
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorogenic sensor platform for the histone code using receptors from dynamic combinatorial libraries†
Brendan C.
Peacor
,
Christopher M.
Ramsay
and
Marcey L.
Waters
*
Department of Chemistry, University of North Carolina at Chapel Hill, CB 3290, Chapel Hill, NC 27599, USA. E-mail: mlwaters@unc.edu
First published on 20th October 2016
Abstract
Post-translational modifications (PTMs) on histone tails act in diverse combinations in the ‘histone code’ to control gene expression, with dysregulation observed in a variety of diseases. However, detection and sensing methods are limited, expensive, and/or low-throughput, including MS and antibody based detection. We found that by combining four synthetic receptors developed by dynamic combinatorial chemistry (DCC) in an indicator displacement system, we are able to create a pattern-based sensor platform that can discriminate single PTMs such as methylation and acetylation on a representative histone peptide with 100% accuracy as well as peptides bearing both dimethyl and trimethyl lysine in the presence of arginine methylation, which has not previously been demonstrated, and can even correctly distinguish the position of lysine methylation individually or in the presence of other PTMs. To extend this approach, a full panel of thirteen analytes containing different combinations of PTMs were classified with 96 ± 1% overall accuracy in a 50% left-out analysis, demonstrating the robustness and versatility of the sensor array. Finally, the sensor platform was also used to demonstrate proof of concept for enzymatic assays by analysing the mock reaction of a threonine kinase, successfully identifying analytes representative of substrate conversion both with and without neighboring PTMs. This work provides a rapid platform for the analysis of peptides bearing complex modifications and highlights the utility of receptors discovered though DCC that display variations in binding affinity and selectivity.
Introduction
The expression and control of the genetic landscape relies on several different factors, most notably the installation and deletion of a dynamic network of post-translational modifications (PTMs) on the DNA organizing histone proteins in nucleosomes.1 These PTMs are covalent chemical markers, such as those shown in Fig. 1, that are responsible for the signaling of a variety of biological functions. Moreover, dysregulation of PTMs has been observed in numerous diseases and cancers.2–4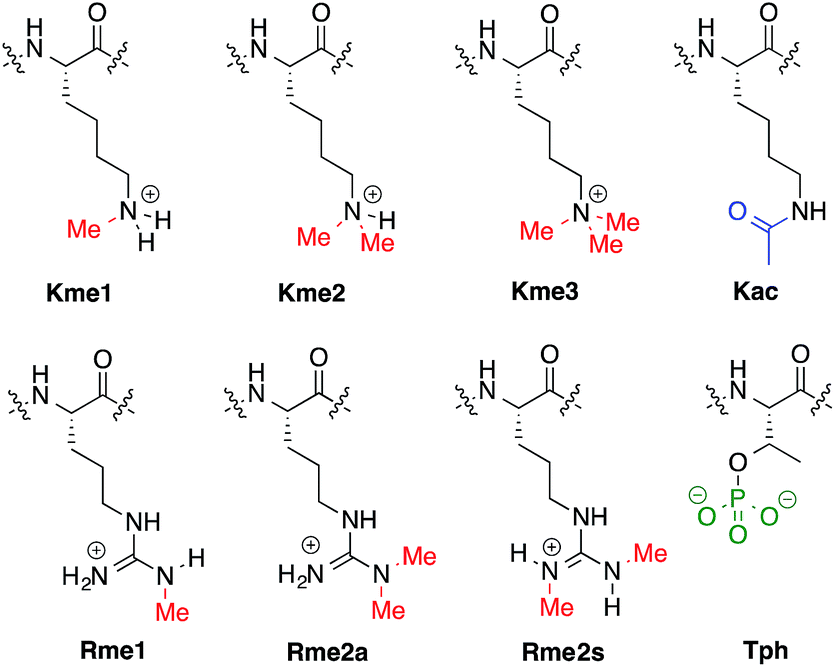 | ||
| Fig. 1 PTMs on the side chains of Lys, Arg, and Thr. | ||
PTMs are well known to interact through a number of ‘cross-talk’ events to change the chromatin landscape, called the histone code.5 Modification of residues on a single histone tail can regulate different events depending on not only the site and identity of the modification, but the presence of neighboring PTMs. For example, phosphorylation of serine 10 on the histone H3 tail inhibits the binding of the HP1 reader protein to the adjacent trimethyl lysine at position 9.6 In contrast, phosphorylating threonine 11 instead results in the recruitment of the methyltransferase enzyme responsible for methylation of lysine 4.7 Other studies have shown that proteins responsible for binding to specific marks interact more strongly when particular patterns of other modifications are installed, highlighting the critical need to be able to study and analyze the entirety of chromatin modifications.8–10
Because of the observed complexity of the histone code, assays that are capable of recognizing not just the identity, but also the context of a modification are in high demand. Traditionally, the field has utilized mass spectrometry and antibody based approaches.11–13 However, the former suffers from the requirements of expensive equipment and the latter has issues with false-negatives and off target effects.14 Indeed, recent work in several labs has shown that antibodies, which passed all standard tests for specificity, had false negatives due to the presence of neighbouring modifications, or had a 20% failure rate in chromatin immunoprecipitation experiments.15,16 Furthermore, reading out the exact positions of combinations of PTMs is difficult with these methods. Because of this, histone tail microarrays have become a popular method to evaluate the histone code hypothesis, but the limitations of antibodies described above has hampered their application. Thus, new sensing methods are needed.
Sensor arrays make use of differential receptors to sense multiple analytes using pattern recognition, often termed an “artificial nose”.17 Coupling of a binding event with a signal such as a change in color or fluorescence allows for the readout and characterization of a range of analytes (Scheme 1a). This expanded classification range is due to small differences in interaction with the suite of sensors, which provides a unique pattern for each analyte (Scheme 1b). The resulting sensor output is then resolved using statistical software to generate an easily interpreted readout of analyte classification (Scheme 1c). This method of combinatorial sensor arrays has been used to great success for a wide range of analytes, including amino acids,18 nucleotides, carbohydrates, compounds in a wide range of beverages, flavonoids,19 and even drugs in biological media.20 Recently, several synthetic receptors have been developed for the different methylation states of Lys and Arg by our group and others.21–27,40 In a previous report, a sensor array was developed for single histone protein modifications using one of these synthetic receptors, with one example of sensing a peptide containing both Kme3 and Tph.28 However, there currently is no general, rapid fluorogenic method for sensing multiple PTMs within the same peptide, with multiple methylations being a significant unmet challenge.
 | ||
| Scheme 1 Schematic representations of (a) indicator displacement assay, (b) sensor array showing a different pattern for each analyte, and (c) statistical output. | ||
We report here a sensor array using a suite of small molecule receptors for the sensing and classification of peptides bearing single and multiple histone PTMs in aqueous solution. We find that using four unique receptors we are able to distinguish between the different methylation states of lysine and arginine, for which the receptors were designed. Moreover, due to small differences in binding affinity, this set of receptors can also correctly classify the positions of methylated Lys and Arg, and even other modifications for which the receptors were not developed, such as acetylation, and phosphorylation. Furthermore, these receptors successfully identify peptides that bear combinatorial modifications, including multiple methylations within the same sequence, providing facile readout of complex analytes representative of the histone code. This is the first example that is able to differentiate both multiple modifications in the same peptide as well as the position of those modifications, including analytes with multiple methylations.
Results and discussion
Previously we have reported a set of small molecule receptors for methylated Lys and Arg peptides in aqueous solution (Fig. 2).21–24 These receptors were developed and synthesized using dynamic combinatorial chemistry (DCC), which is a reversible process that relies on the thermodynamic templation of building blocks into favorable receptors due to binding to guests.29–31 Importantly, it allows for rapid variation of each building block in the receptor, therefore easily generating a number of host molecules. The receptors have been shown to bind to methylated histone peptides with low micromolar to high nanomolar affinities in aqueous solution through cation–π and electrostatic interactions. Moreover, each receptor has a distinct pattern of binding affinities and selectivities, and is sensitive to the neighboring charge.23,24,41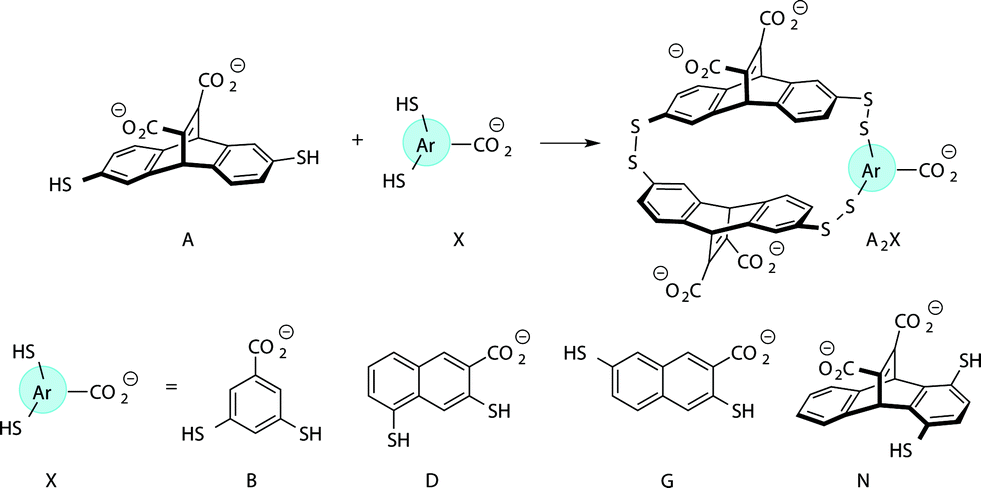 | ||
| Fig. 2 Receptors used in the sensor array for histone PTMs. Each receptor displays a different pattern of inter-action for the methylated forms of lysine and arginine. | ||
Four of our previously reported receptors were chosen for the sensor array, which exhibit a range of binding affinities and selectivities for methylated Lys and Arg (Fig. 2). All four receptors, A2B, A2D, A2G, and A2N, bind Kme3 in the context of a histone tail sequence (Ac-WGGG-QTAR-Kme3-STG-NH2) with affinities in the low micromolar to high nanomolar range, and selectivities over unmodified Lys of 8-fold to >40-fold.21–24 Additionally, receptor A2D binds Rme2a with low micromolar affinity and with 12-fold selectivity over Arg. To develop a sensor platform for modified histone tails, indicator displacement assays were established for each receptor. Indicator displacement assays rely on the competitive binding of an environmentally sensitive dye or fluorophore and an analyte, wherein a difference in signal is measured based on the occupancy of the fluorophore either in solution or bound to the receptor.32,33
After screening several dyes, we identified lucigenin (LCG) as the optimal dye for all four receptors. LCG fluorescence is quenched in the presence of each of the receptors, but presence of an analyte will displace the dye and result in a “turn-on” fluorescence signal. Titrations indicated that LCG binds to each receptor with low micromolar affinity (see Fig. S1–S4†). Additionally, when a peptide bearing either unmethylated lysine or dimethylated lysine were titrated into the A2N·LCG sensor, fluorescence signal was recovered in proportion to the degree of methylation, as shown in Fig. S5.† We obtained the best signal differentiation using glycine buffer at pH 9 with no added salt using 1 μM of LCG. With a single set of buffer and dye conditions, the assay was easily performed in a multi-well plate in a high-throughput manner.
With the goal of studying peptides bearing multiple modifications, we chose to study the histone 3 (H3) tail, residues 1–12, which is known to have multiple PTM sites, with a C-terminal tyrosine for accurate peptide concentration determination. Using the four receptors and the fluorophore LCG, we measured twenty replicates of each peptide analyte in a high-throughput, low volume 384 well plate. This allowed analysis of each analyte in approximately 30 minutes using low quantities of peptide and receptor in a total volume of 9 μl. Five initial peptides were chosen to determine the suitability of the four-receptor sensor array for detecting a number of disparate single histone modifications, including Kme3, Kac, Rme2a, and Tph, as well as the unmodified parent peptide (Table 1). Based on previous studies investigating sequence selectivity of the receptors,22,23,34 we expected that the sensor array would provide differential signals based on net charge of the peptide as well as methylation state. Thus, while the acetyl and phosphoryl modifications have been previously unexamined with these receptors, we hypothesized that they would weaken the overall interaction with the receptor due to reduction in net positive charge, thus providing a differential signal relative to the parent peptide, H3 1–12.
| Abbreviation | Peptide sequence | Net charge |
|---|---|---|
| a The first number in the abbreviation indicates the position in the sequence. | ||
|
Ac-ARTKQTARKSTGY-NH2 | +4 |

|

|
+4 |

|

|
+3 |

|

|
+4 |
|
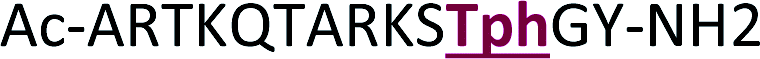
|
+2 |
The fluorescence output from the sensor array exhibited a differential response for each peptide, as expected (Fig. 3). The unmodified peptide exhibited a degree of fluorescence recovery due to the nonspecific electrostatic interaction of the H3 1–12 sequence. The signal was larger for Kme3 with all receptors, as each receptor preferentially binds to this modification. Likewise, A2D had a stronger response to the aR2Me2 mark, which was expected as it is the only receptor that had previously shown favorable interaction to the PTM. Additionally, the acetylation and phosphorylation decreased the fluorescence output of the array due to perturbation of binding, either by loss of a favorable charge interaction or addition of an interfering negative charge proximal to the anionic exterior of the receptors.
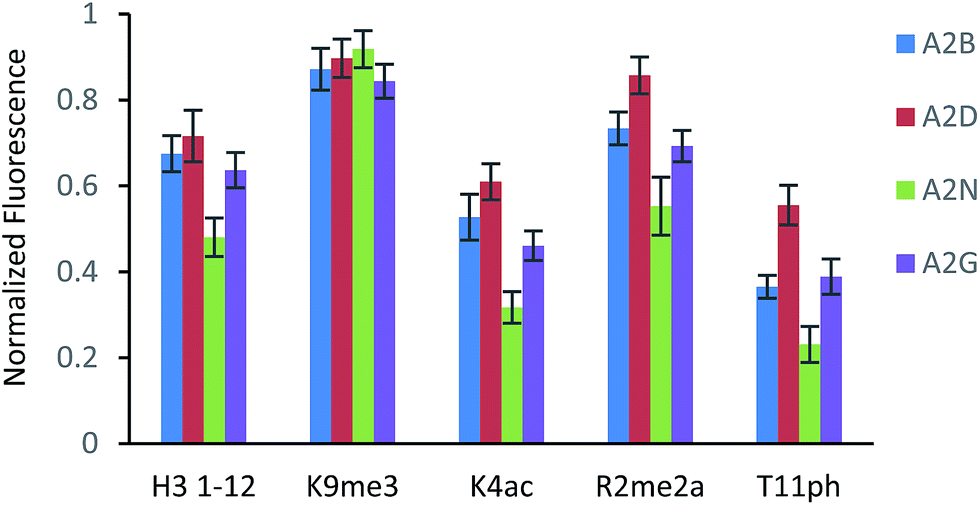 | ||
| Fig. 3 Fluorescence response for each sensor to the given peptide analyte (15 μM). Each sensor utilized a fixed concentration of LCG (1 μM) and receptor (A2B – 10 μM; A2D – 5 μM; A2N – 15 μM; A2G – 10 μM). Each sensor array was run in 50 mM glycine buffer, pH 9.15 at room temperature. Error bars represent 20 replicates. Fluorescence is normalized to a control sample of LCG (F/F∞). | ||
To classify the peptide analytes, discriminate analysis (DA) was applied to the fluorescence output of the sensor array. DA is a powerful statistical method able to determine the classification capability of a given sensor array.20 The statistical software converts the raw data, fluorescence in the case of our IDAs, into a series of eigenvectors, which act as the canonical scores for each axis, allowing graphical representation.35 DA puts the emphasis on clustering repetitive samples, such as multiple analyte trials. This is followed by separating the various clusters of samples, or classes, to achieve the best statistical relevance.36 Importantly, it is capable of deconvoluting complex sensor output into easily visualized 2D or 3D graphs, as shown in Fig. 4 for the singly modified peptide analytes.
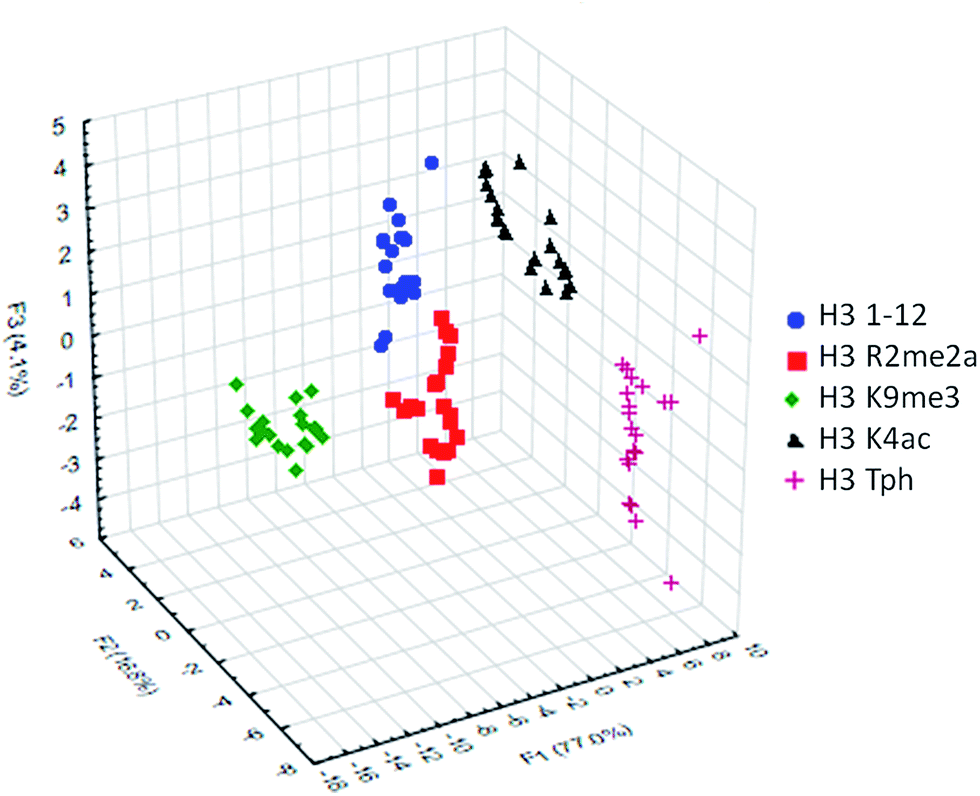 | ||
| Fig. 4 LDA response of the sensor array to five singly modified analytes (20 replicates). Classification accuracy at 100%, jackknife accuracy at 100%. | ||
Using this approach, the array of four sensors was able to successfully classify each of the five analytes with 100% accuracy corresponding to the initial inputted analyte labels within the sample set of 20 replicates. Additionally, a ‘leave-one-out’ analysis was performed in which one analyte was removed from the data set prior to the discriminate analysis and was then re-input as a blind test point to ensure the classification still holds, known as a jackknife experiment, a technique commonly used for validating statistical results.18,37 This result again provided 100% accuracy for the tested samples, demonstrating the utility of the sensor array to discriminate not just methylated peptides, for which the receptors were designed, but other modifications as well, due to small changes in affinity arising from variation in electrostatic interactions.
With single modifications correctly classified, we examined whether the sensor array could discriminate between more complex peptides bearing multiple modifications. Because of the implications of the histone code, we wanted to examine if we could classify peptides with multiple, potentially competitive or cooperative marks. The first of these studied was peptides bearing multiple methylations on the same tail sequence, as shown in Table 2.








We expected that each of these peptides would cause a large fluorescence recovery due to increased binding affinity of our receptors for the methylated forms of lysine. However, each peptide should have a slightly different affinity for the four receptors due to neighboring modifications affecting small changes in the non-covalent binding interactions.
As seen in Fig. 5 above, the sensor array was able to correctly classify 99% of the data points into their respective peptide clusters, with 98% of the data still correct in the follow-up jackknife test, with the outliers centered around the two peptides bearing dimethylation at K4 or K9, indicating that the classification is 100% correct for type of modification, but with slightly diminished sensitivity to the sequence position of the modification. These results are significant because, save for two data points in the jackknife result, the sensor platform is sensitive to not just degree of methylation, but its position as well, suggesting further utility in studying the histone code. Indeed, MS/MS methods would otherwise be necessary to characterize the position of the modification.
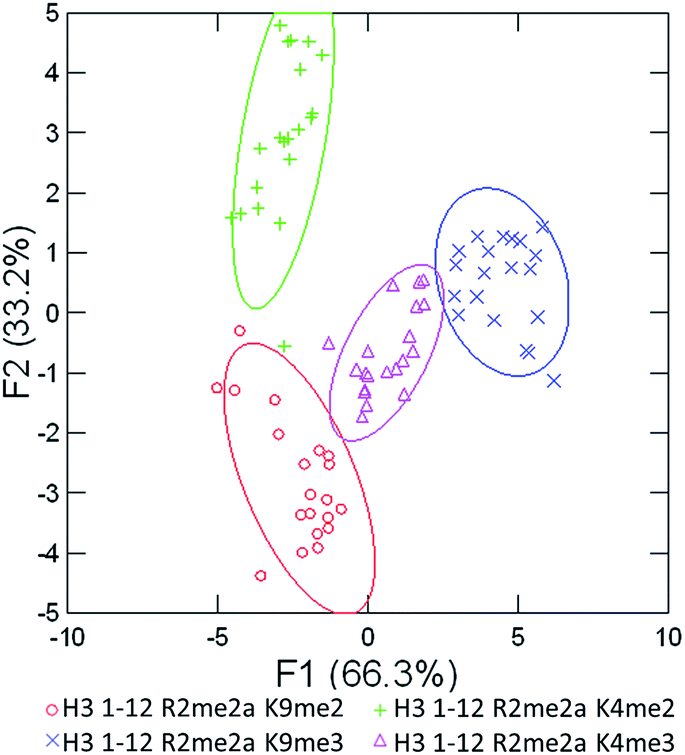 | ||
| Fig. 5 LDA response of the sensor array to four peptides bearing multiple methylation PTMs at varying sites. Confidence ellipses at 85%, classification accuracy of 99%, jack-knife test at 98%. | ||
As mentioned previously, we have found that these small molecule receptors can be sensitive to neighboring charged residues through nonspecific electrostatic interactions.41 Therefore, we wanted to determine if an interfering modification, which we would expect weakens binding, still allows for the recognition and classification of histone peptides. One of the most common modifications of peptides is the phosphorylation of the hydroxyl-side chains of neutral amino acids, adding in a negative charge. We hypothesized that while the phosphorylation will decrease binding, especially when immediately near a methylation site, it will not affect each receptor identically, providing the requisite pattern of binding interactions and subsequent dye displacement. Thus, we studied the effect of threonine 11 phosphorylation on the classification of lysine 4 and 9 methylated peptides, as shown in Table 3.








As seen in Fig. 6, the sensor array was once again able to classify each peptide with 100% accuracy. This is significant because in this case we introduced an unfavorable interaction which we hypothesized weakened overall binding, yet still allowed for differential recognition. Interestingly, the two peptides that were most difficult to differentiate contain K9me3 and K4me2. This may be that the close proximity of T11ph to K9me3 weakens the binding to K9me3.
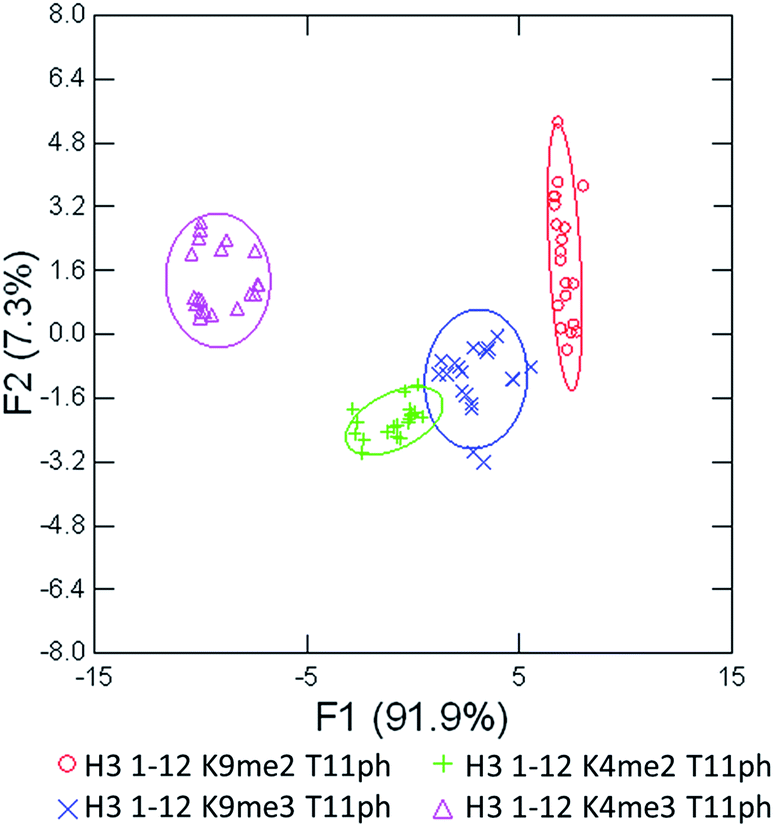 | ||
| Fig. 6 LDA response of the sensor array to four peptides bearing methylation and phosphorylation. Confidence ellipses at 85%, classification accuracy of 100%, jackknife test at 100%. | ||
To examine the sensor array's ability to distinguish a wide range of PTMs, we performed the statistical analysis on the full panel of peptides described above. This provides the sensor system with thirteen different analytes, some bearing only slight differences in modifications or neighboring sequence. The platform was tasked not just with identifying the type of modification, but also the site of modification and whether or not a second PTM is present. The results of this study are shown below in Fig. 7.
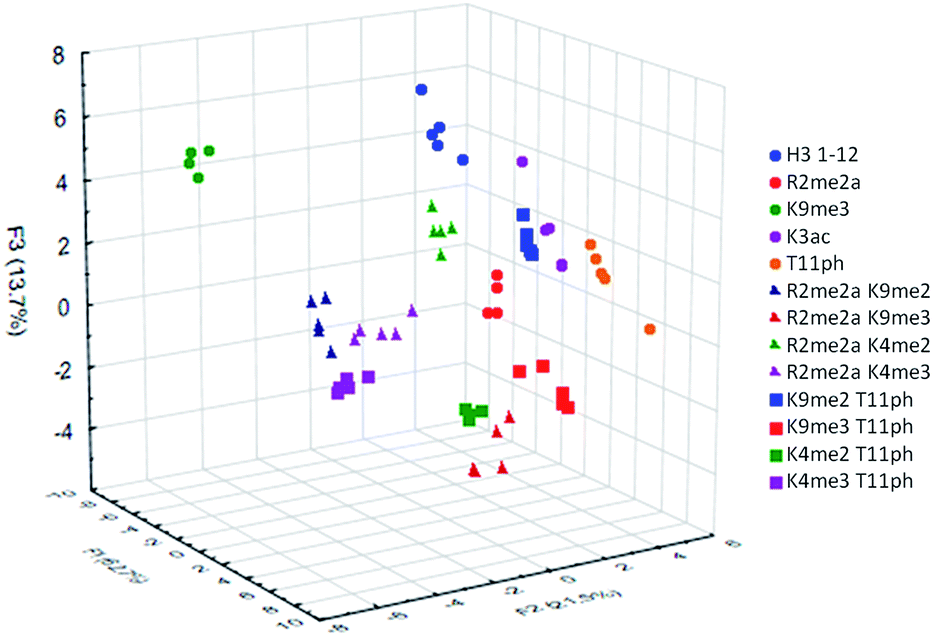 | ||
| Fig. 7 LDA response of the sensor array to all thirteen analytes. Twenty replicates were performed for each analyte, but only the first five are plotted here to simplify the resulting visual output. Observed classification accuracy of 96%, jackknife classification of 95%. | ||
The sensor array was successfully able to distinguish each of the thirteen peptide analytes with a 96% overall accuracy. Importantly, each analyte had an individual classification accuracy of greater than 90%, showing that there were only a few replicates miss-classified rather than a miss-assignment of one entire analyte class.
In addition to the “leave-one-out” analysis, we wanted to perform a more rigorous test of the assays robustness for the thirteen analytes. This was accomplished by removing 50% of the replicates of each analyte from the initial analysis, followed by resubmission of these points as a blind set. This gave the platform half the number of data points to distinguish between analytes and then used the remainder to confirm accuracy. The data points for each set were randomized and that analysis was performed twenty times, giving an overall classification accuracy of the test set at 96 ± 1%.
Having successfully demonstrated that the sensor array works well in the end-point analysis and classification of modified peptides, we wanted to conduct an initial study of the applicability to studying histone PTM enzymatic reactions in vitro. Current research has shown the application of single sensor IDAs to the study of enzymatic reactions, such as diamine oxidase38 and lysine methyltransferase enzymes.39 However, these assays typically rely on the design and optimization of a single receptor with sufficient discrimination between substrate and product to be effective.38 A multiple sensor array, could be ‘trained’ to sense the product of the reaction and be rapidly applied to enzymatic assays, even with complex substrates bearing multiple modifications, as we would expect in studying the histone code. We explored this hypothesis by performing a mock kinase assay based on phosphorylation of threonine 11 in the histone H3 tail. Our proof of concept ‘training set’ used four mimic time-points of an enzymatic reaction corresponding to 0, 33, 66, and 100 percent conversion of H3 1–12 to the phosphorylated product, shown in Fig. 8.
 | ||
Fig. 8 LDA of the mock enzymatic kinase reaction monitoring conversion of H3 1–12 (Ac-ARTKQTARKSTGY-NH2) to H3 1–12 T11ph (Ac-ARTKQTARKSTphGY-NH2). The substrate and product were both at 15 μM, with 33% conversion at 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 μM substrate:product and 66% at 10:5 μM. Arrows were added to represent the path of phosphorylation, confidence ellipses at 90%. :10 μM substrate:product and 66% at 10:5 μM. Arrows were added to represent the path of phosphorylation, confidence ellipses at 90%. | ||
Using four receptors, the assay is able to distinguish between each time point of the mock enzymatic reaction with 100% accuracy within the assay set, and 100% confirmation in the jackknife. This would allow a kinase reaction to be carried out on H3 1–12 to test enzyme activity in a rapid fashion. The enzymatic reaction could be performed followed by transfer to a 384 well plate and addition of the sensor components, minimizing the possibility of competitive receptor inhibition and removing the requirement of a complex detection cocktail.
The true utility of the sensor array approach to enzymatic reactions is the ability to generate a training set for a large variety of possible substrates. To demonstrate this, we performed another mock kinase reaction using a more complex substrate peptide, H3 1–12 trimethylated at lysine 9, shown in Fig. 9.
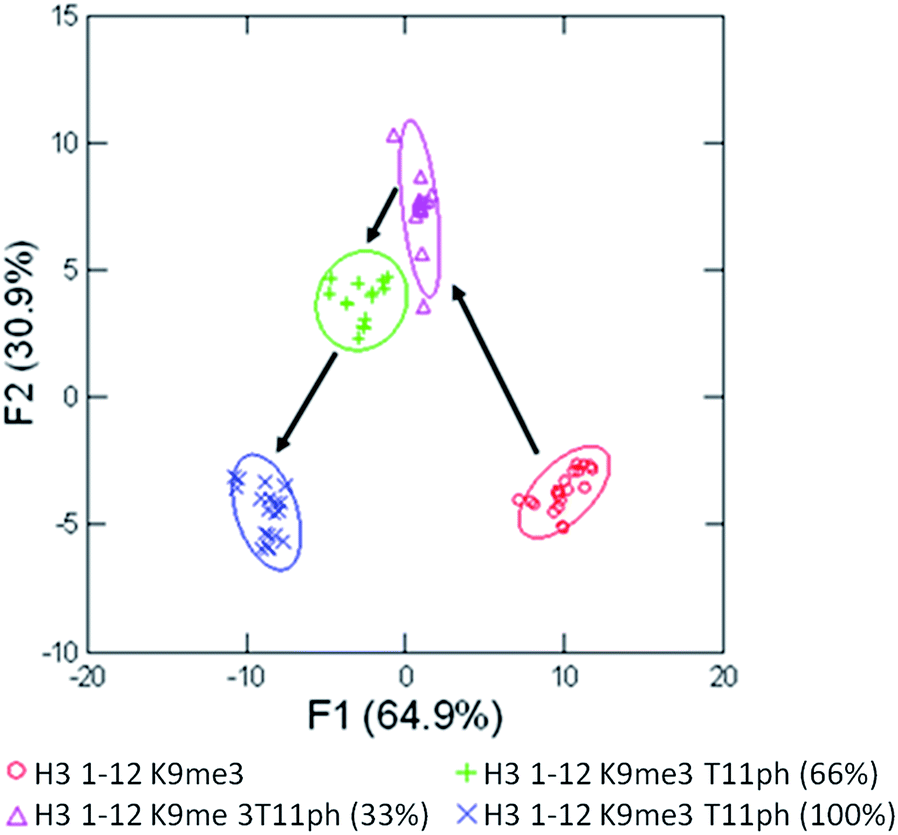 | ||
| Fig. 9 LDA of the mock enzymatic kinase reaction monitoring conversion of H3 1–12 K9me3 (Ac-ARTKQTARKme3STGY-NH2) to H3 1–12 K9me3T11ph (Ac-ARTKQTARKme3STphGY-NH2). Arrows were added to represent the path of phosphorylation, confidence ellipses at 90%. | ||
Once again, the training set of the reaction was able to classify the four major time points with 100% accuracy within the sample set, and 100% confirmation in the jackknife. While this approach of in vitro assay would require a training set for each peptide, there would be minimized development of the assay for individual substrates, since it does not require a specific degree of differentiation between binding of substrate and product. This would allow the sensor array to be applied rapidly to a wide variety of enzymes, including acetyltransferases, arginine demininases, and methyltransferase or demethylases. In each case, the addition or deletion of a favourable binding interaction or the alteration of the landscape surrounding these interactions would allow the qualitative monitoring of the enzymatic regulation of important biological markers.
Conclusion
The application of a sensor array platform to the sensing of combinatorial histone modifications in aqueous solution was achieved through the use of dynamic combinatorial receptors and the organic fluorophore lucigenin. These receptors, previously designed and studied for the binding of methylated lysine and arginine, display slight differences in binding affinity depending on the context of the neighboring sequence and modifications of the peptide. These perturbations in binding affinity allow the sensing of analytes beyond the original designed receptor scope, including the loss of charge by acetylation or the introduction of a repulsive noncovalent interaction in phosphorylation. Through the application of only four receptors under a single set of conditions we were able to successfully classify thirteen different histone peptides. These peptides varied based on the identity, number, and position of the PTM. Singly modified tails as well as peptides bearing a more complex PTM landscape were successfully classified, which is a challenge for traditional PTM analysis15 and has not been accomplished with a previously reported sensor array.28In the case of our sensor array, there is a distinct advantage of using DCC to furnish new receptors. The combinatorial nature of receptor synthesis means that the suite of sensors is easily and rapidly expandable. While each new receptor might display only modest differences in affinity from previous generations, that difference enhances the discriminatory capability of the assay. This modifiability, coupled with the high-throughput and low material cost of running the sensor array should provide an easily adaptable toolkit for the study of a wide scope of histone modifications.
Acknowledgements
We thank Professor John Lavigne and Erin Gatrone for helpful discussions. This material is based upon work supported by the National Science Foundation under grant no. CHE-1306977. We also gratefully acknowledge funding from the W. M. Keck Foundation for this work.References
- T. Kouzarides, Cell, 2007, 128, 693 CrossRef CAS PubMed.
- B. D. Strahl and C. D. Allis, Nature, 2000, 403, 41 CrossRef CAS PubMed.
- S. B. Hake, A. Xiao and C. Allis, Br. J. Cancer, 2004, 90, 761 CrossRef CAS PubMed.
- G. G. Wang, C. D. Allis and P. Chi, Trends Mol. Med., 2007, 13, 363 CrossRef CAS PubMed.
- A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381 CrossRef CAS PubMed.
- W. Fischle, B. S. Tseng, H. L. Dormann, B. M. Ueberheide, B. A. Garcia, J. Shabanowitz, D. F. Hunt, H. Funabiki and C. D. Allis, Nature, 2005, 438, 1116 CrossRef CAS PubMed.
- J. Y. Kim, T. Banerjee, A. Vinckevicius, Q. Luo, J. B. Parker, M. R. Baker, I. Radhakrishnan, J. J. Wei, G. D. Barish and D. Chakravarti, Mol. Cell, 2014, 54, 613 CrossRef CAS PubMed.
- S. B. Rothbart, B. M. Dickson, M. S. Ong, K. Krajewski, S. Houliston, D. B. Kireev, C. H. Arrowsmith and B. D. Strahl, Genes Dev., 2013, 27, 1288 CrossRef CAS PubMed.
- A. P. Lothrop, M. P. Torres and S. M. Fuchs, FEBS Lett., 2013, 587, 1247 CrossRef CAS PubMed.
- S. Ramón-Maiques, A. J. Kuo, D. Carney, A. G. W. Matthews, M. A. Oettinger, O. Gozani and W. Yang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18993 CrossRef PubMed.
- M. Mann and O. N. Jensen, Nat. Biotechnol., 2003, 21, 255 CrossRef CAS PubMed.
- L.-M. P. Britton, M. Gonzales-Cope, B. M. Zee and B. a. Garcia, Expert Rev. Proteomics, 2011, 8, 631 CrossRef CAS PubMed.
- T. S. Furey, Nat. Rev. Genet., 2012, 13, 840 CrossRef CAS PubMed.
- S. B. Rothbart, B. M. Dickson, J. R. Raab, A. T. Grzybowski, K. Krajewski, A. H. Guo, E. K. Shanle, S. Z. Josefowicz, S. M. Fuchs, C. D. Allis, T. R. Magnuson, A. J. Ruthenburg and B. D. Strahl, Mol. Cell, 2015, 59, 502 CrossRef CAS PubMed.
- S. B. Rothbart, S. Lin, L.-M. Britton, K. Krajewski, M.-C. Keogh, B. a. Garcia and B. D. Strahl, Sci. Rep., 2012, 2, 489 Search PubMed.
- T. A. Egelhofer, A. Minoda, S. Klugman, K. Lee, P. Kolasinska-Zwierz, A. A. Alekseyenko, M. Cheung, D. S. Day, S. Gadel, A. a Gorchakov, T. Gu, P. V. Kharchenko, S. Kuan, I. Latorre, D. Linder-Basso, Y. Luu, Q. Ngo, M. Perry, A. Rechtsteiner, N. C. Riddle, Y. B. Schwartz, G. A. Shanower, A. Vielle, J. Ahringer, S. C. R. Elgin, M. I. Kuroda, V. Pirrotta, B. Ren, S. Strome, P. J. Park, G. H. Karpen, R. D. Hawkins and J. D. Lieb, Nat. Struct. Mol. Biol., 2011, 18, 91 CAS.
- J. J. Lavigne and E. V. Anslyn, Angew. Chem., Int. Ed., 2001, 40, 3118 CrossRef CAS.
- T. Minami, N. A. Esipenko, B. Zhang, L. Isaacs and P. Anzenbacher, Chem. Commun., 2014, 50, 61 RSC.
- A. P. Umali, S. E. LeBoeuf, R. W. Newberry, S. Kim, L. Tran, W. a. Rome, T. Tian, D. Taing, J. Hong, M. Kwan, H. Heymann and E. V. Anslyn, Chem. Sci., 2011, 2, 439 RSC.
- Y. Liu, T. Minami, R. Nishiyabu, Z. Wang and P. Anzenbacher, J. Am. Chem. Soc., 2013, 135, 7705 CrossRef CAS PubMed.
- L. A. Ingerman, M. E. Cuellar and M. L. Waters, Chem. Commun., 2010, 46, 1839 RSC.
- L. I. James, J. E. Beaver, N. W. Rice and M. L. Waters, J. Am. Chem. Soc., 2013, 135, 6450 CrossRef CAS PubMed.
- N. K. Pinkin and M. L. Waters, Org. Biomol. Chem., 2014, 12, 7059 CAS.
- J. E. Beaver, B. C. Peacor, J. V. Bain, L. I. James and M. L. Waters, Org. Biomol. Chem., 2015, 13, 3220 Search PubMed.
- C. S. Beshara, C. E. Jones, K. D. Daze, B. J. Lilgert and F. Hof, ChemBioChem, 2010, 11, 63 CrossRef CAS PubMed.
- K. D. Daze, M. C. F. Ma, F. Pineux and F. Hof, Org. Lett., 2012, 14, 1512 CrossRef CAS PubMed.
- M. A. Gamal-Eldin and D. H. Macartney, Org. Biomol. Chem., 2013, 11, 488 CAS.
- S. A. Minaker, K. D. Daze, M. C. F. Ma and F. Hof, J. Am. Chem. Soc., 2012, 134, 11674 CrossRef CAS PubMed.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590 CrossRef CAS PubMed.
- P. T. Corbett, J. K. M. Sanders and S. Otto, Chem.–Eur. J., 2008, 14, 2153 CrossRef CAS PubMed.
- P. Corbett, J. Leclaire, L. Vial, K. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652 CrossRef CAS PubMed.
- B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118 CrossRef CAS.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941 CrossRef CAS PubMed.
- N. Pinkin, A. Power and M. L. Waters, Org. Biomol. Chem., 2015, 13, 10939 CAS.
- S. Stewart, M. A. Ivy and E. V. Anslyn, Chem. Soc. Rev., 2014, 43, 70 RSC.
- J. H. Friedman, J. Am. Stat. Assoc., 1989, 84, 165 CrossRef.
- K. L. Bicker, J. Sun, M. Harrell, Y. Zhang, M. Pena, P. Thompson and J. J. Lavigne, Chem. Sci., 2012, 3, 1147 RSC.
- W. M. Nau, G. Ghale, A. Hennig, H. Bakirci and D. M. Bailey, J. Am. Chem. Soc., 2009, 131, 11558 CrossRef CAS PubMed.
- M. Florea, S. Kudithipudi, A. Rei, M. J. González-Úlvarez, A. Jeltsch and W. M. Nau, Chem.–Eur. J., 2012, 18, 3521 CrossRef CAS PubMed.
- J. E. Beaver and M. L. Waters, ACS Chem. Biol., 2016, 11, 643–653 CrossRef CAS PubMed.
- N. K. Pinkin, I. Liu, J. D. Abron and M. L. Waters, Chem. Eur. J., 2015, 21, 17981–17987 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc03003c |
| This journal is © The Royal Society of Chemistry 2017 |