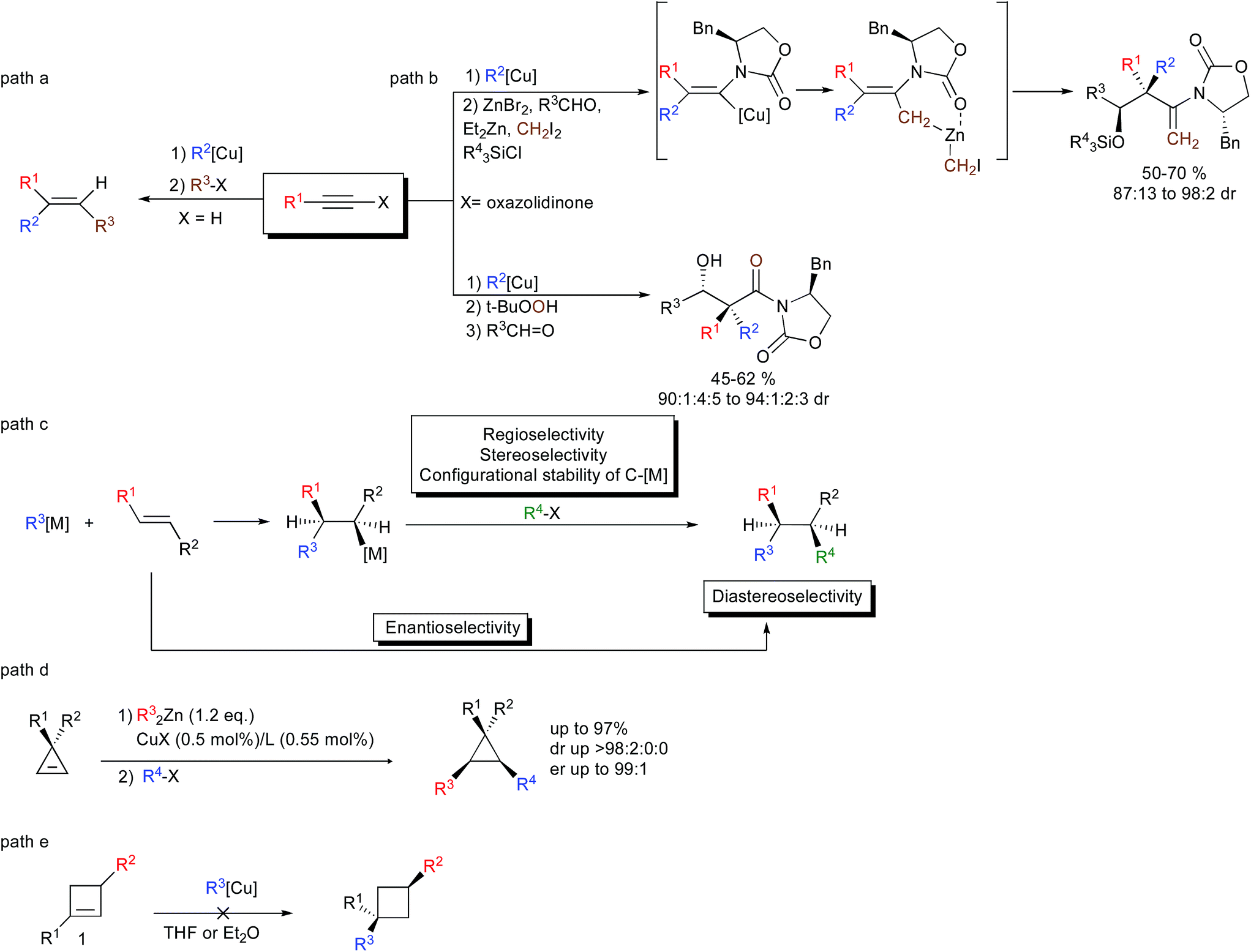 | ||
| Scheme 1 Carbometalation reactions. | ||
As stereodefined cyclobutyl metal species en route to polysubstituted cyclobutane derivatives still represent an important building block in the field of small ring chemistry,14 we therefore decided to pursuit our efforts to functionalize cyclobutene species into polysubstituted metalated cyclobutanes through carbometalation reaction, and more particularly through the Dzhemilev reaction. It should be emphasized that all starting cyclobutenes 1 were prepared by a rhodium-catalyzed intermolecular [2 + 2] cycloaddition of terminal alkynes with electron-deficient alkenes.15
Results and discussion
In this context, and as alluded previously, we were particularly interested in the possibility to reach our goal through the diastereoselective zirconocene-catalyzed carbomagnesiation reaction (Dzhemilev reaction) as described in Scheme 2.16 | ||
| Scheme 2 Proposed zirconocene-catalyzed diastereoselective ethylmagnesiation reaction of cyclobutene. | ||
In this transformation, the addition of ethylmagnesium bromide to a catalytic amount of dichlorobis(η5-cyclopentadienyl)zirconium(IV) [Cp2ZrCl2] should provide the zirconacyclopropane I that would react in situ with the double bond of the cyclobutene 1 to form either the addition product II or III. Each of these two possible regioisomers could be present as potentially two diastereoisomers (IIanti versusIIsyn and IIIanti versusIIIsyn). So, not only the regioselectivity of the zirconocene-catalyzed carbomagnesiation of substituted cyclobutene 1 should be controlled (IIversusIII) but also the diastereoselectivity of the reaction (syn versus anti). Assuming that from the four possible regio- and diastereoisomers only the isomer IIanti will be produced, the reaction of ethylmagnesium bromide with the zirconacyclopentane IIanti would then provide the ate-complex IV that may either lead to the cyclobutyl magnesium V or cyclobutyl zirconocene species VI after transmetalation. The selectivity of the transmetalation is critical as the carbon attached to the zirconocene will then be subsequently reduced to regenerate the catalytic zirconacyclopropane species I (see for example the preparation of VIIIvia the reduction depicted in VII). If one assumes that the reaction would only provide V, then the cyclobutyl magnesium derivative VIII may be expected, whereas if the transmetalation occurs to provide VI, the cyclobutyl ethylmagnesium species IX is anticipated. Finally, if one still assumes that the catalytic cycle would only provide VIII, the configurational stability of this cyclobutyl magnesium species as well as its reactivity towards electrophiles needs to be investigated in detail.
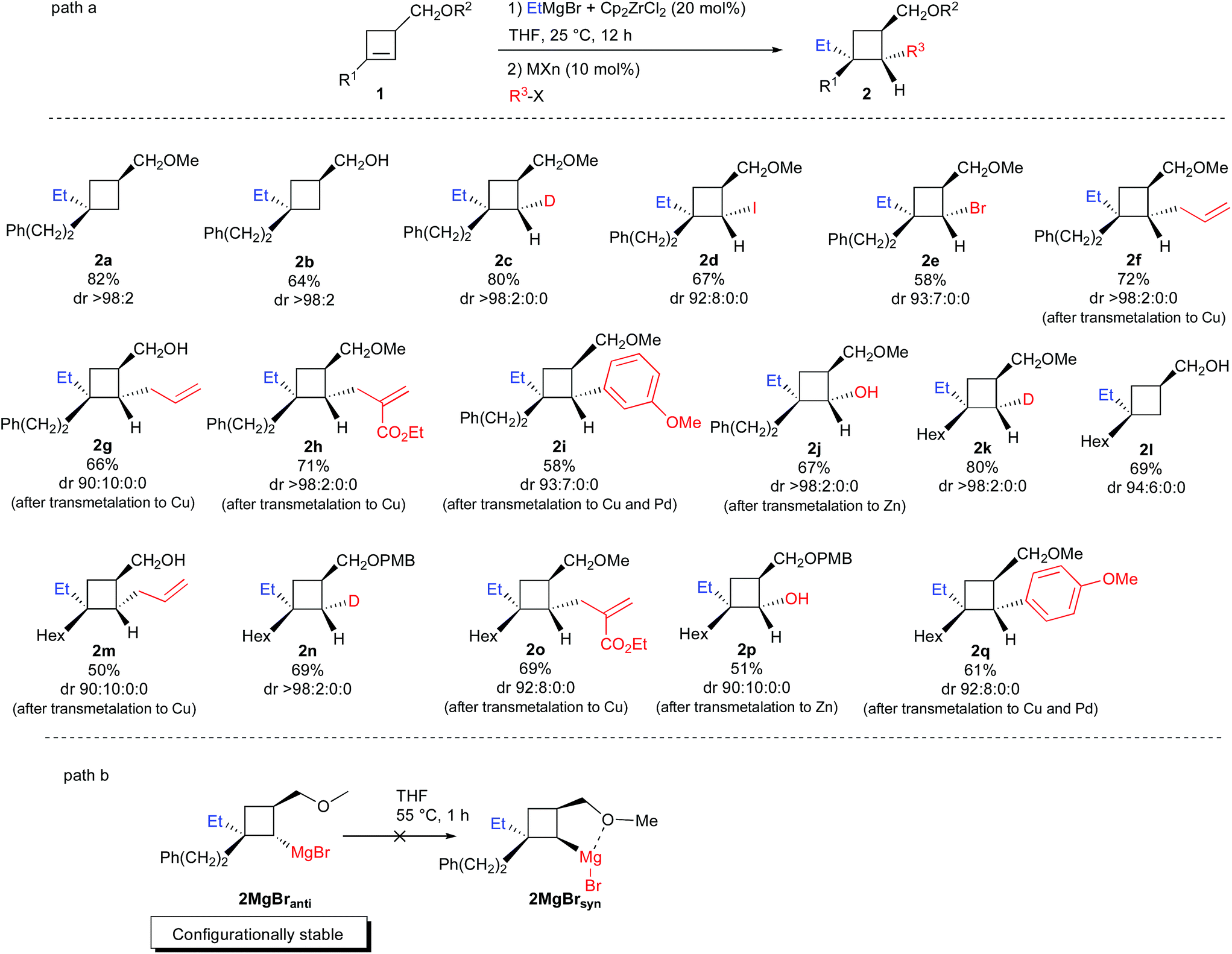
Therefore, the Dzhemilev ethylmagnesiation of cyclobutene catalyzed by dichlorobis(η5-cyclopentadienyl)zirconium(IV) proceeds by a rather convoluted process with potentially several cyclic intermediates and the unique formation of VIII, requiring a complete control of all the elementary steps. We initially focused our attention on the diastereoselective zirconocene-mediated carbomagnesiation of cyclobutene 1a (R1 = (CH2)2Ph, R2 = Me) in THF and we were pleased to observe that the addition reaction proceeds selectively under mild conditions (25 °C, 12 h) to provide 2a in 82% isolated yield with >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 diastereoselectivity (Scheme 3). The relative configuration of cyclobutane 2a was determined by the Nuclear Overhauser Effect (NOE) and from this analysis, we could confirm that the zirconocene-catalyzed ethylmagnesiation is not only highly regioselective (formation of IIversusIII in a 92:8 ratio) but also fully anti-diastereoselective (unique formation of IIanti versusIIsyn, Scheme 2). As the reaction of a cyclobutene possessing an ether group (1a, R1 = (CH2)2Ph, R2 = Me) or an alcohol (1b, R1 = (CH2)2Ph, R2 = H) could potentially present a complementary sense of stereoinduction (reversal of stereoselectivity due to association of magnesium alkoxides with the zirconocene reagent),17 the same reaction was performed with 1b and the product 2b was obtained with the same diastereoisomeric ratio and with the same relative configuration albeit in slightly lower yield (64%, 2b was then transformed into 2a and the stereochemistry was corroborated).
:2 diastereoselectivity (Scheme 3). The relative configuration of cyclobutane 2a was determined by the Nuclear Overhauser Effect (NOE) and from this analysis, we could confirm that the zirconocene-catalyzed ethylmagnesiation is not only highly regioselective (formation of IIversusIII in a 92:8 ratio) but also fully anti-diastereoselective (unique formation of IIanti versusIIsyn, Scheme 2). As the reaction of a cyclobutene possessing an ether group (1a, R1 = (CH2)2Ph, R2 = Me) or an alcohol (1b, R1 = (CH2)2Ph, R2 = H) could potentially present a complementary sense of stereoinduction (reversal of stereoselectivity due to association of magnesium alkoxides with the zirconocene reagent),17 the same reaction was performed with 1b and the product 2b was obtained with the same diastereoisomeric ratio and with the same relative configuration albeit in slightly lower yield (64%, 2b was then transformed into 2a and the stereochemistry was corroborated).
 | ||
| Scheme 3 Zirconocene-catalyzed diastereoselective ethylmagnesiation reaction of cyclobutene and reaction with electrophiles. | ||
Therefore, the uniform sense of stereoinduction of the reaction with 1a and 1b implies that the reaction is fully controlled by steric factors. Decreasing the basicity of the reaction medium and using Et2O as solvent instead of THF does not change the stereochemical outcome of the reaction and the major isomer 2a was still observed with, however, higher quantity of product resulting from the formation of the opposite regioisomer III (IIanti:IIIsyn = 87:13, not shown in Scheme 3). Now that the regio- and diastereoselectivity of the zirconocene-catalyzed ethylmagnesiation reaction of cyclobutene 1a is controlled, we were interested to understand the following step, namely the transmetalation step, and determine if cyclobutylmagnesium species VIII or its ethyl metalated cyclobutyl isomer IX would be formed. To answer this question, treatment of 1a with ZrCp2Cl2 (20 mol%) and ethylmagnesium bromide was stirred at room temperature overnight and quenched with D2O/DCl. The cyclobutane 2c was selectively obtained in 80% yield as a unique diastereoisomer (Scheme 3) suggesting complete selectivity in the transmetalation reaction. Only the cyclobutylmagnesium bromide VIII was therefore obtained through the reduction depicted in VII. Similarly, when the intermediate cyclobutylmagnesium species was trapped with I2 or NBS, 2d and 2e were isolated in 67% and in 58% yield, respectively, in an excellent diastereoisomeric ratio. The relative configuration of the cyclobutane 2d was determined by NOE (see ESI†).
The unique stereochemistry of these functionalized cyclobutanes indicates that the cyclobutylmagnesium bond is configurationally stable at room temperature under this experimental condition. As the rate for inversion of configuration of the C–MgBr should be higher for cyclobutyl than for cyclopropyl, we were interested to check if the inversion of the organometallic species of 2MgBranti into 2MgBrsyn (Scheme 3, path b) could occur at higher temperature, as a chelated system would be preferentially formed. When the zirconocene-catalyzed ethylmagnesiation reaction was performed on 1a (R1 = (CH2)2Ph, R2 = Me) at room temperature for 12 h in THF followed by warming at 55 °C for 1 h and finally quenching with I2, the same isomer 2d was obtained with an identical diastereoisomeric ratio suggesting that the cyclobutylmagnesium bromide species is configurationally stable despite the potential stabilizing intramolecular chelation (Scheme 3, path b). The same configurational stability was observed when the isomerization was tested on 1d (R1 = Hex, R2 = H).
Having a configurationally stable C–MgBr bond in the cyclobutylmagnesium bromide structure VIII, we were then concerned by the stereochemistry of transmetalation with copper salt. Would the resulting cyclobutylcopper species be produced with retention18 or inversion19 of configuration and would it also present some configurational stability? Thus, to the intermediate VIII, prepared as previously described from 1a with ZrCp2Cl2 (20 mol%) and ethylmagnesium bromide, the corresponding cyclobutylcopper species was obtained by addition of CuI and LiCl (10 and 20 mol% respectively) at 0 °C for 15 min. Then, addition of allylbromide at 0 °C provided the allylated product 2f in 72% yield with the same diastereoisomeric ratio of 98:2:0:0. The relative configuration of the cyclobutane 2f was determined by NOE and indicates that the transmetalation reaction proceeds with retention of configuration to lead to a configurationally stable cyclobutyl carbon–copper bond.20 The transmetalation reaction was also performed on the intermediate resulting from the zirconocene-catalyzed carbomagnesiation reaction on alcohol 1b (R1 = (CH2)2Ph, R2 = H) to check if a potential reversal of stereoselectivity with the copper salt may occur due to association with magnesium alkoxide. Further addition of allylbromide leads to the same major diastereoisomer 2g in slightly lower ratio but still indicating that the transmetalation proceeds again with retention of configuration and that the C–Cu bond is configurationally stable. The reaction is not restricted to allylbromide and a functionalized electrophile could also be added successfully (formation of 2h) with similar diastereoisomeric ratio and yield. For functionalization with a sp2-carbon center, an additional transmetalation to Pd is required and 2i could be obtained in 58% yield and 93:7:0:0 diastereoisomeric ratio when the cyclobutylmagnesium species VIII is first transmetalated to copper salt [CuI (10 mol%) and LiCl (20 mol%) at 0 °C for 15 min] and then with Pd(Ph3)4 (10 mol%) followed by addition of 3-bromoanisole. Finally, due to the importance of diastereoisomerically pure cyclobutanol in synthesis,21 the simple oxidation of cyclobutyl zinc, easily obtained by transmetalation of VIII with ZnCl2 (1 equiv.) followed by addition of O2 gave the cyclobutanol 2j in 67% yield and outstanding diastereoisomeric ratio.22 The zirconocene-catalyzed carbometalation reaction of cyclobutenes could be extended to various starting materials (R1 = PhCH2CH2; Hex; R2 = Me, H, PMB) and in all cases, the carbometalation remains highly diastereoselective and all subsequent transmetalation and reactions with electrophiles proceeded with retention of configuration (Scheme 3, formation of 2k to 2q). It should be noted that subsequent functionalization of cyclobutylmagnesium species VIII with an sp2-carbon center after transmetalation to Pd is not restricted to bromoarene, as 2q could be obtained in 61% yield and in 92:8:0:0 diastereoisomeric ratio by addition of 4-iodoanisole.
Conclusions
In conclusion, the zirconocene-catalyzed ethylmagnesiation of cyclobutene proceeds with a very high regio- and diastereoselectivity to give configurationally stable cyclobutylmagnesium species. The transmetalation reaction of the latter with copper and palladium salts proceeded with complete preservation of the stereochemical integrity. Reactions with various electrophiles led to different polysubstituted cyclobutyl derivatives in excellent yields and diastereoisomeric ratios.Acknowledgements
This research was supported by the European Research Council under the European Community's Seventh Framework Program (ERC grant agreement no. 338912) and by the Israel Science Foundation administrated by the Israel Academy of Sciences and Humanities (140/12). S. R. R. acknowledges the Planning and Budgeting Committee (PBC) for the financial assistance. I. M. is holder of the Sir Michael and Lady Sobell Academic Chair.Notes and references
- (a) I. Marek and A. Basheer, in Stereoselective Synthesis; Science of Synthesis, ed. J. G. de Vries, Thieme, 2011, p. 325 Search PubMed; (b) K. Itami and J.-I. Yoshida, in The Chemistry of Organomagnesium Compounds, ed. Z. Rappoport and I. Marek, Wiley, Chichester, 2008 Search PubMed; (c) A. G. Fallis and P. Forgione, Tetrahedron, 2001, 57, 5899 CrossRef CAS; (d) I. Marek and J.-F. Normant, in Metal-Catalyzed Cross-Coupling Reactions, ed. P. J. Stang and F. Diederich, Wiley-VCH, Weinheim, 1998, p. 271 Search PubMed; (e) N. Chinkov, D. Tenne and I. Marek, in Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and A. de Meijere, Wiley-VCH, Weinheim, 2004, p. 395 Search PubMed; (f) P. Knochel, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, New York, 1991, vol. 4, p. 865 Search PubMed.
- (a) N. Yoshikai and E. Nakamura, Chem. Rev., 2012, 112, 2339 CrossRef CAS PubMed; (b) F. Chemla and F. Ferreira, in The Chemistry of Organocopper Compounds, ed. Z. Rappoport and I. Marek, Wiley, Chichester, 2011 Search PubMed; (c) A. Basheer and I. Marek, Beilstein J. Org. Chem., 2010, 6, 77 CrossRef PubMed; (d) B. H. Lipshutz and S. Sengupta, in Organic Reactions, Wiley, Hoboken, 2004 Search PubMed; (e) J.-F. Normant and A. Alexakis, Synthesis, 1981, 841 CrossRef CAS.
- (a) E. Negishi and Z. Tan, Top. Organomet. Chem., 2004, 8, 139 CrossRef; (b) E. Negishi and S. Huo, in Titanium and Zirconium in Organic Synthesis, ed. I. Marek, Wiley-VCH, Weinheim, 2002, p. 1 Search PubMed; (c) E. Negishi, D. E. Van Horn and T. Yoshida, J. Am. Chem. Soc., 1985, 107, 6639 CrossRef CAS; (d) E. Negishi, ARKIVOC, 2011, 34 CAS; (e) P. Wipf and S. Ribe, Org. Lett., 2000, 2, 1713 CrossRef CAS PubMed.
- (a) Z. Nairoukh and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 14393 CrossRef CAS PubMed; (b) Y. Minko, M. Pasco, L. Lercher and I. Marek, Nat. Protoc., 2013, 8, 749 CrossRef PubMed; (c) Y. Minko, M. Pasco, L. Lercher, M. Botoshansky and I. Marek, Nature, 2012, 490, 522 CrossRef CAS PubMed; (d) J. P. Das, H. Chechik and I. Marek, Nat. Chem., 2009, 1, 128 CrossRef CAS PubMed.
- (a) S. Klein, I. Marek, J. F. Poisson and J. F. Normant, J. Am. Chem. Soc., 1995, 117, 8853 CrossRef CAS; (b) C. M. Coleman and D. F. O'Shea, J. Am. Chem. Soc., 2003, 125, 4054 CrossRef CAS PubMed; (c) A. Kessler, C. M. Coleman, P. Charoenying and D. F. O'Shea, J. Org. Chem., 2004, 69, 7836 CrossRef CAS PubMed; (d) B. Cottineau and D. F. O'Shea, Tetrahedron Lett., 2005, 46, 1935 CrossRef CAS; (e) B. Cottineau and D. F. O'Shea, Tetrahedron, 2007, 63, 10354 CrossRef CAS; (f) A.-M. L. Hogan and D. F. O'Shea, J. Am. Chem. Soc., 2006, 128, 10360 CrossRef CAS PubMed; (g) J. Clayden, M. Donnard, J. Lefranc, A. Minassi and D. J. Tetlow, J. Am. Chem. Soc., 2010, 132, 6624 CrossRef CAS PubMed; (h) A. M. Fournier and J. Clayden, Org. Lett., 2012, 14, 142 CrossRef CAS PubMed; (i) S. Norsikian, I. Marek, S. Klein, J.-F. Poisson and J.-F. Normant, Chem.–Eur. J., 1999, 5, 2055 CrossRef CAS.
- M. Morton, Anionic Polymerization: Principles and Practice, Academic Press, New York. 1983 Search PubMed.
- (a) A. H. Hoveyda and J. P. Morken, Angew. Chem., Int. Ed. Engl., 1996, 35, 1262 CrossRef; (b) D. Y. Kondakov and E. Negishi, J. Am. Chem. Soc., 1995, 117, 10771 CrossRef CAS; (c) J. P. Morken, M. T. Didiuk and A. H. Hoveyda, J. Am. Chem. Soc., 1993, 115, 6997 CrossRef CAS; (d) A. F. Houri, M. T. Didiuk, Z. Xu, N. R. Horan and A. H. Hoveyda, J. Am. Chem. Soc., 1993, 115, 6614 CrossRef CAS; (e) N. Suzuki, D. Y. Kondakov and T. Takahashi, J. Am. Chem. Soc., 1993, 115, 8485 CrossRef CAS; (f) T. Takahashi, M. Hasegawa, N. Suzuki, M. Saburi, C. J. Rousset and E. Negishi, J. Am. Chem. Soc., 1991, 113, 8564 CrossRef CAS.
- (a) I. Marek, J. Chem. Soc., Perkin Trans. 1, 1999, 535 RSC; (b) G. Eppe, D. Didier and I. Marek, Chem. Rev., 2015, 115, 9175 CrossRef CAS PubMed; (c) E. Negishi, Pure Appl. Chem., 2001, 73, 239 CrossRef CAS.
- D. S. Müller and I. Marek, Chem. Soc. Rev., 2016, 45, 4552 RSC.
- D. S. Müller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414 CrossRef PubMed.
- (a) T. Dudev and C. Lim, J. Am. Chem. Soc., 1998, 120, 4450 CrossRef CAS; (b) S. Grimme, J. Am. Chem. Soc., 1996, 118, 1529 CrossRef CAS; (c) P. R. Khoury, J. D. Goddard and W. Tam, Tetrahedron, 2004, 60, 8103 CrossRef CAS; (d) R. D. Bach and O. Dmitrenko, J. Am. Chem. Soc., 2004, 126, 4444 CrossRef CAS PubMed.
- D. Didier, P.-O. Delaye, M. Simaan, B. Island, G. Eppe, H. Eijsberg, A. Kleiner, P. Knochel and I. Marek, Chem.–Eur. J., 2014, 20, 1038 CrossRef CAS PubMed.
- M. Guisán-Ceinos, A. Parra, V. Martín-Heras and M. Tortosa, Angew. Chem., Int. Ed., 2016, 55, 6969 CrossRef PubMed.
- For recent examples, see: (a) I. Colomer, R. C. Barcelos and T. J. Donohoe, Angew. Chem., Int. Ed., 2016, 55, 4748 CrossRef CAS PubMed; (b) J. M. Hoyt, V. A. Schmidt, A. M. Tondreau and P. J. Chirik, Science, 2015, 349, 960 CrossRef CAS PubMed; (c) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918 CrossRef CAS PubMed; (d) N. N. Noucti and E. J. Alexanian, Angew. Chem., Int. Ed., 2015, 54, 5447 CrossRef CAS PubMed; (e) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef CAS PubMed; (f) D. Audisio, M. Luparia, M. T. Oliveira, D. Klütt and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 7314 CrossRef CAS PubMed; (g) M. Luparia, M. T. Oliveira, D. Audisio, F. Frébault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631 CrossRef CAS PubMed; (h) W. Liu and T. V. RajanBabu, J. Org. Chem., 2010, 75, 7636 CrossRef CAS PubMed; (i) F. Frébault, M. Luparia, M. Oliveira, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 5672 CrossRef PubMed; (j) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783 RSC; (k) H. Ito, T. Toyoda and M. Sawamura, J. Am. Chem. Soc., 2010, 132, 5990 CrossRef CAS PubMed; (l) E. Tsoglin, H. Chechik, G. Karseboom, N. Chinkov, A. Stanger and I. Marek, Adv. Synth. Catal., 2009, 351, 1005 CrossRef CAS.
- K. Sakai, T. Kochi and F. Kakiuchi, Org. Lett., 2013, 15, 1024 CrossRef CAS PubMed.
- For representative examples, see: (a) U. M. Dzhemilev, R. M. Sultanov, R. G. Gaimaldinov and G. A. Tolstikov, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1991, 40, 1229 CrossRef; (b) U. M. Dzhemilev and O. S. Vostrikova, J. Organomet. Chem., 1985, 285, 43 CrossRef CAS; (c) U. M. Dzhemilev, O. S. Vostrikova and G. A. Tolstikov, J. Organomet. Chem., 1986, 304, 17 CrossRef CAS; (d) U. M. Dzhemilev, O. S. Vostrikova and R. M. Sultanov, Izv. Akad. Nauk SSSR, Ser. Khim, 1983, 218 CAS; (e) K. S. Knight, Y. Liu, R. Hara and T. Takahashi, Chem. Commun., 1998, 1989 Search PubMed; (f) T. Takahashi, D. Y. Kondakov and N. Suzuki, Chem. Lett., 1994, 23, 259 CrossRef; (g) T. Takahashi, T. Seki, Y. Nitto, M. Saburi, C. J. Rousset and E. Negishi, J. Am. Chem. Soc., 1991, 113, 6266 CrossRef CAS; (h) D. P. Lewis, P. M. Muller, R. J. Whitby and R. V. H. Jones, Tetrahedron Lett., 1991, 32, 6797 CrossRef CAS.
- (a) A. H. Hoveyda, Z. Xu, J. P. Morken and A. F. Houri, J. Am. Chem. Soc., 1991, 113, 8950 CrossRef CAS; (b) A. H. Hoveyda and Z. Xu, J. Am. Chem. Soc., 1991, 113, 5079 CrossRef CAS; (c) J. P. Morken, M. T. Didiuk and A. H. Hoveyda, J. Am. Chem. Soc., 1993, 115, 8485 CrossRef.
- V. Tarwade, X. Liu, N. Yan and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 5382 CrossRef CAS PubMed.
- For a review, see: A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS PubMed.
- (a) R. W. Hoffmann and B. Holzer, J. Am. Chem. Soc., 2002, 124, 4204 CrossRef CAS PubMed; (b) M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48 CrossRef CAS PubMed; (c) Y.-M. Wang, N. C. Bruno, A. L. Placeres, S. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 10524 CrossRef CAS PubMed; (d) F. Meng, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 11304 CrossRef CAS PubMed; (e) F. Meng, H. Jang and A. H. Hoveyda, Chem.–Eur. J., 2013, 19, 3204 CrossRef CAS PubMed; (f) Y. Miki, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 10830 CrossRef CAS PubMed; (g) M. Leibeling, K. A. Shurrush, V. Werner, L. Perrin and I. Marek, Angew. Chem., Int. Ed., 2016, 55, 6057 CrossRef CAS PubMed.
- (a) T. Nishimura, K. Ohe and S. Uemura, J. Am. Chem. Soc., 1999, 121, 2645 CrossRef CAS; (b) T. Nishimura and S. Uemura, J. Am. Chem. Soc., 1999, 121, 11010 CrossRef CAS; (c) N. Ishida, W. Ikemoto and M. Murakami, J. Am. Chem. Soc., 2014, 136, 5912 CrossRef CAS PubMed; (d) N. Ishida, S. Sawano and M. Murakami, Nat. Commun., 2014, 5, 3111 Search PubMed; (e) T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340 CrossRef CAS PubMed; (f) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed; (g) I. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 CAS.
- (a) M. Simaan, P.-O. Delaye, M. Shi and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 12345 CrossRef CAS PubMed; (b) P.-O. Delaye, D. Didier and I. Marek, Angew. Chem., Int. Ed., 2013, 52, 5333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c6sc02617f |
| This journal is © The Royal Society of Chemistry 2017 |