
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering monolayer poration for rapid exfoliation of microbial membranes†
Alice
Pyne‡
ab,
Marc-Philipp
Pfeil‡
ac,
Isabel
Bennett
 b,
Jascindra
Ravi
a,
Patrizia
Iavicoli
d,
Baptiste
Lamarre
a,
Anita
Roethke
e,
Santanu
Ray
a,
Haibo
Jiang
f,
Angelo
Bella
a,
Bernd
Reisinger
e,
Daniel
Yin
ac,
Benjamin
Little
g,
Juan C.
Muñoz-García
c,
Eleonora
Cerasoli
a,
Peter J.
Judge
c,
Nilofar
Faruqui
a,
Luigi
Calzolai
d,
Andre
Henrion
e,
Glenn J.
Martyna
h,
Chris R. M.
Grovenor
i,
Jason
Crain
ag,
Bart W.
Hoogenboom
b,
Anthony
Watts
c and
Maxim G.
Ryadnov
*a
b,
Jascindra
Ravi
a,
Patrizia
Iavicoli
d,
Baptiste
Lamarre
a,
Anita
Roethke
e,
Santanu
Ray
a,
Haibo
Jiang
f,
Angelo
Bella
a,
Bernd
Reisinger
e,
Daniel
Yin
ac,
Benjamin
Little
g,
Juan C.
Muñoz-García
c,
Eleonora
Cerasoli
a,
Peter J.
Judge
c,
Nilofar
Faruqui
a,
Luigi
Calzolai
d,
Andre
Henrion
e,
Glenn J.
Martyna
h,
Chris R. M.
Grovenor
i,
Jason
Crain
ag,
Bart W.
Hoogenboom
b,
Anthony
Watts
c and
Maxim G.
Ryadnov
*a
aNational Physical Laboratory, Teddington, Middlesex TW11 0LW, UK. E-mail: max.ryadnov@npl.co.uk
bLondon Centre for Nanotechnology and Department of Physics and Astronomy, University College London, London WC1E 6BT, UK
cDepartment of Biochemistry, University of Oxford, Oxford OX1 3QU, UK
dEuropean Commission, Joint Research Centre, Institute for Health and Consumer Protection, Ispra (VA), Italy
ePhysikalisch-Technische Bundesanstalt, 38116 Braunschweig, Germany
fCentre for Microscopy, Characterisation and Analysis, The University of Western Australia, Crawley, Western Australia 6009, Australia
gSchool of Physics and Astronomy, University of Edinburgh, Edinburgh EH9 3JZ, UK
hIBM T. J. Watson Research Center, Yorktown Heights, NY 10598, USA
iDepartment of Materials, University of Oxford, Oxford OX1 3PH, UK
First published on 26th September 2016
Abstract
The spread of bacterial resistance to traditional antibiotics continues to stimulate the search for alternative antimicrobial strategies. All forms of life, from bacteria to humans, are postulated to rely on a fundamental host defense mechanism, which exploits the formation of open pores in microbial phospholipid bilayers. Here we predict that transmembrane poration is not necessary for antimicrobial activity and reveal a distinct poration mechanism that targets the outer leaflet of phospholipid bilayers. Using a combination of molecular-scale and real-time imaging, spectroscopy and spectrometry approaches, we introduce a structural motif with a universal insertion mode in reconstituted membranes and live bacteria. We demonstrate that this motif rapidly assembles into monolayer pits that coalesce during progressive membrane exfoliation, leading to bacterial cell death within minutes. The findings offer a new physical basis for designing effective antibiotics.
Introduction
Membrane poration is fundamental to cell function.1 Bacterial exotoxins (cytolysins) porate phago- and hemocytes,2 cytolytic mediators (perforins) form transmembrane channels in infected and neoplastic cells to activate intrinsic suicide programs,3 and large nuclear pore complexes that cross the nuclear envelope facilitate transport in and out of the cell nucleus.4 The spread of antimicrobial resistance re-emphasizes poration as a means of counteracting opportunistic infections, with a specific focus on evolutionarily conserved host defense or antimicrobial peptides (AMPs).5 The peptides, of which there are >1000 reported to date,6 preferentially bind to anionic microbial membranes in which they fold into amphipathic structures by partitioning cationic and hydrophobic amino-acid side chains.7 This property enables them to assemble into transmembrane structures that coalesce into permanent, transient or expanding pores.8–10 Poration mechanisms vary but all appear to involve an inactive surface-bound (“S”) state followed by a pore-like insertion (“I”) state.11 Peptide monomers are distributed both on and in membranes and tend to bind more strongly to the edges of forming pores,12 promoting pore expansion and merger.10 Membrane thinning and positive curvature may precede poration,13 which at non-antimicrobial concentrations of AMPs can induce small transient pores without causing cell lysis.14 At antimicrobial concentrations, the exposed outer monolayer may alone support the progressive migration of peptide monomers.10,15 The rate and mode of migration, with or without poration, can then be facilitated by the relative orientation of the monomers to the membrane surface, which can allow them to remain positioned in the monolayer16 or in the interface.17 Indeed, AMPs adopt different orientations in membranes that range from flat and perpendicular to oblique or tilted,12,16,18 while peptide lengths often exceed the thickness of the bilayer or are too short to span it.7 Consequently, tilted conformations may offer structural motifs that are predisposed to monolayer rather than bilayer poration. Transmembrane pores form hydrophilic channels that interface the extra- and intra-cellular environments, both of which are aqueous. These pores can equilibrate, stabilize and expand. In contrast, monolayer pores expose the hydrophobic core of the bilayer to water. Such an energetically unfavorable configuration, which may only partially be compensated by peptide-water networks formed in the monolayer,19 would cause complex changes that microbial membranes cannot accommodate thus inducing a cost of fast and strong antimicrobial effects. Here we test this prediction and provide the first direct evidence of the phenomenon.Results and discussion
Design rationale
Our approach adapts the principles of “inverse protein folding”20 by designing an antimicrobial sequence that folds with the formation of a desired structure, a monolayer pore. The sequence used here is an archetypal AMP that was converted from an antimicrobial helical insert (amhelin)10 into a tilted antimicrobial insert (tilamin). Both peptides comprise neutral polar or small (N), polar cationic (C) and hydrophobic (H) residues that arrange into a contiguous amphipathic helix of ∼3.15 nm in length (0.54 nm per helical turn)21 (Fig. 1A). This arrangement allows the helix to span microbial phospholipid bilayers of ∼3–4 nm in thickness.22–24 However, unlike transmembrane amhelin,10 the tilamin helix has to insert only half-way through the bilayer (Fig. 1B). To achieve this, tilamin was designed to exploit charge interplays with phospholipids25 while adopting a tilted orientation in the outer leaflet of the bilayer (Fig. 1B).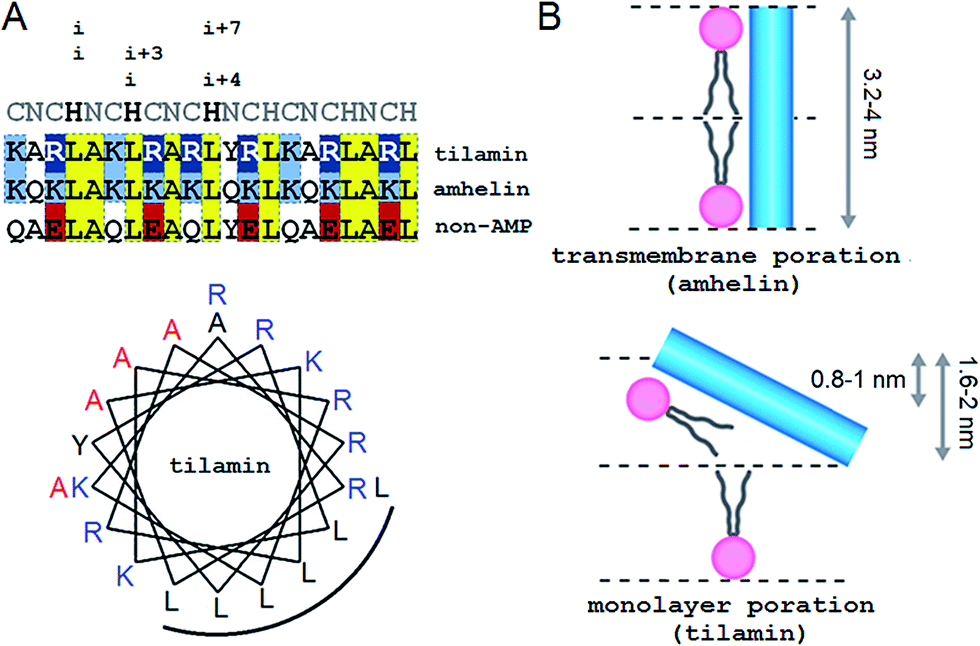 | ||
| Fig. 1 Tilted antimicrobial helix (tilamin): design and proposed mechanism of membrane insertion. (A, top) Peptide sequences aligned with helical repeats, CNCHNCH, in which hydrophobic residues are separated at i, i + 3 and i, i + 4 helical spacings. Cationic lysines and arginines are in blue and dark blue, respectively, anionic residues in red and absolute identities in yellow. (A, bottom) Tilamin sequence configured onto an α-helical wheel with 3.6 residues per turn. Cationic residues are in blue and the hydrophobic face is underlined. Alanines deuterated for GALA scans are in red. Note: the helical wheel does not reflect the exact spread of the side chains. (B) Schematics of pore edges showing depths of insertion for proposed monolayer and transmembrane poration mechanisms. For clarity, only one peptide (blue cylinder) and one phospholipid per layer are shown (aliphatic chains in grey, headgroups in pink). | ||
These peptide–phospholipid charge interplays derive from differences between cationic lysine and arginine residues, which are primary electrostatic binders to anionic microbial membranes. These amino acids have different chemistries and different free energies of hydration, but both have to overcome the same free energy barrier when crossing membranes in protonated forms. For neutral lysine, which in membranes can remain deprotonated, the barrier is lower than that for neutral arginine.25 Therefore, protonated arginines are favored at all stages of membrane insertion, and starting with surface binding they maintain extensive hydrogen-bonding networks that trap more phosphate and water.16,25 Each arginyl residue can provide five hydrogen-bond donors stabilizing peptide–phosphate clusters and thereby enhancing the affinity between the peptide and the membrane. To enable the formation of electrostatic networks along the helix while inserting in membranes, arginines were evenly distributed in the tilamin sequence. A high concentration of arginines was allowed in the central part of the peptide to arrest its interfacial binding, which was further stabilized by the short hydrophobic face (Fig. 1A).10 To avoid undesired hemolytic effects common for venom peptides (melittin) that have broad hydrophobic faces,7,10 the charge-hydrophobic contributions to the binding were balanced at a 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by splitting the polar face into two unequal sub-faces via a neutral alanyl cluster placed opposite to the hydrophobic face. A bulky and hydrophobic tyrosine with propensity for slight β-bends was incorporated into the alanyl cluster in the central part of the helix to help the termini adapt to membrane curvature. The resulting sequence comprises three helical heptads, CNCHNCH, which arrange the same-type residues (i and i + 7 spacing) next to one another when viewed along the helical axis. This spacing contains i, i + 3 and i, i + 4 helical patterns characteristic of canonical coiled coils, known to maintain interfacial contacts in a highly cooperative manner,24 and thus able to support cooperative tilamin assembly in lipid bilayers. Since tilamin is a helical design, all amino acids used have high helical propensity. Finally, an anionic version of tilamin was designed as a non-AMP control (Fig. 1A). This control is important for two reasons. Firstly, the peptide has helical propensity that is comparable to that of tilamin. But, secondly, it lacks the ability to bind to membranes and thence cannot undergo folding-mediated insertion into bacterial lipid bilayers, which renders it non-antimicrobial. Thus, all three designed peptides are cross-referenced controls, with each responsible for a specific mode of interactions with microbial membranes: from no interactions (non-AMP) to folding-mediated transmembrane (amhelin) and monolayer (tilamin) poration.
:1 ratio by splitting the polar face into two unequal sub-faces via a neutral alanyl cluster placed opposite to the hydrophobic face. A bulky and hydrophobic tyrosine with propensity for slight β-bends was incorporated into the alanyl cluster in the central part of the helix to help the termini adapt to membrane curvature. The resulting sequence comprises three helical heptads, CNCHNCH, which arrange the same-type residues (i and i + 7 spacing) next to one another when viewed along the helical axis. This spacing contains i, i + 3 and i, i + 4 helical patterns characteristic of canonical coiled coils, known to maintain interfacial contacts in a highly cooperative manner,24 and thus able to support cooperative tilamin assembly in lipid bilayers. Since tilamin is a helical design, all amino acids used have high helical propensity. Finally, an anionic version of tilamin was designed as a non-AMP control (Fig. 1A). This control is important for two reasons. Firstly, the peptide has helical propensity that is comparable to that of tilamin. But, secondly, it lacks the ability to bind to membranes and thence cannot undergo folding-mediated insertion into bacterial lipid bilayers, which renders it non-antimicrobial. Thus, all three designed peptides are cross-referenced controls, with each responsible for a specific mode of interactions with microbial membranes: from no interactions (non-AMP) to folding-mediated transmembrane (amhelin) and monolayer (tilamin) poration.
Measurement strategy: a molecular-cellular scale continuum
To experimentally test the hypothesis of monolayer poration requires a combinatory approach comprising different measurement methods that are best performed in a continuum across relevant length scales. Given that individual methods have their own limitations and advantages, each measurement can address a specific peptide property, physico-chemical, mechanistic or biological, and at given length and temporal scales. In such a continuum, all methods are posed to generate findings that complement one another, which help to compile information as exhaustive as necessary for the demonstration of the phenomenon. With this in mind, we fully characterized the behavior of tilamin in biological and model membrane environments by performing complementary measurements in a molecular-to-cellular scale continuum, while providing computational atomistic and molecular descriptions of monolayer pore formation. Fig. 2 schematically illustrates our measurement strategy.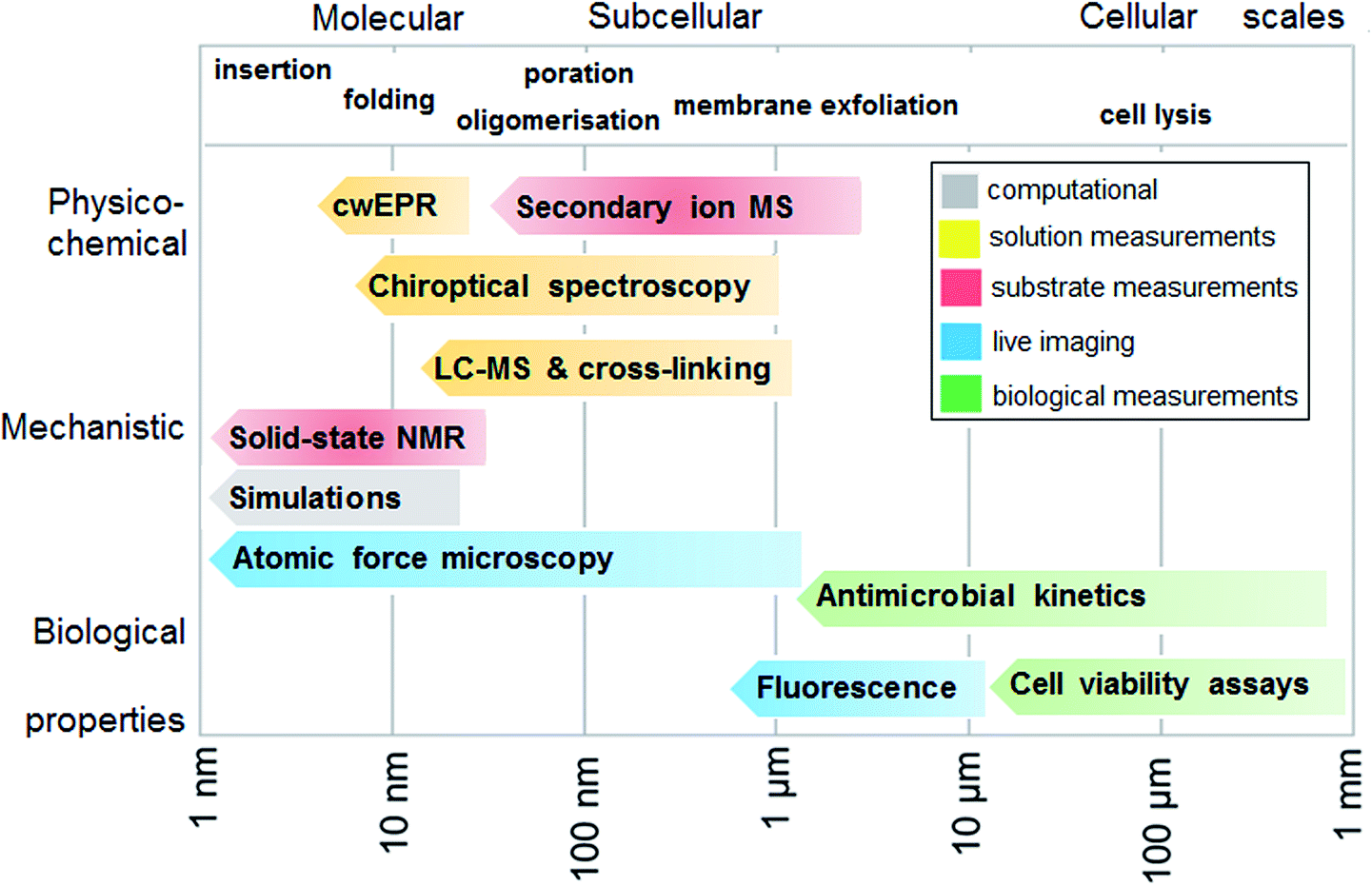 | ||
| Fig. 2 Measurement continuum. A schematic representation of a molecular-to-cellular scale measurement continuum used to demonstrate antimicrobial monolayer poration. Key: cwEPR: continuous wave electron paramagnetic resonance, MS: mass spectrometry, LC-MS: liquid-chromatography mass spectrometry, NMR: nuclear magnetic resonance spectroscopy. | ||
Biological activity
Consistent with design, tilamin was found to be non-hemolytic and antimicrobial, with Minimum Inhibitory Concentrations (MIC) typical of polypeptide antibiotics, while the non-AMP was biologically inactive (Table S1 in ESI†).Tilamin exhibited comparably strong activities against all the bacteria tested with no apparent preference for Gram positive or Gram negative bacteria. By contrast, the activities of amhelin and a naturally occurring AMP, cecropin B,26 were less consistent across different bacteria: Gram positive bacteria tended to be more tolerant to these two peptides and both these peptides were inactive against S. aureus (Table S1†). The differences did not appear systematic and varied from one bacterium type to another, which may be attributed to reversible or phenotypic tolerance to antibiotics.27,28 Similar trends were observed for commercial membrane-active polypeptide antibiotics, daptomycin and polymyxin B, which can preferentially target Gram positive and Gram negative membranes, respectively (Table S1†). Although daptomycin was inactive against Gram negative bacteria, while polymyxin B was toxic to all of these, E. faecalis proved to be tolerant to both antibiotics (Table S1†). Further, tilamin and daptomycin were the only agents with appreciable activity against S. aureus, though daptomycin proved to be marginally hemolytic (Table S1†). Given that among the tested antibiotics only tilamin was non-differentially antimicrobial, the observed variations suggest that the peptide should exhibit a distinctive antimicrobial mechanism.7 In native environments AMPs are secreted to respond to microbial challenge within their proteolytic life time (minutes). Therefore, the mechanism should become apparent within the same timescale. MIC assays are not suitable in this regard as these are optical density measurements performed over much longer periods of time (24 hours) and take no account of changes at the cellular level. To mitigate this, we sought additional evidence.
Firstly, we monitored bacterial cell death by live-dead assays using fluorescence microscopy.10,26 Tilamin caused up to 80% lysis rates at the MIC within the first 30 min for E. coli (Fig. S1†). These were noticeably faster than those of cecropin B and amhelin (both <30%) in the same timeframe, while comparable rates were only seen for amhelin at concentrations three times above MIC (Fig. S1†). Secondly, to better understand the mechanistic basis of the killing kinetics we monitored individual cell lysis by time-lapse atomic force microscopy (AFM) in water. Gram negative bacteria, such as E. coli, present ideal models for this purpose.29 These cells have two distinct membranes (outer and inner or cytoplasmic) with the bilayer thicknesses of 3.7–4.1 nm.23 A thin and highly porous peptidoglycan layer (4 nm) separates the two membranes,30 but cannot protect the inner membrane.31 Moreover, the layer exhibits high affinity to cationic AMPs thereby facilitating their entry to the cytoplasmic membrane.32
Therefore, if tilamin causes poration it should affect both membranes. Consequently, in the context of monolayer poration, one should distinguish the effect of the peptide on the outer leaflets, which are present in both outer and inner membranes, from that on the outer membranes. This may prove to be important as the outer membranes are affected first, which is likely to be associated with their more apparent or advanced removal during tilamin treatment.
Monolayer poration in live bacteria and progressive removal of the outer membranes
Indeed, this was observed. AFM revealed the progressive removal of the outer membrane of E. coli within the first minutes of tilamin treatment (Fig. 3, S2A and B†). Fluorescence microscopy confirmed a nearly complete lysis of E. coli cells within 15 min, which agreed well with the results from the live-dead assays (Fig. S2C†). A clear separation between the inner and outer membranes was also evident. The height of the separation matched the expected depth of the outer cell-wall layer when the outer membrane and peptidoglycan are combined (∼8 nm) (Fig. 3A). In both outer and inner membranes, pore-like features with depths of ∼2 nm were observed during imaging (Fig. 3B, C and S3†). Consistent with our prediction of monolayer poration, these pores were small, but abundant, forming rapidly and continuously. This formation imposed an energetically unfavorable configuration on the bilayer, which subsequently caused localized membrane rupture (Fig. 3A–C, S2B and S3†). The latter manifested as deeper holes or ridges piercing through the membrane layers (Fig. S3B and C†), as the bacterial membranes disintegrated within minutes (Fig. 3D, area 2). Taken together, the results suggest that tilamin elicits rapid antimicrobial responses via monolayer poration, leading to the fast and progressive disintegration of bacterial membranes.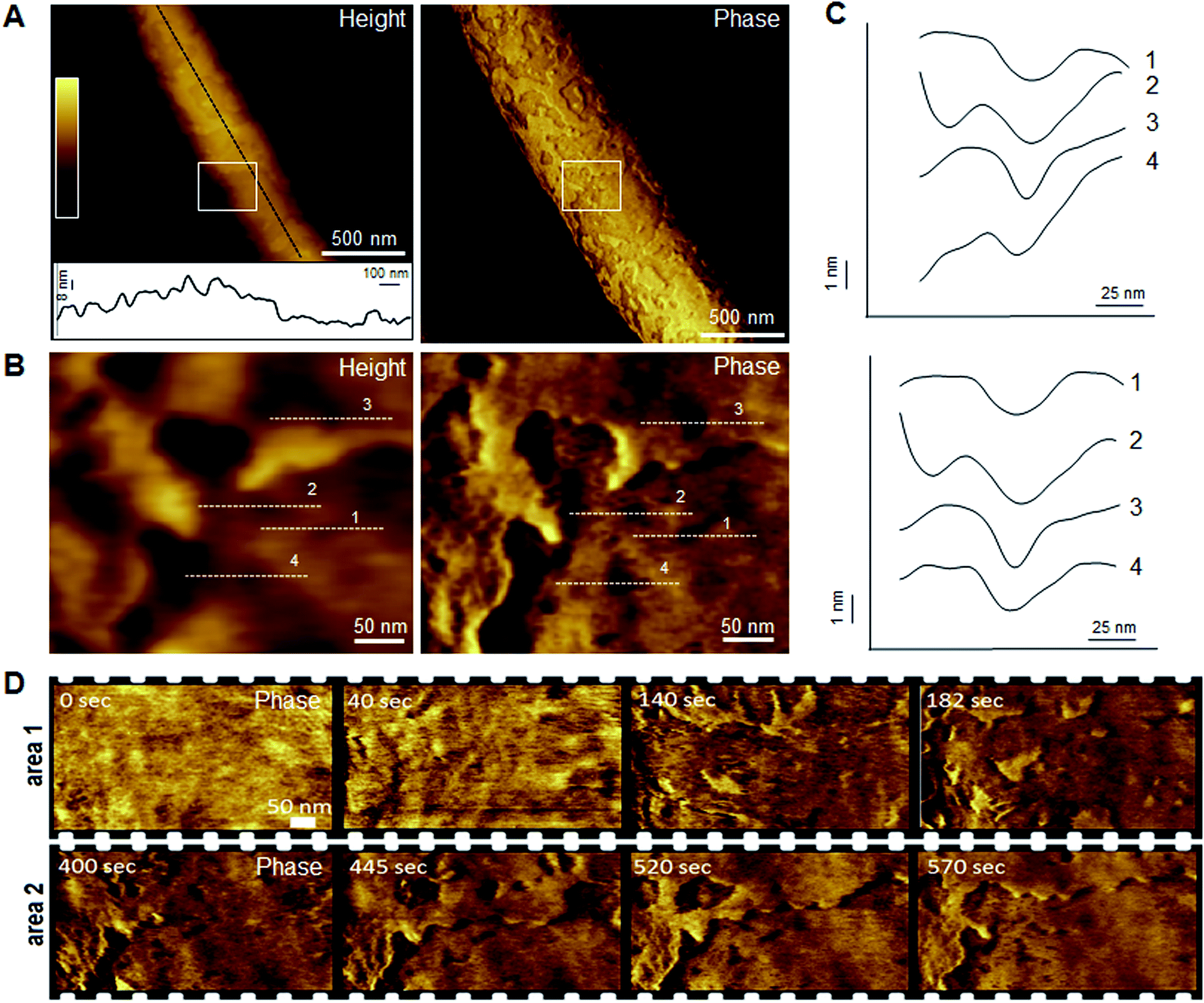 | ||
| Fig. 3 Monolayer poration and progressive membrane removal in live bacteria. (A) Topographic (height) and phase AFM images of live E. coli in water taken at the first 780 s incubation with tilamin. Inset in height image: a height profile taken along the black dotted line on the back of the bacterium, showing 8 nm-deep steps from the outer cell-wall layer to the inner membrane. The outer layer includes the outer membrane (3.7–4.1 nm)23 and a periplasmic peptidoglycan layer (4 nm). (B) High resolution topographic and phase images of the areas marked by white boxes in (A), after correction for background curvature in the height data (see ESI†). These images show exfoliation of the outer membrane and poration in both outer and inner membrane. (C) Height profiles taken along the highlighted lines in (B) (top), and after additional local background subtraction (bottom). These data show the depth profiles of individual pores. (D) High resolution phase images of E. coli during incubation with tilamin, as a function of time (for the corresponding height images, see Fig. S2†). Two different areas are shown. Time-point labels indicate time since injection. Colour scale for height data, see (A) 140 nm (A) and 19 nm (B). Incubation conditions: 3.5 μM, pH 7.4, room temperature (rt). | ||
Up to this point, tilamin was shown to porate both membranes, but had to affect the outer membranes to induce visible poration in the inner membranes. As a result, the outer membranes underwent a more rapid disintegration, prompting a question of exact pore dynamics in isolated lipid bilayers. Similarly, the impact of affecting intracellular processes that are crucial to cell viability cannot be fully excluded in live bacteria measurements.7 It should be noted however that intracellular targeting is the consequence of membrane permeabilization and may not outcompete poration within the observed timescale of membrane rupture. Indeed, traditional antibiotics, which typically engage with intracellular targets, exhibit much slower killing kinetics that are strictly proportional to the rate of bacterial growth.33 This is unlike membrane-permeabilizing AMPs, which attack both growing and mature bacteria.7
Considering the two points together, it was deemed appropriate to relate the killing and poration rates in live bacteria with the membrane-porating effects of tilamin in isolation from inter-membrane and intracellular processes. To achieve that, we probed the action of tilamin in reconstituted membranes using lipid compositions that yield fluid-phase membranes at booth room and physiological temperatures.34
Probing outer-leaflet poration in reconstituted membranes
Reconstituted membranes were used to investigate the effect of membrane composition on the binding and activity of tilamin. Anionic unilamellar vesicles (AUVs) and zwitterionic unilamellar vesicles (ZUVs) provided microbial and mammalian membrane mimetics, respectively. 1,2-Dilauroyl-sn-glycero-3-phosphocholine (DLPC) was used to assemble ZUVs, whereas its 3:1 molar mixtures with 1,2-dilauroyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DLPG) were used to assemble AUVs. As gauged by circular dichroism spectroscopy, tilamin was appreciably helical in AUVs but did not fold in aqueous buffers and in ZUVs (Fig. S4A†). Linear dichroism spectroscopy, which probes relative peptide orientation to membrane surfaces,35 confirmed tilamin binding to AUVs. The observed band patterns were similar to those reported for membrane-inserting aureins and melittins35,36 indicating helix orientation more parallel to the membrane normal (Fig. S4B†). As expected, the non-AMP did not fold or order under any of the conditions used (Fig. 1A and S4†). The data hence confirms folding-mediated poration by tilamin in response to microbial membrane binding.
To visualize the process, supported lipid bilayers (SLBs) were prepared by the surface deposition of the dilauroyl (DLPC/DLPG, 3:1 molar ratio) AUVs on appropriate substrates.10 These preparations had the additional advantage – compared to whole bacteria – of being flat (to within ∼0.1 nm) in their unperturbed state, allowing more accurate depth measurements for tilamin-induced pores by AFM. AFM analyses of SLBs that were fixed after the 15 minutes of tilamin treatment revealed pore-like structures of varying diameters (Fig. 4A). The pores tended to fuse into heterogeneous networks suggesting much faster diffusion through the supported membrane matrix than that of expanding transmembrane pores of amhelin.10
 | ||
| Fig. 4 Monolayer poration in reconstituted membranes. Topographic (AFM) and chemical (NanoSIMS) imaging of DLPC/DLPG (3:1, molar ratio) SLBs. (A) In-air AFM topographic images with a cross-section along the highlighted line (green) shown for the inset. Colour scales: 3 nm for the topography images; and 15 degrees for the phase image. Incubation conditions: 10 μM, pH 7.4, rt. (B) Topography of supported lipid bilayers measured by AFM in aqueous solution during incubation with tilamin where 0 min corresponds to the time of tilamin injection. Colour scale: 3 nm. Incubation conditions: 1.8 μM, pH 7.4, rt. (C) SIMS images of 12C15N−/12C14N− ratio obtained for the tilamin-treated DLPC/DLPG (3:1, molar ratio) SLBs, and expressed as hue saturation images, which are the sums of sequential images to enhance the statistical significance of the measured ratios. The rainbow scale changes from blue (natural abundance of 0.37%) to pink (10%). Insets for tilamin-treated bilayers highlight pore clusters (middle and right), and no clusters in untreated bilayers (left). Incubation conditions: 10 μM, pH 7.4, rt. | ||
Complementary to these findings, AFM in aqueous solution and in real time showed that the networks were complete within the first 15 min of incubation at the concentrations used (Fig. 4B). The pore depths of ∼1.4 nm were remarkably consistent in the networks and appeared independent of pore sizes and incubation times (>2 hours) (Fig. 4A, B, S5A and B†). This was in contrast to amhelin (Fig. 1A and B) serving for tilamin as a transmembrane control, which can bind to and fold in the microbial membranes, but which porates them by spanning the whole bilayer (Fig. S6†).10 Comparable results of monolayer poration were obtained for thicker SLBs (∼4 nm) assembled as 3:1 molar mixtures of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) lipids (Fig. S5C†). In these SLBs, similarly sized pores of 1.2–1.8 nm in depth were formed in abundance at the same time scales as observed on live bacteria and for the DLPC/DLPG (3:1, molar ratio) SLBs. Quantitative analyses of pore size dynamics measured at different depth thresholds showed close distributions of overlapping pore diameters supporting the coalescence of individual pores into larger structures and networks (Fig. S7†). In addition, the coalescence did not have an appreciable effect on pore depths, with depth values being universal across all pores, individual and coalescing (Fig. S7†). These depths agree with the expected difference between a bilayer surface and the hydrophobic surface of an inner monolayer as measured by AFM,37 and thus indicate the local removal of the outer leaflet of the membrane (Fig. 1B).
The pore networks were prone to convert into regions of higher pore densities, the component specificity of which was explicitly confirmed by nanoscale resolution secondary-ion mass spectrometry (NanoSIMS). Fig. 4C shows 12C15N−/12C14N− ratio images obtained from the surfaces of the DLPC/DLPG (3:1, molar ratio) SLBs treated with a 15N-labelled tilamin. Ratios above natural abundance values (0.37%) were evident for larger pore clusters where the peptide content was the highest (10%). The lateral resolution of NanoSIMS, <100 nm38 did not permit the detailed visualization of smaller pores. Outside of the larger clusters, these appeared as high ratio constellations spread across the imaged areas thus supporting the mechanism of continuous and heterogeneous peptide migration (Fig. 4C). Consistent with this, mass spectrometry and gel electrophoresis analyses of tilamin-treated DLPC/DLPG (3:1) AUVs that were subjected to amine-specific cross-linking returned broad mass distributions (Fig. S8A–C†).39 The lowest oligomeric forms that could be assigned comprised 4–7 monomers. These oligomers correlated well with the constellations observed by NanoSIMS and the smallest pores of 5–10 nm measured by AFM in aqueous solution (Fig. S8D†). Quantified size distributions at different depths below the membrane surface to ensure consistency in measurements gave mean radii of the smallest pores in the range of 1–6 nm, i.e. mean diameters of 2–12 nm (Fig. S9†).
Membranes as primary targets for tilamin
As shown in live bacteria, monolayer poration leads to the rapid lysis of bacterial cells (Fig. 3 and S1–S3†). Yet, microorganisms incorporate other structures and processes that can be potential targets for tilamin. To further emphasize that lipid bilayers themselves constitute the primary targets for tilamin, we performed a synthetic version of the live-dead bacterial assays using giant unilamellar vesicles (GUVs) and monitored tilamin action in real time by fluorescence microscopy. GUVs represent minimum membrane models of biological cells and are composed of a phospholipid bilayer membrane and an aqueous interior.40 The same lipid composition of POPC/POPG (3:1, molar ratio) was used. The resulting vesicles have no interior targets, thereby excluding an intracellular mechanism, and lack any other membrane components, which provides the necessary focus on the lipid bilayers. A co-staining strategy was applied to visualize the vesicles by selectively staining their membranes (CellMask™) and interior (sulforhodamine B) (Fig. 5 and Video S1†). Tilamin was found to rapidly disintegrate the GUVs under the same timescales as observed in the other experiments (Fig. 5, S10A and Video S2†). Carboxyfluorescein-labelled peptide co-localised with the membrane dye, but not with the interior dye, confirming the membrane targeting mechanism of tilamin (Fig. 5, S10B and Video S3†). To form shallow pores (Fig. 4) tilamin has to splay the lipids in the outer monolayer and induce positive membrane curvature that can result in budding.41 The effect was strikingly apparent for tilamin-treated vesicles (Fig. 5, S10A and B†). It appears that by intercalating in the outer leaflet tilamin introduces hydration shells that support long-range orientational correlations between lipids in areas (pores) affected by the peptide. This enables these areas to bulge, resembling effects observed along the boundaries of liquid-ordered and liquid-disordered lipid phases40 (Fig. 5 and S10†). This effect then evolved into abrupt dye release, as opposed to a gradual leakage (Fig. S10A, Videos S2 and S3†), with the continuous bulging of collapsed GUVs also evident (Fig. S10C†). The latter suggests a scaffolding mechanism, reminiscent to that of amphiphysin N-BAR domains splaying lipids in the outer monolayer,41 which, possibly via monolayer stalks, may promote the merger of positively curved domains at the sites of coalescing tilamin pores. The observations on tilamin-induced poration and rupture of phospholipid bilayers in live and reconstituted membranes are strikingly consistent. The dimensions of the observed pores and the rates of their formation were comparable in bacterial and synthetic membranes, while the conserved depth profiles of the pores and the tilamin-induced budding in GUVs suggest a particular orientation of the peptide in the outer leaflet of the bilayer.
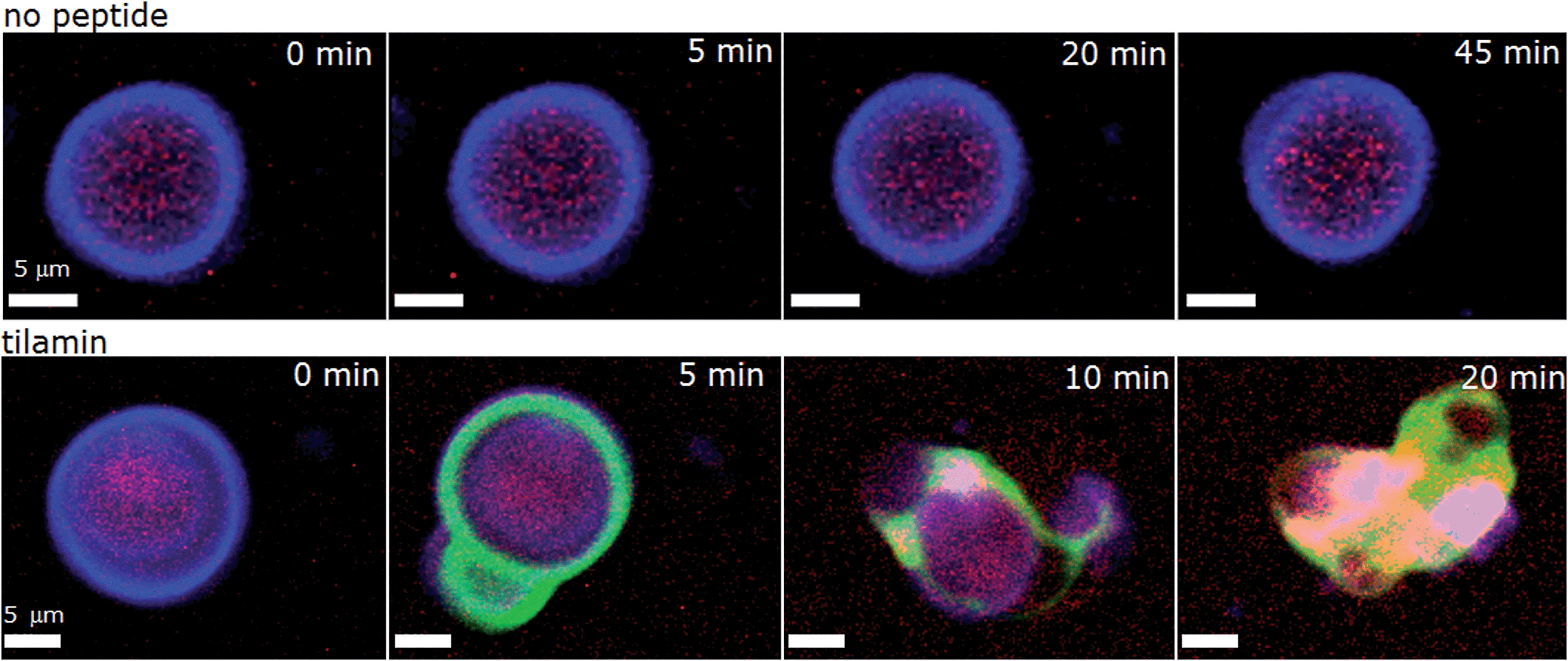 | ||
| Fig. 5 Tilamin-induced lysis of giant unilamellar vesicles. Fluorescence micrograph of GUVs assembled from POPC/POPG (3:1, molar ratio) and incubated in phosphate-buffered saline without (upper) and with (lower) tilamin (10 μM, L/P 100). Key: violet is CellMask™ Deep Red staining GUV membranes, red is for sulforhodamine B staining the interior of the GUVs, green is for carboxyfluorescein-labelled tilamin. See also corresponding Videos S1 and S3.† | ||
Monolayer poration correlates with insertion mode
To clarify the insertion mode of tilamin, four tilamin sequences, each deuterated at a single alanine (Fig. 1A), were subjected to the geometric analysis of labelled alanines (GALA) by oriented solid state NMR (ssNMR) spectroscopy42,43 (Fig. S11†). An overall tilt of 70° (equivalent to 110° for the opposite terminus) with respect to the membrane normal was derived from 2H quadrupolar splittings obtained for tilamin in DLPC/DLPG (3:1, molar ratio) AUVs (Table S2†). With the peptide dynamics being within the timescale of the NMR measurements, the tilted helix appeared as an equilibrium ensemble of close angles implying adaptability to variations in bilayer thickness. Indeed, quadrupolar splittings obtained in the thicker POPC/POPG (3:1, molar ratio) AUVs, gave comparable tilts (Tables S2–S4, Fig. 6A and S12†).
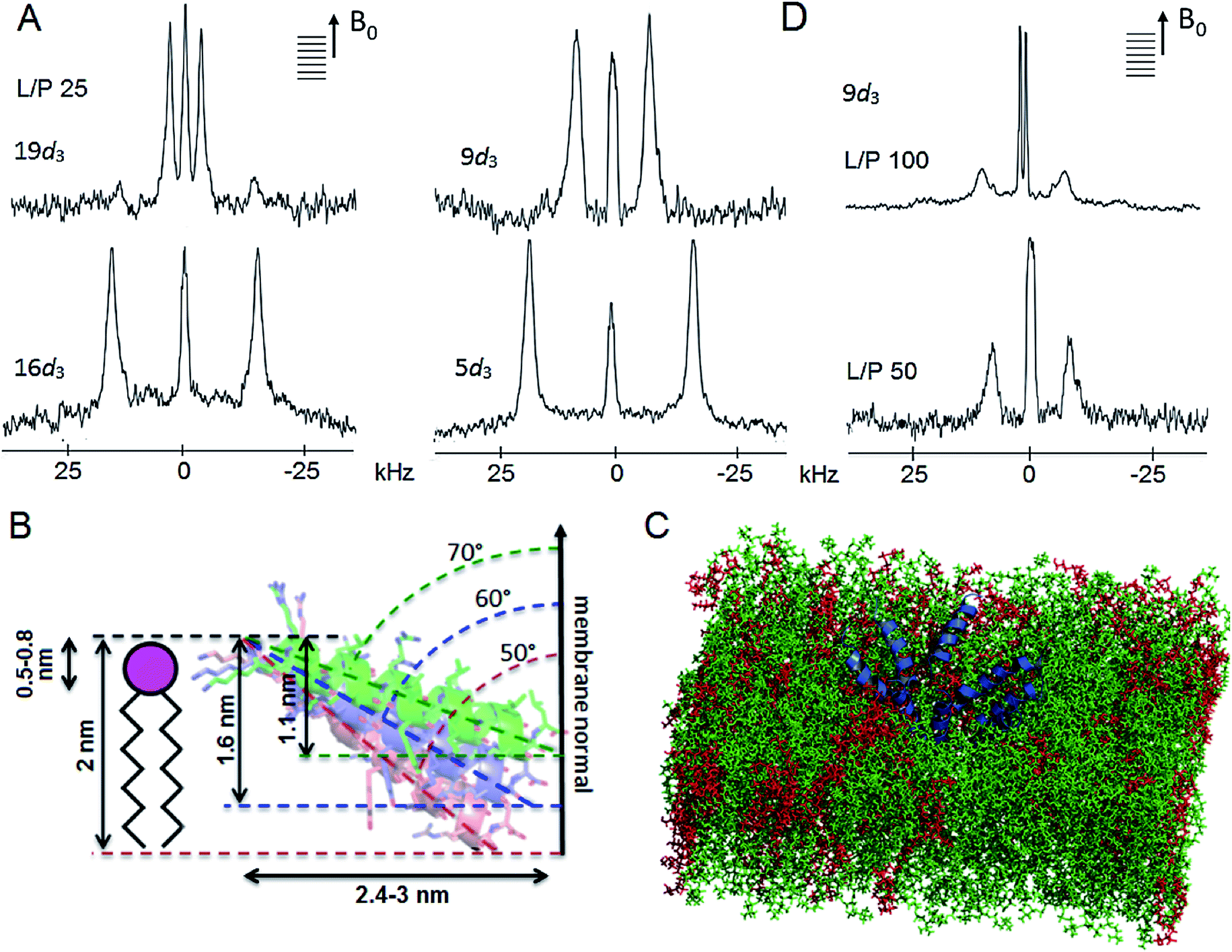 | ||
| Fig. 6 Tilamin insertion mode. (A) Oriented 2H NMR quadrupolar splittings (ΔvQ) in palmitoyl (POPC/POPG, 3:1 molar ratio) membranes for tilamin deuterated (Ala-d3) at positions 19, 16, 9, 5 at a lipid-peptide (L/P) ratio 25. (B) A GALA-derived and MD-derived ensemble of helix tilt angles for tilamin in the outer leaflet of the phospholipid bilayer showing tilt angles with corresponding insertion depths and horizontal spans of the tilted helix. The helices are not to scale. The helix insertion by the C-terminus (solution 2) is shown (Table S4†). (C) A rudimentary low-oligomer pore constructed by coarse-grained molecular dynamics simulations and converted to atomistic coordinates in a newly equilibrated palmitoyl (POPC/POPG, 3:1 molar ratio) membrane. Membrane slabs are oriented to view the peptide tilts. Peptides, POPC and POPG lipids are in blue, green and red, respectively. (D) Oriented 2H NMR quadrupolar splittings (ΔvQ) in palmitoyl (POPC/POPG, 3:1 molar ratio) membranes for tilamin deuterated (Ala-d3) at position 9 at L/P ratios 100 and 50. | ||
The root-mean-square deviation (RMSD) plots combining the 2H data with 15N spectra for tilamin labelled at the central tyrosine residue, which provided additional orientational constraints,44 gave overlapping regions of orientational and tilt angles revealing two possible solutions of tilamin insertion (Fig. S13A, B and Table S4†). Both solutions tilted by ∼20° relative to the bilayer plane, which makes helix insertion by the N- or C-terminus equally possible. The inserting end of tilamin for solution 1 (110°) is the N-terminus, whereas for solution 2 (70°) it is reversed (Fig. S13C and Table S4†). Simulated 2H quadrupolar splittings using Spinach software45 returned close fits with the experimental data confirming the solutions (Table S5, Fig. S14A and B†). Complementary to this, atomistic molecular dynamics simulations of a single tilamin helix inserted into the DLPC/DLPG (3:1) bilayer produced similar angle distributions (110–130°) after simulations of 500 ns (Fig. S14C†). Taken together these results are notable for two reasons.
Firstly, the NMR-derived insertion angles match the depth profiles of monolayer pores measured by AFM. With the tilamin helix (3.15 nm) at tilts of 70°, 60° and 50° (or, equivalently, 110°, 120° and 130° for the opposite terminus) with the respect to the membrane normal, its projection onto the membrane normal gives the insertion depths of 1.1, 1.6 and 2 nm, respectively (Fig. 6B and S15†). These orientations yield helix projections of 2.4–3 nm in length in the membrane plane (Fig. 6B), thus giving a minimum of 5–6 nm for a pore with two opposing helices spanning its diameter. The span constitutes a half of a minimal pore facing an opposite helix, i.e. 5–6 nm (Fig. 6B and S15†), which is consistent with the smallest pores of 5–10 nm assembled from low peptide oligomers (Fig. S8†). Such low-oligomer tilamin pores in POPC/POPG (3:1 molar ratio) bilayers were simulated by coarse-grained molecular dynamics, which revealed that transmembrane configurations were not stable: individual peptides tended to move towards one leaflet generating relatively symmetrical monolayer pores, which in newly equilibrated atomistic model membranes46 appeared as partially stabilized in the upper leaflet (Fig. 6C).
Secondly, the palmitoyl POPC/POPG (3:1 molar ratio) and live cell membranes contain unsaturated lipids, which alone cannot support an orientational order,40,41 and their thicknesses exceed the span of the folded tilamin. Therefore, the membranes are unable to fully support transmembrane orientation of the peptide without distorting the bilayer. The dilauroyl DLPC/DLPG (3:1, molar ratio) membranes comprise saturated lipids, are more densely packed and their thickness (3.2 nm) can match the complete transmembrane span of the folded tilamin.10,22 Despite such differences, tilamin promotes the same poration mechanism in all the membranes used. The tilted helix thus appears as an equilibrium configuration that adapts in the outer layer of the bilayer by maintaining close charge interplays with phospholipids. Supportive of this conjecture ssNMR spectra obtained in deuterated palmitoyl POPC/POPG (3:1 molar ratio) membranes showed greater disordering effects in lipid headgroup regions compared to tail regions (Fig. S16†). In accord with this, continuous wave electron paramagnetic resonance (cwEPR) showed appreciable reduction in amplitude of motion about the lipid chain axis at the headgroup regions (Fig. S17†). At decreasing L/P ratios (increasing peptide) tilamin induced clear segmental ordering in lipids that were spin-labelled at the carboxyl ends (nitroxide spin labelled phosphatidylcholine at position 5, 5-PCSL, versus 12-PCSL) (Fig. S17†). Similarly, the same decreasing L/P ratios led to reduced 2H quadrupolar splittings reflecting a more pronounced membrane disruption upon binding more peptide (Table S2, Fig. 6D, S16 and S18†).
Taken together the reduced amplitude of motion, observed at the nanosecond timescale of cwEPR, and the disordering of acyl chains, occurring at the microsecond timescale of 2H NMR, suggest that tilamin tilts into the bilayer and perturbs its upper leaflet across a range of bio-molecular timescales. Atomistic molecular dynamics simulations performed over 500 ns confirmed the nature of tilamin binding as preferential interactions between cationic residues and the anionic (PG) components of the leaflet (Fig. S19†). The simulations also inferred a relative position of the tyrosine side chain in the bilayer interface, which was consistent with the tilamin conformation: the residue showed binding preferences similar to those of the adjacent cationic residues (Fig. 1A, S20A†). Given its central location in the peptide and in the tilt (0.55–1 nm depth; and 1.2–1.5 nm span) the tyrosine should be buried in the upper leaflet (Fig. 6B). Gratifyingly, the intrinsic fluorescence of tyrosine in both membrane types gave characteristic red-shifted increases suggesting a mixed type of interactions in the distorted leaflet, likely involving hydrogen bonding to phosphate groups, hydrophobic interactions to tail regions and cation–π interactions with the choline moieties of DLPC (Fig. S20B†).
Mechanistic rationale for antimicrobial monolayer poration
The results support our prediction for tilamin-induced poration of the outer leaflet of phospholipid bilayers as a molecular mechanism for membrane disintegration resulting in rapid and strong antimicrobial effects. Such a mechanism is consistent with other poration mechanisms of biological significance. For example, bacterial cassette transporters induce pore-like perturbations in the cytosolic leaflet of the membrane bilayer,47 whereas neurotoxic prion proteins tend to penetrate only a half of mammalian membranes.48 Further, evidence exists for the ability of individual bilayer leaflets to independently regulate membrane permeability in living systems.49In this light, the mechanism of tilamin supports a distinct antimicrobial process. This process is manifested in the rapid assembly of antimicrobial peptides in the outer leaflet of the bilayer forming pore-like structures by inserting in a tilted orientation. The resulting shallow pores impose energy penalties that bacterial membranes cannot accommodate, but which are compensated for by rapid membrane thinning and exfoliation at the sites of pore formation. Such a state of monolayer poration, an “M” state, is distinguished from hydrophilic transmembrane pores, which can equilibrate, stabilize and develop laterally (Fig. 7).
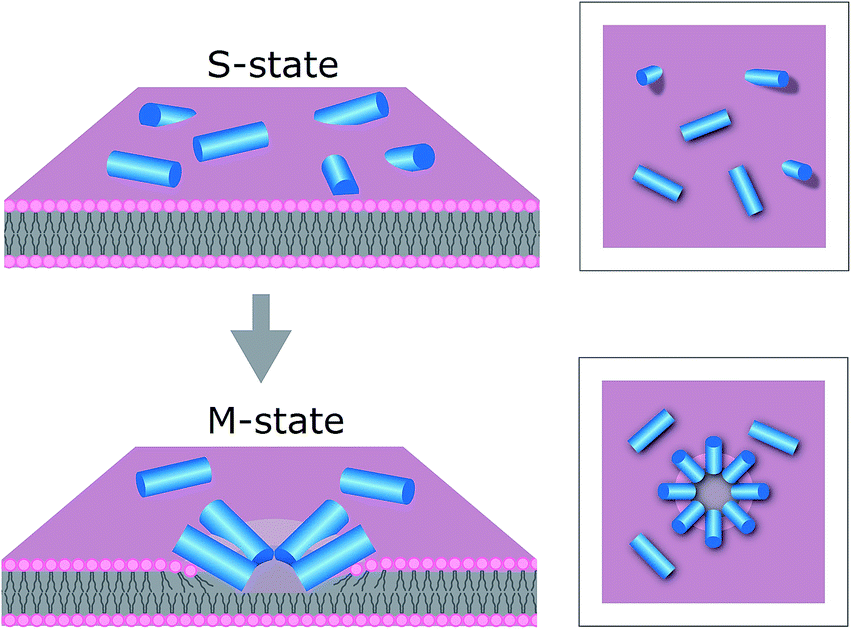 | ||
| Fig. 7 Proposed mechanism for antimicrobial monolayer poration. Peptides (blue cylinders) bind to the membrane surface – “S”-state (upper), and insert with forming pores in the outer leaflet of the bilayer – “M” state (lower). | ||
A transmembrane pore can also be viewed as a combination of two porated monolayer interfaces merging into the more favourable configuration of a hydrophilic hole.14 However, this process would require peptides to attack from both the extra- and intra-cellular sides of the bilayer, implying that peptide translocation and engagement with the inner leaflet should occur at rates faster than those necessary for disrupting the outer leaflet – the first and primary target of antimicrobial peptides. Although possible at non-antimicrobial concentrations,14 this scenario may not reflect the biological rationale of AMPs, which are overexpressed by cells at inflammatory sites,50 and whose primary role is to kill microbial cells before proteolytic degradation. The faster AMPs rupture microbial membranes, the more efficient they are. Shallow pores formed in the outer leaflet need not expand or stabilize to lyse microbial cells. The energetically unfavourable configuration they cause by exposing the hydrophobic core of the bilayer to water is unlikely to be mitigated by resealing51 or trans-bilayer (flip-flop) lipid re-arrangements52 observed for transmembrane pores in synthetic bilayers, because the exfoliation of the outer leaflet leaves only an inner leaflet, both sides of which, intra- and extracellular, are exposed to water. Subsequently, this causes the localised membrane rupture, accompanied by the formation of larger and deeper lesions and holes, which constitute the functional hallmark of monolayer poration. For this reason, in live cells monolayer pores appeared as short-lived, but forming continuously and in abundance. Pores in SLBs can be expected to form within the same timescale, which they did, but their stability is defined by a higher activation barrier needed to flip or remove lipids of the lower leaflet from the substrate. Indeed, supported lipid monolayers can remain stable over days in water despite it not being an equilibrium configuration,37 which suggests that monolayer pores in SLBs become kinetically arrested in a sub-gel phase.38
Nonetheless, in all cases – live cells, SLBs and GUVs – tilamin helices are likely to disrupt not only the membrane but also the hydrogen-bond network of associated water, which may also be reflected in the central doublet in the 2H NMR spectra being less resolved with decreasing L/P ratios (Fig. 6D). The helices then can be viewed as hydration shells that progressively intercalate in the outer leaflet and cause orientational correlations between lipids in the areas of increased tilamin concentrations – shallow pores (Fig. 4 and 5). By contrast, phospholipid bilayers alone, and particularly those composed of unsaturated lipids that have a greater variety of configurations, lack such long-range correlations.40 Hydrated tilamin helices might lower the free energy penalty for exposing the hydrophobic core in shallow pores, which may explain the relative stability of these pores within the timescales observed. However, the pores form with abundance and coalesce triggering sharp increases in positive membrane curvature that lead to significant membrane thinning and bulging.41 The abrupt and total rupture of the membrane is an invariable consequence.
With respect to the mode of action, the rates of monolayer poration appeared to directly correlate with bacterial kill rates showing appreciable membrane thinning and exfoliation and antimicrobial effects in the same timescales. The action proved to be folding-dependent and, as seen by NMR spectroscopy, involves molecular dynamics at faster timescales, supporting monolayer poration as a rapid and non-specific antimicrobial mechanism. Upon reaching critical concentrations on the membrane surfaces tilamin splays the lipids in the outer leaflet of the bilayer, where it oligomerizes with the formation of abundant shallow pores. Tilamin oligomers instantaneously remove the outer layer of Gram negative cell walls, while progressively porating their cytoplasmic membranes. Thicker peptidoglycans on the surface of Gram positive bacteria, which lack an outer membrane, might be expected to induce the partial folding and pre-oligomerisation of the peptide before it can reach the cytoplasmic membrane.53 Yet, this had no effect on antimicrobial activity, as tilamin did not differentiate between Gram positive and Gram negative types (Table S1†). In this regard, the non-differentiating mechanism of tilamin that proved to be equally effective in live bacteria and reconstituted anionic membranes can present a generic mode of action that is independent of bacterial lipid compositions. Indeed, lipids in different microbial membranes can vary significantly.54 This underlies an intrinsic limitation of synthetic membranes regardless of the chosen lipids and therefore requires complementary live-bacteria measurements. Most bacterial cells tend to maintain anionic PG components in their membranes, whereas the choice for zwitterionic lipids to distribute the negative charge is seemingly arbitrary among different organisms. For example, E. coli is known to prefer phosphatidylethanolamine (PE), but S. aureus is lacking the lipid completely.55 In P. aeruginosa it mixes with phosphatidylcholines (PC), while double-membrane spirochetes (B. burgdorferi) opt it out for PC.54 Perhaps even more importantly, emerging resistant strains learn to incorporate lipids that are alien to their wild types thus exploiting combinations that may provide them with a competitive advantage.56
Conclusions
In summary, we have predicted, engineered and demonstrated antimicrobial monolayer poration. Specifically, our findings support a molecular mechanism whereby a tilted peptide insertion triggers rapid and continuous membrane thinning that is accompanied by the formation of monolayer pores causing local membrane rupture. This mechanism was visualised in live bacteria as well as reconstituted lipid bilayers composed of saturated and unsaturated phospholipids used in two formats – in solution and on supported lipid bilayers. In all cases the mechanism consistently involved the rapid formation of coalescing pores splaying the outer leaflet of the lipid bilayers. The mechanism may advance our understanding of analogous naturally occurring phenomena including prion porations and protein oligomerisation dynamics in membrane leaflets,47–49 while the described correlations between the designed insertion mode, pore formation and antimicrobial effects emphasize antimicrobial peptides as a physical means of innate host defense. As an archetypal molecular motif tilamin folds and assembles in microbial membranes in a specific orientation, which defines the rate and extent of membrane disintegration, thus providing a mechanistic basis for designing more effective antibiotics.Acknowledgements
We thank the Bechinger group for their advice and support for the work, Hasan Alkassem for his help with AFM imaging and the EPSRC IRC in Early-Warning Sensing Systems for Infectious Diseases (EP/K031953/1) for use of research facilities. We acknowledge funding from the United Kingdom's Department of Business, Innovation and Skills, Engineering and Physical Sciences Research Council (EP/G036675/1 and EP/M506448/1), Biotechnology and Biological Sciences Research Council (BB/J006254/1) and the European Metrology Research Programme (EMRP) projects. The EMRP is jointly funded by the EMRP participating countries within EURAMET and the European Union.Notes and references
- R. J. Gilbert, M. Dalla Serra, C. J. Froelich, M. I. Wallace and G. Anderluh, Trends Biochem. Sci., 2014, 39, 510 CrossRef CAS PubMed.
- C. Leung, et al. , eLife, 2014, e04247, DOI:10.7554/eLife.04247.
- R. H. Law, et al. , Nature, 2010, 468, 447 CrossRef CAS PubMed.
- A. Bestembayeva, et al. , Nat. Nanotechnol., 2015, 10, 60 CrossRef CAS PubMed.
- J. L. Fox, Nat. Biotechnol., 2013, 31, 379 CrossRef CAS PubMed.
- C. D. Fjell, R. E. W. Hancock and A. Cherkasov, Bioinformatics, 2007, 23, 1148 CrossRef CAS PubMed.
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37 CAS.
- K. Matsuzaki, S. Yoneyama and K. Miyajima, Biophys. J., 1997, 73, 831 CrossRef CAS PubMed.
- S. Qian, W. Wang, L. Yang and H. W. Huang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17379 CrossRef CAS PubMed.
- P. D. Rakowska, et al. , Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8918 CrossRef CAS PubMed.
- H. W. Huang, Biochemistry, 2000, 39, 8347 CrossRef CAS PubMed.
- M. Mihajlovic and T. Lazaridis, Biochim. Biophys. Acta, 2010, 1798, 1494 CrossRef CAS PubMed.
- K. J. Hallock, D.-K. Lee and A. Ramamoorthy, Biophys. J., 2003, 84, 3052 CrossRef CAS PubMed.
- M. T. Lee, T. L. Sun, W. C. Hung and H. W. Huang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14243 CrossRef CAS PubMed.
- H. W. Huang, Biochim. Biophys. Acta, 2006, 1758, 1292 CrossRef CAS PubMed.
- M. B. Ulmschneider, M. S. P. Sansom and A. Di Nola, Biophys. J., 2006, 90, 1650 CrossRef CAS PubMed.
- G. Illya and M. Deserno, Biophys. J., 2008, 95, 4163 CrossRef CAS PubMed.
- B. Scott Perrin, et al. , J. Am. Chem. Soc., 2014, 136, 3491 CrossRef PubMed.
- L. Thøgersen, B. Schiøtt, T. Vosegaard, N. C. Nielsen and E. Tajkhorshid, Biophys. J., 2008, 95, 4337 CrossRef PubMed.
- K. Yue and K. A. Dill, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4163 CrossRef CAS.
- K. S. Akerfeldt, J. D. Lear, Z. R. Wasserman, L. A. Chung and W. F. DeGrado, Acc. Chem. Res., 1993, 26, 191 CrossRef CAS.
- N. Kucerka, M. P. Nieh and J. Katsaras, Biochim. Biophys. Acta, 2011, 1808, 2761 CrossRef CAS PubMed.
- K. Mitra, I. Ubarretxena-Belandia, T. Taguchi, G. Warren and D. M. Engelman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4083 Search PubMed.
- M. Mihajlovic and T. Lazaridis, Biochim. Biophys. Acta, 2012, 1818, 1274 CrossRef CAS PubMed.
- L. Li, I. Vorobyov and T. W. Allen, J. Phys. Chem. B, 2013, 117, 11906 CrossRef CAS PubMed.
- L. Ryan, et al. , J. Biol. Chem., 2013, 288, 20162 CrossRef CAS PubMed.
- F. Corona and J. L. Martinez, Antibiotics, 2013, 2, 237 CrossRef CAS PubMed.
- J. Haaber, C. Friberg, M. McCreary, R. Lin, S. N. Cohen and H. Ingmer, mBio, 2015, 6, e02268 CAS.
- G. E. Fantner, R. J. Barbero, D. S. Gray and A. M. Belcher, Nat. Nanotechnol., 2010, 5, 280 CrossRef CAS PubMed.
- L. Gan, S. Chen and G. J. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18953 CrossRef CAS PubMed.
- K. Matsuzaki, K. Sugishita, M. Harada, N. Fujii and K. Miyajima, Biochim. Biophys. Acta, 1997, 1327, 119 CrossRef CAS.
- N. Malanovic and K. Lohner, Biochim. Biophys. Acta, 2016, 1858, 936 CrossRef CAS PubMed.
- E. Tuomanen, R. Cozens, W. Tosch, O. Zak and A. Tomasz, J. Gen. Microbiol., 1986, 132, 1297 CAS.
- W.-C. Lin, C. D. Blanchette, T. V. Ratto and M. L. Longo, Biophys. J., 2006, 90, 228 CrossRef CAS PubMed.
- M. R. Hicks, J. Kowalski and A. Rodger, Chem. Soc. Rev., 2010, 39, 3380 RSC.
- A. Damianoglou, A. Rodger, C. Pridmore, T. R. Dafforn, J. A. Mosely, J. M. Sanderson and M. R. Hicks, Protein Pept. Lett., 2010, 17, 1351 CrossRef CAS PubMed.
- N. N. Gosvami, E. Parsons, C. Marcovich, M. L. Berkowitz, B. W. Hoogenboom and S. Perkin, RSC Adv., 2012, 2, 4181 RSC.
- M. L. Kraft, P. K. Weber, M. L. Longo, I. D. Hutcheon and S. G. Boxer, Science, 2006, 313, 1948 CrossRef CAS PubMed.
- S. Toraya, T. Nagao, K. Norisada, S. Tuzi, H. Saitô, S. Izumi and A. Naito, Biophys. J., 2005, 89, 3214 CrossRef CAS PubMed.
- S. Semrau and T. Schmidt, Soft Matter, 2009, 5, 3174 RSC.
- F. Campelo, H. T. McMahon and M. M. Kozlov, Biophys. J., 2008, 95, 2325 CrossRef CAS PubMed.
- E. Strandberg, et al. , Biophys. J., 2004, 86, 3709 CrossRef CAS PubMed.
- P. C. A van der Wel, E. Strandberg, J. A Killian and R. E Koeppe, Biophys. J., 2002, 83, 1479 CrossRef.
- C. Aisenbrey and B. Bechinger, Biochemistry, 2004, 43, 10502 CrossRef CAS PubMed.
- H. J. Hogben, M. Krzystyniak, G. T. P. Charnock, P. J. Hore and I. Kuprov, J. Magn. Reson., 2011, 208, 179 CrossRef CAS PubMed.
- J. B. Klauda, et al. , J. Phys. Chem. B, 2010, 114, 7830 CrossRef CAS PubMed.
- D. Y. Haubertin, H. Madaoui, A. Sanson, R. Guérois and S. Orlowski, Biophys. J., 2006, 91, 2517 CrossRef CAS PubMed.
- J. Zhong, W. Zheng, L. Huang, Y. Hong, L. Wang, Y. Qiu and Y. Sha, Biochim. Biophys. Acta, 2007, 1768, 1420 CrossRef CAS PubMed.
- H. O. Negrete, R. L. Rivers, A. H. Goughs, M. Colombini and M. L. Zeidel, J. Biol. Chem., 1996, 271, 11627 CrossRef CAS PubMed.
- T. Ganz, Integr. Comp. Biol., 2003, 43, 300 CrossRef CAS PubMed.
- M.-T. Lee, W.-C. Hung, F.-Y. Chen and H. W. Huang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5087 CrossRef CAS PubMed.
- K. Matsuzaki, O. Murase, N. Fujii and K. Miyajima, Biochemistry, 1996, 35, 11361 CrossRef CAS PubMed.
- M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 55, 27 CrossRef CAS PubMed.
- C. Sohlenkamp and O. Geiger, FEMS Microbiol. Rev., 2016, 40, 133 CrossRef PubMed.
- R. F. Epand, G. Wang, B. Berno and R. M. Epand, Antimicrob. Agents Chemother., 2009, 53, 3705 CrossRef CAS PubMed.
- S. Gruenheid and H. Le Moual, FEMS Microbiol. Lett., 2012, 330, 81 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, microscopy, spectroscopy, molecular dynamics and spectrometry data. See DOI: 10.1039/c6sc02925f |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |