
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWelcoming natural isotopic abundance in solid-state NMR: probing π-stacking and supramolecular structure of organic nanoassemblies using DNP†
Katharina
Märker‡
ab,
Subhradip
Paul‡§
ab,
Carlos
Fernández-de-Alba
ab,
Daniel
Lee
ab,
Jean-Marie
Mouesca
ab,
Sabine
Hediger
abc and
Gaël
De Paëpe
*ab
aUniv. Grenoble Alpes, INAC, F-38000 Grenoble, France. E-mail: gael.depaepe@cea.fr
bCEA, INAC, F-38000 Grenoble, France
cCNRS, MEM, F-38000 Grenoble, France
First published on 19th October 2016
Abstract
The self-assembly of small organic molecules is an intriguing phenomenon, which provides nanoscale structures for applications in numerous fields from medicine to molecular electronics. Detailed knowledge of their structure, in particular on the supramolecular level, is a prerequisite for the rational design of improved self-assembled systems. In this work, we prove the feasibility of a novel concept of NMR-based 3D structure determination of such assemblies in the solid state. The key point of this concept is the deliberate use of samples that contain 13C at its natural isotopic abundance (NA, 1.1%), while exploiting magic-angle spinning dynamic nuclear polarization (MAS-DNP) to compensate for the reduced sensitivity. Since dipolar truncation effects are suppressed to a large extent in NA samples, unique and highly informative spectra can be recorded which are impossible to obtain on an isotopically labeled system. On the self-assembled cyclic diphenylalanine peptide, we demonstrate the detection of long-range internuclear distances up to ∼7 Å, allowing us to observe π-stacking through 13C–13C correlation spectra, providing a powerful tool for the analysis of one of the most important non-covalent interactions. Furthermore, experimental polarization transfer curves are in remarkable agreement with numerical simulations based on the crystallographic structure, and can be fully rationalized as the superposition of intra- and intermolecular contributions. This new approach to NMR crystallography provides access to rich and precise structural information, opening up a new avenue to de novo crystal structure determination by NMR.
Introduction
Organic self-assembled structures find applications in fields ranging from medicine to molecular electronics.1 Their huge popularity stems from the fact that large and complex structures can be formed from comparably small building blocks that are easy to fabricate and functionalize. Non-covalent interactions like hydrogen bonding, ionic and aromatic interactions as well as hydrophobic effects play a key role in the self-assembly process and govern the shape, and physical properties of the supramolecular structure.2,3 Therefore, their thorough understanding is crucial for the rational design of new and improved structures. This requires precise structural knowledge, both on the molecular and the supramolecular level.Solid-state nuclear magnetic resonance (ssNMR) spectroscopy is the method of choice for structural investigations at atomic resolution when single-crystal X-ray diffraction (XRD) cannot be successfully applied. However, NMR suffers from low sensitivity, which is aggravated by the low natural isotopic abundance (NA) of the key nuclei 13C and 15N (1.1% and 0.37%, respectively). Hence, the probability of encountering a particular 13C–13C spin pair, e.g. for 2D correlation experiments and/or internuclear distance measurements, is as low as ∼0.01%. Therefore, structure determination of NA samples by ssNMR has to rely heavily on complementary techniques like powder XRD and density functional theory (DFT), the combination of which is usually termed NMR crystallography.4
In this work, we prove the feasibility of a novel strategy for solving 3D structures of organic nanoassemblies, which we introduced recently.5–7 Our approach crucially requires recording NMR data on the samples at their low NA, firstly for spectral assignment,5,7–9 and secondly for extraction of structural information (using for instance dipolar recoupling experiments).5,6,10 Dramatically reduced sensitivity resulting from the low NA is compensated for by using magic-angle spinning dynamic nuclear polarization (MAS-DNP). As clearly demonstrated in this work, experiments at the NA of 13C yield unprecedented spectra, which would be impossible to obtain with isotopically labeled material. For instance, long distance polarization transfers can occur thanks to significantly reduced dipolar truncation11,12 effects, and the corresponding 13C peak resolution is substantially improved, owing to the absence of broadening from one-bond JCC-couplings.
The capabilities of our approach are demonstrated on a self-assembled organic structure, with an emphasis on aromatic interactions. We focus on the cyclic diphenylalanine peptide (cyclo-FF, Fig. 1a) which self-assembles into nanotubes or nanowires.13,14 Cyclo-FF nanowires possess semiconducting properties and strong blue fluorescence,14 and surface coating with cyclo-FF nanotubes can provide highly hydrophobic self-cleaning surfaces, ultra-capacitors for energy storage, and microfluidic chips.13 Apart from hydrogen bonding between the backbone amides, aromatic interactions between the phenyl rings are the main driving forces in cyclo-FF self-assembly, with both parallel and perpendicular π-stacking occurring.14,15 In addition to this structure determining function, aromatic moieties often constitute the main functional part of molecular assemblies, owing to the possibilities of energy and charge transport through their π-systems.16–19 Therefore, probing π-stacking interactions is of high interest. Numerous ssNMR studies have been discussed in the literature which rely on probing the aromatic proton spins.20,21 Here we show an alternative powerful approach based on the observation of π-stacking through 13C–13C correlation spectra.
 | ||
| Fig. 1 (a) Chemical structure of cyclo-FF. (b) Scanning electron microscopy image of cyclo-FF nanotubes. (c) 13C CPMAS spectra of cyclo-FF with and without microwave (μw) irradiation. εon/off describes the ratio of peak heights of the two spectra. The ASR is the ratio of the signal-to-noise per unit square root of time between the spectrum recorded with μw irradiation and a CPMAS spectrum taken in a conventional ssNMR experiment. | ||
The experiments presented here were also made possible by the use of the dipolar recoupling pulse sequences S3 and [S3] (“bracketed S3”),22,23 which facilitate the recoupling of carbons with large chemical shift anisotropy (CSA), such as carbonyl and aromatic carbons. Based on this, we achieved well-resolved 13C–13C dipolar correlation spectra of aromatic carbons, and polarization transfer dynamics that can be completely rationalized by numerical simulation. Contributions from numerous intermolecular contacts and notably several π-stacking interactions were detected. We discuss the information content of the polarization transfer curves and corresponding treatment as the superposition of intra- and/or intermolecular spin pair contributions. Furthermore, the limits for neglecting dipolar truncation for 13C at NA are examined. The data and the in-depth analysis provided here demonstrate the benefits of working with 13C at its NA. Taking all factors into account, we show that these experiments are sensitive to distances up to ∼7 Å for cyclo-FF, which is supported by the excellent agreement of experimental data with simulated polarization transfers based on the crystal structure.
Experimental
DNP sample preparation of cyclo-FF
All chemicals were purchased from Sigma-Aldrich and used without further purification, unless otherwise stated. The cyclo-FF DNP sample was prepared by adapting the existing protocol:6 cyclo-FF powder (20 mg, Bachem, Weil am Rhein, Germany) was dissolved under gentle heating in hexafluoroisopropanol (750 μl). The solution was added dropwise and under continuous sonication to a solution of AMUPol24 (0.3 mg, SATT Sud Est, Marseille, France), trehalose dihydrate (0.5 mg) and D8-glycerol (2 mg) in D2O (2.5 ml) and D4-methanol (0.5 ml), causing instant precipitation of cyclo-FF nanotubes. The mixture was transferred into a Petri dish and dried slowly under reduced pressure, while repeatedly stirring it with a spatula. After sufficient drying time (10–13 h), a wet paste was obtained and packed into a 3.2 mm thin-wall zirconia rotor. This process was repeated once more to fill up the rotor. It was observed that the slow drying process and the retaining of moisture at all times are beneficial for optimal MAS-DNP sensitivity.DNP-enhanced ssNMR experiments
The experiments were performed on a wide-bore AVIII 400 MHz (1H Larmor frequency) Bruker DNP-NMR spectrometer equipped with a 263 GHz gyrotron, a μw transmission line and a low temperature 3.2 mm MAS-DNP probe in double-resonance mode operating at ∼105 K. All spectra were recorded at a MAS frequency of 13.889 kHz with a recycle delay of 6.5 s. More details on the experimental conditions and data extraction can be found in the ESI.†Simulation of polarization buildup curves
All simulations were done using the SPINEVOLUTION package.25 The atomic coordinates were taken from the X-ray structure of cyclo-FF,15 and the CSA tensors (ESI Table S1†) were calculated using DFT-GIPAW, except the isotropic chemical shifts which were taken from experimental data. The simulations were done with the ZCW26–28 scheme of powder averaging with 233 pairs (α and β angles) of crystal orientations each averaged over 48 γ-angles.DFT-GIPAW
Quantum chemical calculations on the cyclo-FF crystal structure were run using the planewave pseudopotential approach, with the gauge including projector augmented wave (GIPAW) method29 for computation of CSA tensors. The calculations were performed with the PWSCF module version 5.2.1, and GIPAW version 5.1. within the Quantum ESPRESSO package.30 Norm-conserving pseudopotentials of the Troullier–Martins type31 were used for all atoms, employing the PBE exchange–correlation functional.32,33 The crystal structure of cyclo-FF15 was used as an initial input, and hydrogen positions were optimized prior to GIPAW calculations. A planewave cutoff energy of 96 Ry, and a 2 × 2 × 2 Monkhorst–Pack k-point grid34 were used. The results are summarized in the ESI (Table S1†) and were used as an input in SPINEVOLUTION simulations.Optimization of the molecular structure
TINKER molecular modeling software package35 version 7.1 was used for scanning the potential energy surface and ORCA36 version 3.0.2 was used for geometry optimization and computation of 13C CSA tensors. Computational details are given in the ESI.†Results and discussion
Experimental challenges and strategies
MAS-DNP can enhance the sensitivity of ssNMR experiments by transferring polarization from unpaired electron spins to atomic nuclei using high-power high-frequency microwave irradiation.37–39 In recent years, it has enabled numerous experiments that were unfeasible with standard high-field NMR systems, such as the acquisition of signals from diluted species as well as nuclei with low gyromagnetic ratio and/or low NA.40–47 In addition, advanced experiments probing 13C–13C and 15N–13C proximities through 2D correlation spectra recorded at NA are within reach.5,7,8,10In this context, important steps have been taken towards 3D (supramolecular) structure determination at NA with the help of DNP. Takahashi et al. reported for the first time the detection of NA intermolecular 13C–13C cross peaks.6 Later, Mollica et al. recorded double quantum (DQ) buildup curves, which were shown to be sensitive to intermolecular distances and allowed the identification of the correct crystal structure among a set of proposed structures.10 In parallel to their work, we also advanced further towards structure determination, and specifically focus in this paper on the potential of extracting internuclear distances from dipolar recoupling experiments.
The DNP process requires sample preparation that ensures a uniform distribution of the polarizing agent.48 We have recently introduced an efficient, matrix-free (MF) protocol for the preparation of cyclo-FF for DNP.6 It is a solution-based method where nanotubes are formed in the presence of the biradical polarizing agent. This protocol was developed because direct impregnation of cyclo-FF powder with a radical-containing solution does not lead to an efficient DNP process. For experiments presented in this work, we followed the MF protocol, and improved the NMR sensitivity by replacing the previously employed polarizing agent with the AMUPol biradical.24
The 1H–13C cross-polarization (CP) MAS spectra of cyclo-FF with and without microwave irradiation are shown in Fig. 1c. The improved sample preparation results in a higher DNP enhancement (εon/off = 25), and more importantly an increased Absolute Sensitivity Ratio (ASR = 960)5 as compared to the sample used previously (8.8, and 390, respectively). The improved ASR (sensitivity ratio between 100 K DNP and conventional room temperature (RT) experiments) signifies that experiments can be run six times faster than before, which was already 5 orders of magnitude faster than when using conventional ssNMR. Note that the ASR is very high in this case because of the long 1H T1 relaxation time of cyclo-FF at RT (1H T1 > 600 s),6 but that the approach presented here would also be possible on powdered samples with a smaller ASR, combined with a shorter 1H T1 at RT (e.g. ASR ∼ 10, 1H T1 ∼ 3 s).
Measuring distances between 13C nuclei using ssNMR can be achieved through the use of specific radiofrequency (RF) pulse sequences, such as dipolar recoupling sequences.12 Since the dipolar coupling between two spins is inversely proportional to the cube of the distance between them, distances can be extracted from plots of the buildup of transferred polarization as a function of increasing recoupling times.
In the context of dipolar recoupling, the characterization of aromatic systems is particularly challenging because of the large CSA of their 13C nuclei (up to 200 ppm), which is significantly larger than that of most other 13C moieties.49 Since the efficiency of most recoupling pulse sequences decreases dramatically in the presence of large CSA (see ESI Fig. S3†), we chose the S3 and [S3] pulse sequences, which were recently developed by Edén and coworkers, and are robust to high CSA values.22,23 These sequences were originally designed for the very fast MAS regime (νr ≥ 60 kHz) where they work without additional 1H decoupling.22 They were later shown to also be efficient in the moderate spinning regime, if the spinning frequencies are considerably larger than the strength of the CSA (νr ≥ 1.5νCSA) and high power 1H decoupling is applied.23 Considering the CSAs of aromatic carbons (up to ∼20 kHz at 9.4 T) and the available spinning frequencies of our commercial DNP setup (max. 14 kHz), these conditions are by far not met here. Despite this, we show that efficient polarization transfer involving aromatic spins can be achieved with S3 and [S3] under DNP conditions. As an additional benefit, these pulse sequences have very low RF power requirements (νnut = νr/2). Therefore, they allow long recoupling times without any detrimental effects on the hardware, which is mandatory for observation of long distance contacts. This also means that the recoupling bandwidth is limited to ∼7 kHz under our experimental conditions (νr = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 889 Hz), which is however sufficient for the studies reported herein at 9.4 T.
889 Hz), which is however sufficient for the studies reported herein at 9.4 T.
In 13C–13C dipolar recoupling experiments performed on samples at NA, only a small part of the detected signal comes from polarization transfer in 13C–13C spin pairs. The dominating but undesired part originates from uncoupled 13C spins and has to be suppressed, either by the use of selective pulses or by a double quantum filter (DQF). Taking this into account, two complementary experimental strategies for recording polarization buildup curves and extracting structural information are employed. The first one makes use of selective pulses, such that the buildups can be recorded from a series of 1D spectra. This offers the advantage of very short experimental times. In the second, more traditional strategy, a series of DQF 2D double quantum-single quantum (DQ-SQ) correlation spectra is recorded. This (time-consuming) approach is preferred when the involved spins have similar isotropic chemical shifts, such that selective pulses cannot be conveniently applied. This was for instance the case for recoupling experiments between aromatic carbons. In the following sections, we will present and analyze in detail both experimental approaches on cyclo-FF.
The 1D approach: results and discussion
The pulse sequence used in the experiment is shown in Fig. 2a. After an initial CP step from DNP-enhanced protons to carbons, the CO magnetization is selectively flipped back to the z-axis from the transverse plane. During the following recoupling period, this magnetization is transferred to aromatic carbons, using the dipolar recoupling sequence [S3]. After a selective readout pulse on the aromatics, their magnetization is detected.
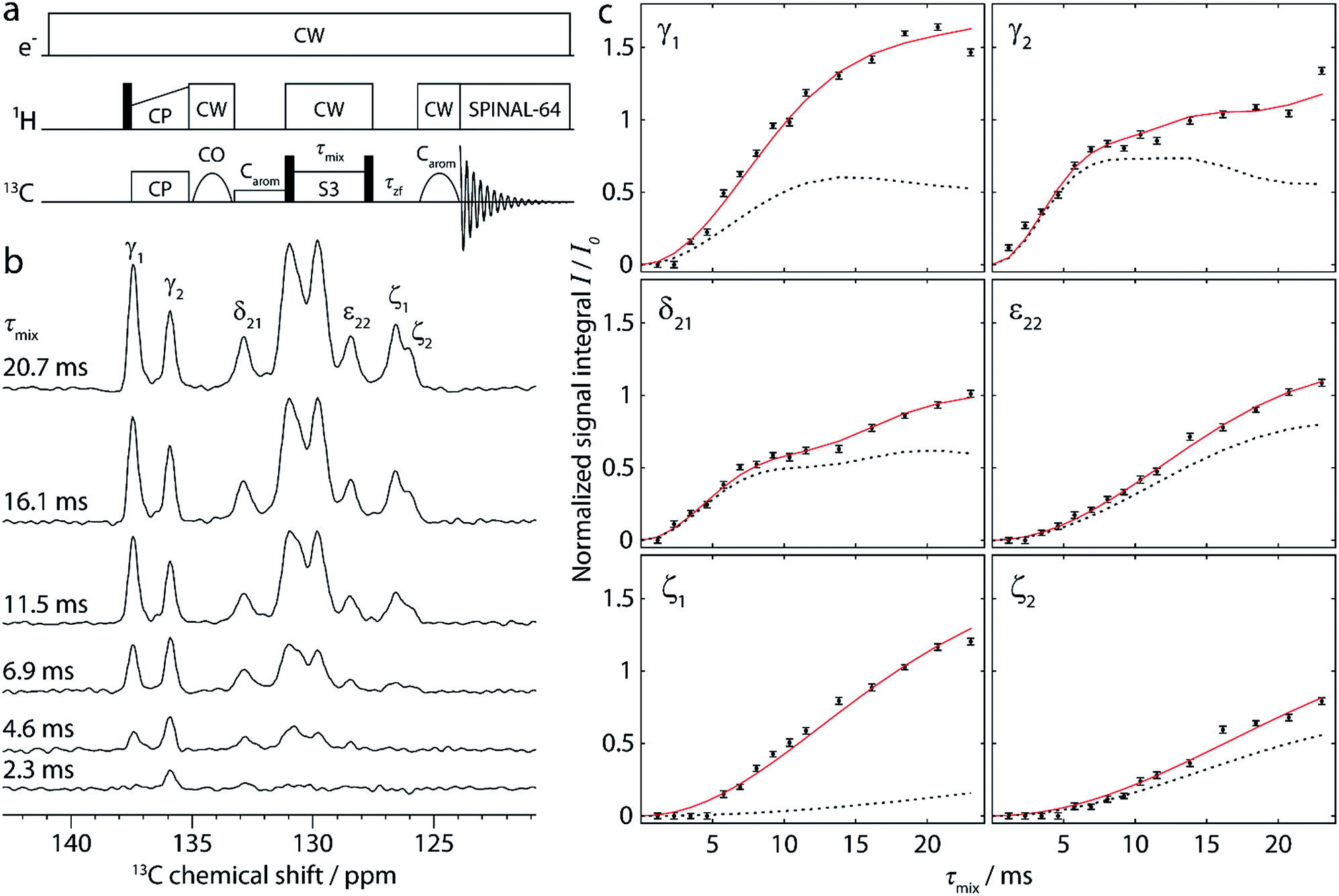 | ||
| Fig. 2 (a) Pulse sequence for the 1D approach, used to obtain buildup curves for polarization transfer from carbonyl to aromatic carbons. [S3] is employed as the dipolar recoupling sequence, with the π/2 bracketing pulses indicated as black bars on the 13C channel. The recoupling time (τmix) is incremented to record the buildup curves. More details are given in the text. (b) Aromatic region of selected 1D spectra, recorded on cyclo-FF with the pulse sequence shown in (a) using different recoupling times τmix, as indicated in the figure. (c) Full experimental polarization buildups (black points) for the six peaks labeled in (b). Error bars denote the average noise integral over 2 ppm. Red solid and black dotted lines show the SPINEVOLUTION simulations of the buildups based on the cyclo-FF crystal structure,15 respectively taking into account all distances up to 7 Å (red) or intramolecular distances only (black). The simulated curves are fitted to experimental points with the amplitude factor A (see Table 1). Note that the experimental signal integral (I) is normalized with respect to the initial polarization of CO spins (I0) in such a way that the initial available polarization in a particular 13CO–13Carom spin pair is 1 (more details in the ESI†). This allows a direct comparison to the output of numerical simulations (the amplitude factor A is expected to be close to 1). | ||
The pulse sequence allows a straightforward measurement of polarization buildup on the aromatic carbons by following their signal intensity as a function of the incremented recoupling period. Due to the CO-selective excitation pulse (followed in addition by a selective saturation pulse on aromatics), no magnetization is present on the aromatics at the beginning of the recoupling period. The selective pulse on the aromatics after recoupling ensures that the large magnetization of uncoupled CO spins remains along the z-axis and does not perturb the detection of the much smaller aromatic signals. This avoids the use of a DQF, which would decrease the sensitivity of the experiment by a factor of 2. It should be noted that this advantage can only be taken for pure double-quantum (e.g. [S3]) recoupling sequences. Sequences like S3, which contain both ZQ and DQ terms, would require the use of a DQF for the observation of polarization transfer. Overall, the 1D approach benefits from immensely decreased experimental times compared to the acquisition of DQF 2D spectra. In addition, data extraction by deconvolution and integration of 1D spectra is relatively straightforward.
A selection of 1D spectra acquired with the sequence of Fig. 2a using different recoupling times (τmix) is presented in Fig. 2b. The full dataset, consisting of 15 1D spectra, was recorded in ∼7 h, corresponding to only 28 min per spectrum. From each spectrum, the integrals of six peaks (Cγ1, Cγ2, Cδ21, Cε22, Cζ1, and Cζ2) were extracted after deconvolution. The large central peaks (128–126 ppm) could not be reliably deconvoluted due to the contributions of too many carbons (Cδ1, Cδ22, Cε1, and Cε21).
Fig. 2c shows the resulting polarization buildup curves for the six peaks (black points). They are normalized with respect to the initial polarization on the carbonyls, taking into account the number of COs per molecule and the fraction of them which is coupled to another 13C spin (1.1%, see ESI for more details†). As a result, the y-axes in Fig. 2c reflect the absolute transferred polarization and can thus be directly compared to numerical simulations.
All buildup curves display polarization transfer efficiencies far above the expected maximum for a single spin pair under these conditions (∼0.38 in numerical simulations). This is a clear indicator that various spin pairs contribute additively to each buildup curve. For instance, both CO1 and CO2 can provide polarization because they are excited simultaneously. Thus, two different intramolecular polarization transfer pathways are possible for each peak, and the sum of their contributions can give rise to a maximum polarization of ∼0.76. This number is still exceeded in the majority of cases, which indicates that intermolecular distances contribute as well (discussed in more detail below).
The six experimental buildup curves possess distinctly different features. For instance, while most peaks build up continuously, the Cγ2 and Cδ21 signal intensities slightly level out before rising again. Moreover, the Cγ2 peak has a faster initial buildup, most probably indicating the presence of a very short CO-Cγ2 distance compared to all other distance contributions here. The Cγ1 peak, on the other hand, has a slower initial buildup, but rises to higher intensity at longer mixing times, suggesting a higher total number of distances involved in the buildup.
The reason for the additive contribution of distances to the buildup curves lies in the NA nature of the sample. Nearly all 13C–13C spin pairs are isolated (surrounded by 12C), such that the polarization transfer between them is not detrimentally affected by the presence of a third 13C spin, even over longer distances. In addition, all different kinds of spin pairs are randomly distributed in the sample. Each spin pair leads to a typical polarization buildup depending on the internuclear distance, and it is the addition of these individual contributions that yields the experimentally observed buildups. Similar observations have been reported by Brouwer et al. in the process of structure determination of zeolites, where they probed the slightly more abundant 29Si spins.50,51
Overall, the buildup curves contain a high level of information. As described above, some qualitative information is already accessible from a relative comparison between the different curves. However, the extraction of quantitative distance information requires the use of numerical simulations for comparison and fitting.
As a first step, the behavior of S3 and [S3] under the experimental conditions employed here was investigated in more detail, since these conditions do not comply with the recommended regime (νr < νCSA instead of νr ≥ 1.5νCSA).23 Numerical simulations (see ESI Fig. S4 and S5†) show that polarization can still be transferred efficiently with S3 and [S3] under our experimental conditions. However, the buildup curves for [S3] exhibit a strong dependency on all CSA parameters. More precisely, the asymmetry and anisotropy affect the overall polarization transfer amplitude, without having a major influence on the shape of the curve. On the other hand, a change of the orientation of the principal axes of the CSA tensor can change this shape significantly. To run accurate simulations, the magnitudes and orientations of the 13C CSA tensors of cyclo-FF were computed with the gauge-including projector-augmented wave (GIPAW)29 approach, using its implementation in the Quantum ESPRESSO30 software package. The crystal structure of cyclo-FF15 served as the initial input for the DFT calculations, and hydrogen atom positions were optimized before the GIPAW calculations.
The expected polarization buildups were then simulated with SPINEVOLUTION, based on the CSA tensor parameters from the DFT computations, atomic positions from the crystal structure, and isotropic chemical shifts from the experiment. The polarization buildups for all relevant spin pairs were simulated separately, and the individual curves were then co-added for each peak. It is important to stress again that, although several different distances contribute to each buildup curve, we consider these contributions to be coming from isolated spin pairs. Deviations from this approximation by the presence of a third spin leading to dipolar truncation at NA will be discussed later in this paper.
Notably, our concept of data analysis differs from those employed previously in similar contexts of 13C–13C10 or 29Si–29Si51 recoupling experiments at NA, which rely on analytical functions to describe polarization transfers. We instead chose to use numerical simulations, since they can easily account for, among other things, isotropic and anisotropic chemical shift effects.
| Cγ1 | Cγ2 | Cδ21 | Cε22 | Cζ1 | Cζ2 | |
|---|---|---|---|---|---|---|
| a Intramolecular distances are shown as bold italic. b Factor for scaling the amplitude of the simulated curves to experimental data as used in Fig. 2c, obtained by minimizing the root mean square deviation (RMSD) between experiment and simulation. | ||||||
| Distances from CO1/Åa | 3.8 | 3.7 | 4.6 | 4.5 | 4.3 | 5.3 |
| 4.9 | 6.0 | 5.9 | 5.1 | 6.9 | ||
| 5.3 | 6.5 | 6.5 | ||||
| 5.5 | 6.8 | |||||
| 6.9 | ||||||
| Distances from CO2/Åa | 4.2 | 3.0 | 3.3 | 5.0 | 4.9 | 5.1 |
| 4.2 | 5.3 | 6.5 | 5.5 | 5.4 | 6.3 | |
| 6.5 | 6.7 | 6.4 | 6.8 | |||
| 6.5 | ||||||
| 6.5 | ||||||
| Total number of distances | 7 | 5 | 5 | 3 | 10 | 5 |
| A | 0.88 | 1.14 | 0.91 | 1.15 | 0.73 | 0.97 |
| RMSD | 0.07 | 0.06 | 0.03 | 0.02 | 0.05 | 0.04 |
The extent of this deviation can be rationalized by the number and magnitude of intermolecular distances, which are listed up to a limit of 7 Å in Table 1. For example, the experimental data for the Cγ1 peak starts to deviate significantly from the simulation for intramolecular distances at mixing times as short as ∼6 ms, and rises to more than three times the simulated intensity. This can be explained by the high number of comparably short intermolecular distances (four distances ≤ 5.5 Å). The Cζ1 buildup shows an even more drastic deviation, which results from the fact that the intramolecular distances here are very long (6.4 and 6.5 Å) compared to other distances. The buildup curve of Cζ1 is therefore almost exclusively determined by intermolecular spin pairs. Deviations between intramolecular simulations and experimental data are less pronounced for the other four peaks, since there the intramolecular spin pairs represent the two shortest distance contributions, separated from the next longer ones by at least 0.5 Å.
This comparison shows that the inclusion of intermolecular spin pairs into the simulation is essential for a thorough explanation of the polarization buildup curves. To obtain more accurate simulated curves, all distances in Table 1 were taken into account. The amplitudes of the calculated curves were adjusted to the experimental points by fitting an individual amplitude factor A for each curve (Iexp = A × Isim). The results of the full simulations, including scaling by A, are shown as red lines in Fig. 2c. The optimized amplitude factors A with corresponding RMSD values are summarized in Table 1.
The full simulation is in excellent agreement with the experimental data. Notably, no systematic deviation at longer mixing time is observed, which would have indicated relaxation-related losses during the recoupling. Even small features like the changes in slope for Cγ2 and Cδ21 are reproduced by simulation. Furthermore, all amplitude scaling factors lie between 0.7 and 1.2, which is reasonably close to the theoretically expected value of 1 (see also next section). This indicates that it is indeed possible to directly relate experimental data to the polarization transfer efficiency. This constitutes a big advantage, as precious information on the approximate number of distances contributing to a buildup curve can be extracted. Furthermore, the fact that the experiments are clearly sensitive to intermolecular contacts is highly encouraging, as these are imperative for supramolecular structure elucidation.
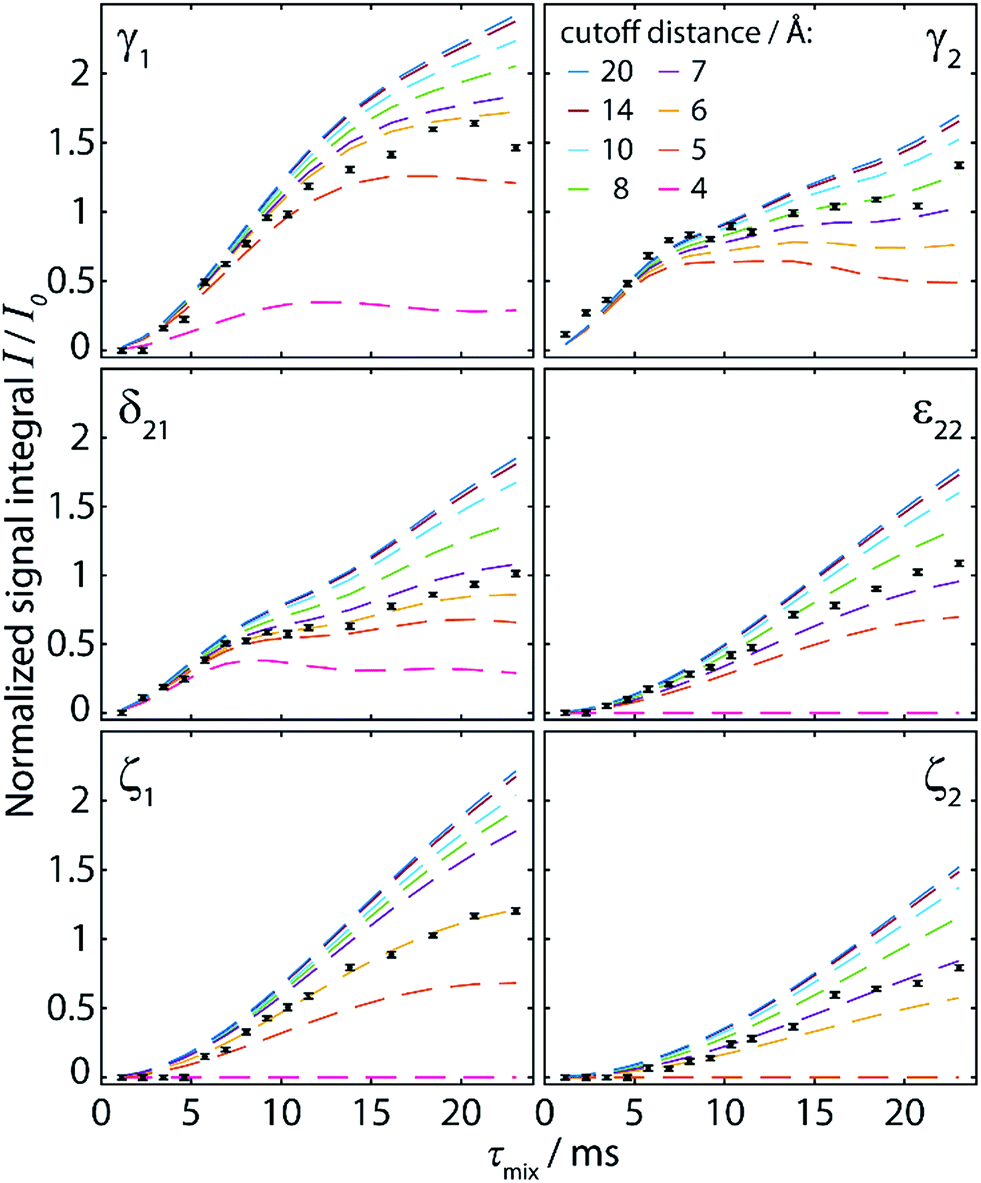 | ||
| Fig. 3 Simulated polarization buildup curves (dashed lines) for the experiment shown in Fig. 2, taking into account the cumulative contribution of all spin pairs whose internuclear distance lies below a cutoff distance increased from 4 Å to 20 Å. The y-axis corresponds to the absolute transferred polarization, and no scaling has been applied to the simulated curves. Experimental points (black points) are identical to those given in Fig. 2c, including normalization with respect to the CO polarization. | ||
A re-evaluation of potential dipolar truncation effects at the NA of 13C becomes necessary when considering the possibility of observing such long distance polarization transfers. To this end, we need to estimate the probability of finding a third 13C spin in a defined volume around the respective 13C–13C two-spin system, which is demarcated by their internuclear distance. At the NA of 13C, this probability is nC × 1.1%, with nC being the number of carbon atoms in the considered volume. Accordingly, the polarization transfer efficiency is expected to be lowered by this percentage. The effect increases with the number of atoms in the volume, and thus with the internuclear distance for the considered polarization transfer. Hence, no polarization transfer can statistically be observed between two spins which are at such a distance that the corresponding volume around the spin pair contains 91 carbons or more, as there will always be a third 13C spin truncating the desired transfer. An exact definition of the “truncating” volume is beyond the scope of this paper, and this crude model also does not consider cases in which polarization transfer is only partially truncated. Nevertheless, from the crystal structure of cyclo-FF, the limit of ∼100 carbon atoms is found within a sphere of radius 8 Å. It is therefore very unlikely that polarization transfers over this distance can be observed in cyclo-FF, even with considerably longer mixing times. This illustrates the limits of the idealized two-spin system model used here to rationalize polarization transfer in NA samples.
Comparison of experimental data (black points in Fig. 3) with the simulated polarization buildup curves for different cutoff distances confirms this crude approximation. Indeed, most of the experimental points for the 6 different polarization buildups lie between the corresponding simulated buildup curves with 6 and 8 Å cutoff distance. In addition, it should be noted that for cutoff distances higher than 7 Å, the shape of the simulated buildup curves does not change significantly anymore, but only their amplitude increases. This finding supports our strategy of including only distances up to 7 Å in the simulations used to characterize the experimental buildups. With this procedure, the shape of the buildup curves can be modelled correctly, using solely an amplitude factor (A) to then fit the simulations to the experimental points. This factor, which should theoretically be unity, takes into account deviations owing to some longer distance transfers, partial dipolar truncation, and the influence of experimental errors and/or instabilities. The different number and distribution of long-distance contributions for each peak also justifies the use of different amplitude factors as opposed to other reported fitting procedures which use a global amplitude factor.10,51 In the case of cyclo-FF, our fitting procedure exclusively returns well defined amplitude factors close to 1 (see Table 1), which confirms the validity of our approach. A more detailed discussion of the behavior of A and the RMSD with increasing cutoff distances can be found in the ESI.†
The 2D approach: results and discussion
Accordingly, we recorded 13C–13C DQ-SQ correlation spectra of cyclo-FF, focusing specifically on the aromatic region. Owing to its excellent CSA compensating properties, S3 was employed as the dipolar recoupling sequence. Fig. 4 shows two of the spectra obtained, using total recoupling times of 2.3 ms (a) and 13.8 ms (b). Nicely resolved correlations among aromatic carbons are observed, highlighting the excellent performance of S3 for recoupling carbons with large CSA. The spectra presented here are the first 13C–13C dipolar correlation spectra showing such well-resolved correlations between aromatic carbons.
 | ||
| Fig. 4 Aromatic region of 13C–13C DQ-SQ correlation spectra of cyclo-FF, obtained with the dipolar recoupling sequence S3 applied during total recoupling times (DQ excitation plus reconversion) of 2.3 ms (a) and 13.8 ms (b). The spectra were recorded in ∼18.5 h (a) and ∼7.4 h (b), respectively. 13C CPMAS spectra are shown above for illustrative purposes. Note the overlapping δ1 and ε1 resonances of one of the phenyl rings compared to the resolved resonances (δ21, δ22, ε21 and ε22) for the second ring. The up- and downfield shifts of ε22 and δ21, respectively, are signs of intermolecular interactions, in this case of parallel π-stacking.6,15 Cross sections along the black dotted lines (δDQ = 264.1 ppm and δDQ = 268.5 ppm) in (a) are displayed below and show the narrowing of linewidths for long distance peaks (upper cross section, linewidths 40–50 Hz) compared to cross-peaks arising from directly bonded carbons (lower cross section, linewidths > 100 Hz). Resolved cross-peaks used for the buildup analysis (see Fig. 5) are labeled in (b). Among them, select pure intermolecular peaks are marked by a red cross, and select peaks resulting from intra- and intermolecular contributions are connected with an orange dashed line. | ||
The spectrum in Fig. 4a was acquired with the shortest possible mixing time, using one S3 block for DQ excitation and reconversion each (2.3 ms in total), and contains mainly one-bond correlations. It can thus be used for complete assignment of all aromatic resonances as illustrated in the figure. In addition, correlation peaks of lower intensity can be observed, which arise from polarization transfers over distances corresponding to two or three bonds within one phenyl ring.
The spectrum acquired with a longer mixing time (13.8 ms, Fig. 4b) looks remarkably different from the previous one. Numerous additional peaks can be observed, e.g. between the Cγ resonances. They arise from long distance correlations, occurring between carbons from different phenyl rings. Notably, the Cγ1–Cγ1 cross-peak demonstrates that polarization transfers over distances of more than 6 Å can be observed (shortest distance between two Cγ1 atoms is 6.2 Å, see Table 2). Moreover, this peak is one of several autocorrelation cross-peaks appearing on the DQ diagonal. As a DQF was implemented in the pulse sequence, they have to originate exclusively from intermolecular correlations, which makes them particularly valuable for the investigation of the supramolecular assembly.
| Cγ1–Cγ1a | Cγ1–Cγ2 | Cγ1–Cδ21 | Cγ1–Cε22 | Cγ2–Cγ2a | Cγ2–Cδ21 | Cγ2–Cε22 | Cδ21–Cδ21a | Cδ21–Cε22 | |
|---|---|---|---|---|---|---|---|---|---|
| a Intermolecular autocorrelation peaks. b Intramolecular distances are shown as bold italic. c Distances in autocorrelation peaks are counted as half for each molecule because they are “shared” between two molecules. d Factor for scaling the amplitude of the simulated curves to experimental data as used in Fig. 5, obtained by minimizing the RMSD between experiment and simulation. | |||||||||
| Distances/Åb | 6.2 | 4.9 | 4.7 | 4.7 | 4.5 | 1.4 | 2.4 | 3.5 | 2.8 |
| 6.2 | 6.2 | 6.0 | 5.2 | 6.2 | 4.2 | 4.5 | 6.2 | 4.2 | |
| 6.6 | 6.6 | 6.4 | 5.4 | 6.2 | 5.0 | 5.5 | 6.2 | 4.7 | |
| 6.8 | 5.8 | 6.4 | 6.7 | 5.7 | |||||
| 6.9 | |||||||||
| Total number of distances per moleculec | 1.5 | 5 | 3 | 4 | 1.5 | 4 | 4 | 1.5 | 4 |
| A | 1.55 | 1.45 | 1.37 | 0.61 | 1.61 | 0.78 | 0.98 | 1.08 | 0.93 |
| RMSD | 0.06 | 0.09 | 0.09 | 0.09 | 0.05 | 0.04 | 0.08 | 0.04 | 0.13 |
Another striking difference between the two spectra in Fig. 4 is a splitting of some cross-peaks along the direct detection dimension, which is only observed at short mixing time. This splitting occurs for correlation peaks that correspond to a one-bond contact, and is caused by the 1JCC-coupling between the two directly bonded carbons. Since the size of the 1JCC-coupling (55–60 Hz for monosubstituted benzenes52) is similar to the peak linewidths (40–60 Hz), it is barely resolved and clearly constitutes the limiting factor for resolution. It results in considerable line broadening and reduced signal-to-noise (S/N) ratios for one-bond cross-peaks. In NA samples, correlation peaks over longer distances are sharper (see cross sections in Fig. 4a) since the detected carbons mostly do not have a directly bonded 13C spin and the size of two-bond and longer-range JCC-couplings is negligible compared to the 13C linewidths obtained here. At the same time, peaks which are split at short mixing time appear also sharper at longer mixing times (e.g. the Cγ1–Cδ21 correlation), as contributions of spin pairs with longer internuclear distances (and therefore free of 1JCC-coupling) are becoming active. They benefit from increased S/N and readily dominate the spectrum.
The spectra presented here illustrate the advantages of NA measurements for the acquisition of 2D spectra. The drastically reduced dipolar truncation allows straightforward observation of long distance correlations, including exclusively intermolecular contacts which appear as diagonal peaks in DQF experiments. Moreover, all correlations except the ones between directly bonded carbons are free from 1JCC-broadening, which can lead to improved resolution, as shown here. Considering these factors, it becomes evident that a spectrum with the resolution and intermolecular information content as shown in Fig. 4b would not be possible to record on a fully isotopically labeled sample.
These experimental points (black points) along with the simulated polarization buildup curves (red solid lines) are shown in Fig. 5 for the different spin pairs. In order to normalize the experimental data, we took the Cγ2–Cε22 and Cε22–Cγ2 cross-peaks at the first point of the buildup (2D spectrum at τmix = 4.6 ms) as reference points. They have the advantage of being free of one-bond J-coupling, and their summed volume (V1 + V2) corresponds to a two-bond distance transfer, which can be compared to the simulated expected transfer efficiency (S). The ratio S/(V1 + V2) was then taken as the normalization factor for all integrated intensities given as experimental points in Fig. 5. Notably, this procedure can be done without knowledge of the crystal structure, because the length of a two-bond distance in a phenyl ring is well-known.
 | ||
| Fig. 5 Normalized experimental (black points) and simulated (red lines) polarization buildup curves for aromatic–aromatic spin pairs in DQ-SQ correlation spectra. Experimental points were obtained from Monte-Carlo integration of 9 different cross-peak pairs as shown in Fig. 4b for ten mixing times ranging from 4.6 ms to 46 ms. Error bars indicate the error in volume extraction using the Monte-Carlo method. Normalization of the experimental points is explained in the text. The simulated curves take into account all distances up to 7 Å and are fitted to experimental points with an amplitude factor A (see Table 2). | ||
Similar observations as in the 1D approach can be made for the buildup curves obtained from 2D spectra, with notably the contribution of multiple distances resulting in higher transferred intensities than the theoretical maximum for a single spin pair (∼0.21). In contrast to the buildup curves obtained with the 1D approach, several correlations here have contributions from distances shorter than 3 Å, originating from spin pairs within one phenyl ring (Cγ2–Cδ21, Cγ2–Cε22, and Cδ21–Cε22). Accordingly, their buildup curves start with high intensity at the first point (reflecting a fast initial buildup), whereas the transferred polarization for all other correlations is significantly lower at such a short mixing time.
For a better evaluation of the experimental data, full simulations of the expected polarization buildup curves were run with SPINEVOLUTION, in the same manner as for the 1D buildup curves. Accordingly, distances up to 7 Å were taken into account (summarized in Table 2). As for the 1D approach, the simulated data was fitted to the experimental points with the amplitude factor A. A good agreement between experiment and simulation is again obtained. The more complex nature of 2D experiments leads however to a lower accuracy when compared to the 1D approach, as revealed by higher RMSDs and larger deviations from unity for the amplitude factor A (see Table 2). This can be explained by the possibility of a direct normalization of 1D spectral intensities as well as by the simpler signal integration of 1D peaks. Moreover, the acquisition of the complete set of 2D spectra requires several days and is hence more susceptible to spectrometer instabilities.
Nevertheless, these experiments constitute a powerful approach for the investigation of π-stacking. It is clearly possible to observe correlations that arise from structurally important π–π interactions, which are illustrated in Fig. 6. For instance, the parallel π-stacking in cyclo-FF (Fig. 6c) is well described by the Cγ2–Cγ2 and the Cδ21–Cδ21 polarization buildup curves. Indeed, the distances describing this interaction are by far the shortest contributions and therefore dominate the buildup curves. For the perpendicular π-stacking, the involved Cγ1–Cε22 distances are illustrated in Fig. 6b, with the shortest distance being intramolecular (4.7 Å) and the next longer distance (5.2 Å) arising from the π–π interaction. This second example illustrates the strength and the challenges of our experiments. They are clearly sensitive to π–π contacts and can be used to probe π-stacking. On the other hand, however, it is challenging to distinguish the “relevant” π–π contacts from other contributions like intramolecular distances. In this respect, peaks on the diagonal are particularly interesting as they do not contain intramolecular contributions.
 | ||
| Fig. 6 (a) Crystal structure of cyclo-FF15 with perpendicular and parallel π–π interactions highlighted in green and red, respectively. (b and c) Close-up of perpendicular (b) and parallel (c) π-stacking. The highlighted distances can be probed by the 2D experiments presented in this paper. Note that the relevant carbon atoms and distances are highlighted for illustrational purposes on the same pair of cyclo-FF molecules. However in a sample at NA, it is highly unlikely to have four 13C isotopes occurring in only two cyclo-FF molecules. The highlighted distances contribute individually to the polarization transfer in different molecule pairs across the sample. | ||
Perspectives in structure determination
After the thorough analysis and rationalization of polarization buildup curves based on the cyclo-FF crystal structure, we now focus on perspectives for the extraction of structural information in the case of an unknown structure. These considerations are based on the experimental approach described in this contribution, which is DNP-enhanced solid-state NMR on samples at NA. As a first step for structure determination of supramolecular systems, determination of the molecular conformation in the supramolecular structure is essential and will be discussed below. We will then move to the perspectives for de novo fitting of polarization buildup curves.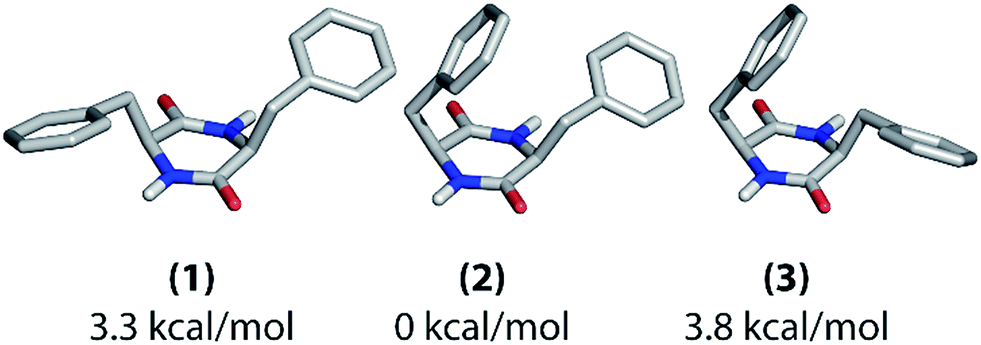 | ||
| Fig. 7 Possible molecular conformations of cyclo-FF as determined with computational structure optimization. Their relative energies with respect to (2) are given below the structures. | ||
These three structures mainly differ in the positioning of the phenyl rings with respect to the central ring. In structure (1), they both point away from the central ring, whereas one of them is bent over it in the other two structures. In structure (2), the phenyl ring which points away is oriented towards the NH group of the central ring, whereas it is oriented towards the carbonyl group in structure (3). Overall, there are two asymmetric (2 and 3) and one nearly C2-symmetric structure (1). Structure (2) has the lowest energy in vacuum.
The different experimental chemical shifts for the two amino acids suggest that an asymmetric structure is present in the nanotubes, such as found in structures (2) and (3). The experimental 1D polarization buildup curves from carbonyl to aromatic carbons should be a suitable way for further probing the positioning of the phenyl rings. The buildups for the two Cγ and the two Cζ resonances, respectively, are clearly different from each other (Fig. 2), which again suggests the presence of structure (2) or (3) (see also ESI Fig. S7†). Discrimination between (2) and (3) should be possible based on the buildups of the Cγ peaks. As described before in this work, the shortest (and mostly intramolecular) distances determine the initial slope of the polarization buildup curves. When considering the shortest CO–Cγ1 and CO–Cγ2 distance, respectively, one can see that they are considerably different in structure (2) (3.9 and 3.0 Å), but of comparable length in structure (3) (3.2 and 3.1 Å). Experimentally, the initial buildups for both peaks are clearly different (Fig. 2b and c), which infers the presence of structure (2) in the solid state. This is also illustrated in Fig. S7† (top panels) where experimental data and intramolecular simulations for the three candidate structures are compared, and the best agreement at short mixing times is obtained for structure (2).
In brief, all these experimental observations and the calculated energy are consistent with a conformation very similar to structure (2) being present in the cyclo-FF self-assembled nanotubes, which is confirmed by its crystal structure. It should be kept in mind, however, that the computational structural search and optimization have been performed on a single molecule in vacuum. Intermolecular interactions were not taken into account, which could potentially change the energetic situation. Despite that, the described method leads to the globally correct structure of cyclo-FF, also signifying that the geometry of the assembly is mainly determined internally on the molecular level.
The example of cyclo-FF shows that the combination of NMR and computational methods can already give an idea of the molecular conformation and provide a valuable entry point for further structural investigations of the supramolecular assembly. The precise methodology of how to model the molecular structure will have to be adjusted to the molecule under investigation, e.g. depending on its size and rigidity/flexibility, and may require more experimental input than shown here.
The strong CSA orientation dependency of the S3 and [S3] sequences (see ESI Fig. S4 and S5†) constitutes a restriction when investigating carbons with a large CSA. Even though the polarization buildup for most orientations does not vary much around an average curve, the occurrence of extreme cases can never be ruled out. Faster spinning would help in this regard, as it decreases the CSA dependency of the polarization transfer and also increases the bandwidth of the sequences. Although the current commercial DNP-NMR system does not allow for faster spinning, technical solutions are under development, such as faster sample spinning with cryogenic helium gas53,54 or the use of smaller rotor diameter MAS-DNP probes.55 On the other hand, the CSA dependency of the buildup curves does not only constitute a restriction, but also contains valuable information on the orientations of the moieties studied, which could be extracted in favorable cases.
Another factor to be considered is the number of distances contributing to one particular buildup curve. Cyclo-FF is a relatively small molecule in a densely packed structure and many distances below 7 Å occur for the same spin pair. In such a case, a de novo fitting of buildup curves becomes very complicated and ambiguous. This aspect is particularly problematic when several contributing distances are of comparable length, as for instance in the CO–Cγ1 polarization buildup. On the other hand, when the distances differ by more than ∼0.5 Å as for CO–Cγ2 and CO–Cδ21 buildups, distinct features occur in the buildup curves and show great promise for a de novo fitting. The extraction of distance information can therefore be expected to be considerably easier for molecules larger than cyclo-FF.
The fitting procedure can also be simplified when some distances are already known, as for example the distances within one phenyl ring or other intramolecular distances in the case where molecular conformation was determined previously. By simulating the expected polarization buildup for the known distances and subtracting it from the experimental curves, the number of distances to be determined can be reduced, leading to a simplification of the fitting procedure.
It is noteworthy that some information can be extracted from experimental polarization buildups without a complete fitting of the curves. For instance, the initial rise of the buildup curve contains information on the shortest distance(s) involved. In addition, the possibility of normalizing experimental data according to the initial polarization as shown in the 1D approach is a great advantage as it directly gives an approximation of the number of distances which contribute to a buildup curve.
For a full structure determination, our experiments can of course be extended to non-aromatic carbons, e.g. probing Cα and Cβ carbons. In this case, not only S3 and [S3], but also other dipolar recoupling sequences can be considered. In particular for the measurement of relatively short distances (<3 Å), alternative sequences should be employed that have a slower polarization buildup and/or shorter sequence blocks. Overall, the quality of our data shows that a de novo fitting of polarization buildup curves is within reach.
Conclusions
The work presented here provides the framework for a novel strategy for structure determination of organic solids, based on DNP-enhanced ssNMR experiments at NA. In particular, we show that experiments at the low NA of 13C are highly worthwhile, regardless of the availability of an equivalent isotopically labeled compound. Different and often even more informative spectra can be obtained from NA samples, owing to the possibilities of long distance polarization transfers, simultaneous undisturbed observation of different distances for the same spin pair and detection of intermolecular contacts. Such NA experiments are made possible by MAS-DNP, which is employed to compensate for the drastically reduced sensitivity.This paper focuses on dipolar recoupling experiments to probe structural information. Two complementary experimental approaches are presented. The first one allows polarization buildup curves to be obtained from a series of 1D spectra, enabled by the use of selective pulses. It has the advantages of very short experimental times (less than half a day for 15 points here) and results in very precise buildup curves. The second approach is more universally applicable and consists in the more traditional way of recording a series of 2D DQ-SQ spectra to follow the buildup of different cross-peaks.
Both approaches are demonstrated on nanotubes of cyclo-FF, investigating structurally relevant distances between carbonyl and aromatic carbons (1D approach) and among the aromatics (2D approach). The efficient recoupling of such carbons with very large CSAs is made possible by the S3 and [S3] pulse sequences, which additionally allow the use of sufficiently long recoupling times required to probe long distances. Using these sequences, the polarization buildup curves obtained with both approaches can be fully rationalized with numerical simulations based on the cyclo-FF crystal structure. Their detailed analysis reveals that each curve consists in the superposition of different distance contributions, reaching up to a remarkable length of ∼7 Å. A theoretical description of these contributions as isolated spin pairs is shown to be sufficient, making the analysis straightforward and simple. In addition, we demonstrate that dipolar truncation plays a role even in samples at NA when polarization transfers over longer distances are considered, and prohibits the detection of very long distances (longer than ∼7 Å in cyclo-FF). In view of the ultimate goal of supramolecular structure determination, the sensitivity of these experiments to intermolecular distances is of extreme importance. Furthermore, we demonstrate the feasibility of probing π-stacking through 13C–13C 2D correlation spectra, shining light on one of the most important non-covalent interactions in supramolecular systems. The wealth of structural information contained in polarization buildup curves measured on organic samples at NA, combined with the simplicity of their theoretical description as added isolated contributions, make up the beauty of this approach to NMR crystallography. It will certainly unfold its power not only for structure validation or refinement as reported by Mollica et al.10 and in our contribution, but can also be expected to enable de novo crystal structure determination by NMR in the future.
Acknowledgements
Hiroki Takahashi and Patrice Rannou are acknowledged for stimulating discussions. This work was supported by the French National Research Agency through the “programme blanc” (ANR-12-BS08-0016-01), the Labex ARCANE (ANR-11-LABX-0003-01) and the European Research Council (ERC-CoG-2015, No. 682895). Funding from the RTB is acknowledged. SEM images were taken at the PFNC platform at MINATEC (CEA Grenoble).References
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- NMR Crystallography, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, WILEY, Chichester, 2009 Search PubMed.
- H. Takahashi, D. Lee, L. Dubois, M. Bardet, S. Hediger and G. De Paëpe, Angew. Chem., Int. Ed., 2012, 51, 11766–11769 CrossRef CAS PubMed.
- H. Takahashi, B. Viverge, D. Lee, P. Rannou and G. De Paëpe, Angew. Chem., Int. Ed., 2013, 52, 6979–6982 CrossRef CAS PubMed.
- K. Märker, M. Pingret, J.-M. Mouesca, D. Gasparutto, S. Hediger and G. De Paëpe, J. Am. Chem. Soc., 2015, 137, 13796–13799 CrossRef PubMed.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- M. Dekhil, G. Mollica, T. T. Bonniot, F. Ziarelli, P. Thureau and S. Viel, Chem. Commun., 2016, 52, 8565–8568 RSC.
- G. Mollica, M. Dekhil, F. Ziarelli, P. Thureau and S. Viel, Angew. Chem., Int. Ed., 2015, 54, 6028–6031 CrossRef CAS PubMed.
- P. R. Costa, Ph.D. thesis, Massachusetts Institute of Technology, Cambridge, MA, 1996.
- G. De Paëpe, Annu. Rev. Phys. Chem., 2012, 63, 661–684 CrossRef PubMed.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS PubMed.
- J. S. Lee, I. Yoon, J. Kim, H. Ihee, B. Kim and C. B. Park, Angew. Chem., Int. Ed., 2011, 50, 1164–1167 CrossRef CAS PubMed.
- M. Gdaniec and B. Liberek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1343–1345 CrossRef.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- H. Dong, H. Zhu, Q. Meng, X. Gong and W. Hu, Chem. Soc. Rev., 2012, 41, 1754–1808 RSC.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- S. P. Brown, Solid State Nucl. Magn. Reson., 2012, 41, 1–27 CrossRef CAS PubMed.
- M. R. Hansen, R. Graf and H. W. Spiess, Chem. Rev., 2016, 116, 1272–1308 CrossRef CAS PubMed.
- G. Teymoori, B. Pahari, B. Stevensson and M. Edén, Chem. Phys. Lett., 2012, 547, 103–109 CrossRef CAS.
- G. Teymoori, B. Pahari and M. Edén, J. Magn. Reson., 2015, 261, 205–220 CrossRef CAS PubMed.
- C. Sauvée, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Angew. Chem., Int. Ed., 2013, 52, 10858–10861 CrossRef PubMed.
- M. Veshtort and R. G. Griffin, J. Magn. Reson., 2006, 178, 248–282 CrossRef CAS PubMed.
- S. K. Zaremba, Ann. Mater. Pura Appl., 1966, 73, 293–317 CrossRef.
- H. Conroy, J. Chem. Phys., 1967, 47, 5307–5318 CrossRef.
- V. B. Cheng, H. H. Suzukawa and M. Wolfsberg, J. Chem. Phys., 1973, 59, 3992–3999 CrossRef CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter, 2001, 63, 245101–245113 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502–395520 CrossRef PubMed.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter, 1991, 43, 1993–2006 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Pseudopotentials were downloaded from http://sites.google.com/site/dceresoli/pseudopotentials in September 2015, X.pbe-tm-new-gipaw-dc.UPF, with X = H, C, N, O.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. W. Ponder and F. M. Richards, J. Comput. Chem., 1987, 8, 1016–1024 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. B. Barnes, G. De Paëpe, P. C. A. Van Der Wel, K.-N. N. Hu, C. G. Joo, V. S. Bajaj, M. L. Mak-Jurkauskas, J. R. Sirigiri, J. Herzfeld, R. J. Temkin and R. G. Griffin, Appl. Magn. Reson., 2008, 34, 237–263 CrossRef CAS PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- D. Lee, S. Hediger and G. De Paëpe, Solid State Nucl. Magn. Reson., 2015, 66–67, 6–20 CrossRef CAS PubMed.
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS PubMed.
- F. Blanc, L. Sperrin, D. A. Jefferson, S. Pawsey, M. Rosay and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 2975–2978 CrossRef CAS PubMed.
- D. Lee, G. Monin, N. T. Duong, I. Zamanillo Lopez, M. Bardet, V. Mareau, L. Gonon and G. De Paëpe, J. Am. Chem. Soc., 2014, 136, 13781–13788 CrossRef CAS PubMed.
- F. Blanc, L. Sperrin, D. Lee, R. Dervişoğlu, Y. Yamazaki, S. M. Haile, G. De Paëpe and C. P. Grey, J. Phys. Chem. Lett., 2014, 5, 2431–2436 CrossRef CAS PubMed.
- P. Wolf, M. Valla, A. J. Rossini, A. Comas-Vives, F. Núñez-Zarur, B. Malaman, A. Lesage, L. Emsley, C. Copéret and I. Hermans, Angew. Chem., Int. Ed., 2014, 53, 10179–10183 CrossRef CAS PubMed.
- A. S. Lilly Thankamony, C. Lion, F. Pourpoint, B. Singh, A. J. Perez Linde, D. Carnevale, G. Bodenhausen, H. Vezin, O. Lafon and V. Polshettiwar, Angew. Chem., Int. Ed., 2015, 54, 2190–2193 CrossRef PubMed.
- T. Gutmann, J. Liu, N. Rothermel, Y. Xu, E. Jaumann, M. Werner, H. Breitzke, S. T. Sigurdsson and G. Buntkowsky, Chem.–Eur. J., 2015, 21, 3798–3805 CrossRef CAS PubMed.
- F. A. Perras, U. Chaudhary, I. I. Slowing and M. Pruski, J. Phys. Chem. C, 2016, 120, 11535–11544 CAS.
- D. A. Hall, Science, 1997, 276, 930–932 CrossRef CAS PubMed.
- C. Ye, R. Fu, J. Hu, L. Hou and S. Ding, Magn. Reson. Chem., 1993, 31, 699–704 CrossRef CAS.
- D. H. Brouwer, P. E. Kristiansen, C. A. Fyfe and M. H. Levitt, J. Am. Chem. Soc., 2005, 127, 542–543 CrossRef CAS PubMed.
- D. H. Brouwer, R. J. Darton, R. E. Morris and M. H. Levitt, J. Am. Chem. Soc., 2005, 127, 10365–10370 CrossRef CAS PubMed.
- L. B. Krivdin and G. A. Kalabin, Prog. Nucl. Magn. Reson. Spectrosc., 1989, 21, 293–448 CrossRef CAS.
- E. Bouleau, P. Saint-Bonnet, F. Mentink-Vigier, H. Takahashi, J.-F. Jacquot, M. Bardet, F. Aussenac, A. Purea, F. Engelke, S. Hediger, D. Lee and G. De Paëpe, Chem. Sci., 2015, 6, 6806–6812 RSC.
- D. Lee, E. Bouleau, P. Saint-Bonnet, S. Hediger and G. De Paëpe, J. Magn. Reson., 2016, 264, 116–124 CrossRef CAS PubMed.
- S. Chaudhari, P. Berruyer, D. Gajan, C. Reiter, F. Engelke, D. Silverio, C. Copéret, M. Lelli, A. Lesage and L. Emsley, Phys. Chem. Chem. Phys., 2016, 18, 10616–10622 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02709a |
| ‡ These authors contributed equally to this work. |
| § Present address: Sir Peter Mansfield Imaging Centre, Department of Physics and Astronomy, University of Nottingham, NG72RD, UK. |
| This journal is © The Royal Society of Chemistry 2017 |