
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic linkers control the thermosensitivity of the emission intensities from Tb(III) and Eu(III) in a chameleon polymer†
Miho
Hatanaka
*ab,
Yuichi
Hirai
c,
Yuichi
Kitagawa
c,
Takayuki
Nakanishi
c,
Yasuchika
Hasegawa
*c and
Keiji
Morokuma
*d
aDepartment of Chemistry, Faculty of Science and Engineering, Kindai University, Higashi-Osaka, Osaka 577-8502, Japan. E-mail: hatanaka@chem.kindai.ac.jp
bPRESTO, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cFaculty of Engineering, Hokkaido University, Sapporo, Hokkaido 060-8628, Japan. E-mail: hasegaway@eng.hokudai.ac.jp
dFukui Institute for Fundamental Chemistry, Kyoto University, Kyoto 606-8103, Japan. E-mail: morokuma@fukui.kyoto-u.ac.jp
First published on 25th August 2016
Abstract
Thermometers whose emission color gradually changes with temperature are called chameleon emitters. In this study, we discuss the mechanism of the thermosensitivity of the emission color of polymers that contain two lanthanides (Ln3+), e.g., [Tb0.99Eu0.01(hfa)3(linker)]n, where the Ln3+(hfa)3 complexes (hfa: hexafluoro acetylacetonato) are connected by a phosphine oxide “linker” molecule. First, the difference in the thermosensitivities of the emissions from Tb3+ and Eu3+ are discussed. With increasing temperature, the green-emission intensity from Tb3+ decreases whereas the red-emission intensity from Eu3+ does not change. This was found to originate from the different reaction barriers for the quenching of the Ln3+ excited state via the intersystem crossing (ISC) between the hfa-centered triplet state and the ground state. Next, the excitation energy transfer (EET) from Tb3+ to Eu3+ is discussed. Although the direct EET between Ln3+ atoms is negligible because of the long distance between them, stepwise EET is found to occur via the linker-centered triplet state with a reasonable barrier. Thus, we propose a new idea—thermosensitivity can be controlled by the linker as well as by the ligand (hfa). To confirm the role of the linker, four phosphine oxides were examined. The thermosensitivity dependence on the linker is validated via experimental measurements.
Introduction
Lanthanide (Ln) compounds are widely used as optical materials and sensors because they show bright visible luminescence originating from intra-4fN transitions.1–7 Typical Ln luminescent materials comprise two parts—photon antenna ligands, which absorb light, and Ln trications (Ln3+), which emit light. The energy levels of the Ln3+ excited states are independent of the surroundings; therefore, to adjust the emission intensity or intensity dependence, the excited states of the photon antenna need to be adjusted.Recently, luminescent materials that comprise more than two Ln3+ ions have gained attention as color-tunable light emitters,8–10 sensors,11–14 and photon up- and down-convertors.15–17 One of the most attractive functional materials is the “chameleon” thermometer, whose emission color changes with temperature. Fig. 1 shows a polymeric chameleon thermometer [Tb0.99Eu0.01(hfa)3(dpbp)]n (hfa: hexafluoro acetylacetonato, dpbp: 4,4′-bis(diphenylphosphoryl)biphenyl), whose emission color gradually changes from green to yellow to red as temperature increases. Note that [Tb0.99Eu0.01(hfa)3(dpbp)]n has drawn attention because of its high quantum yield; thermostability; and wide applicability to fluid dynamics, aeronautical engineering, environment engineering, and energy technology.18 The chameleon thermometer comprises Tb3+ and Eu3+ ions, which emit green and red light, respectively, photon antenna ligands, and phosphine oxide “linker” molecules (dpbp). The hfa ligand has been widely used as a photon antenna for various Ln3+ compounds,19,20 and Tb3+ complexes with hfa ligands have been used in thermometers whose green-emission intensities decrease with increase in temperature.21–23
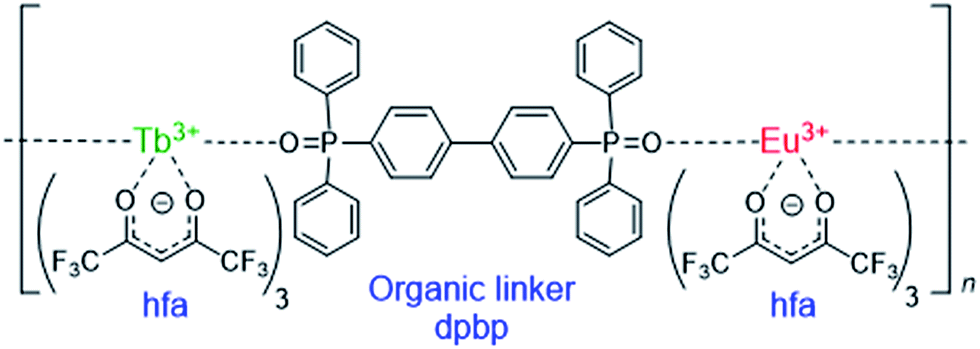 | ||
| Fig. 1 Chameleon thermometer [Ln(hfa)3(dpbp)]n (Ln = Eu and Tb). | ||
The dependence of emission color on temperature has been attributed to the difference between the emission intensities of Tb3+ and Eu3+.18 The thermosensitivities (the ratios of the decrease in emission intensity per 1 K) of [Tb(hfa)3(dpbp)]n and [Eu(hfa)3(dpbp)]n were 0.64% K−1 and <0.05% K−1, respectively, in the temperature range of 200–300 K.18 This means that the emission intensity of Tb3+ decreases as the temperature increases whereas that of Eu3+ is almost independent of the temperature. Therefore, the chameleon emitter comprising 99% Tb3+ and 1% Eu3+, i.e., [Tb0.99Eu0.01(hfa)3(dpbp)]n, shows a green emission from Tb3+ at low temperatures. As the temperature increases, the relative intensity of the red emission increases because of the decrease in the green emission.
If there is no interaction between Tb3+ and Eu3+, the thermosensitivity of the green emission from Tb3+ should be the same for [Tb0.99Eu0.01(hfa)3(dpbp)]n and [Tb(hfa)3(dpbp)]n. However, the thermosensitivity of the green emission was 0.83% K−1 for [Tb0.99Eu0.01(hfa)3(dpbp)]n; this value is larger than that for [Tb(hfa)3(dpbp)]n (0.64% K−1).18 This indicates that excitation energy transfer (EET) takes place from Tb3+ to Eu3+ (as well as to the hfa ligand) in the chameleon thermometer. In previous theoretical studies, EET between Ln3+ atoms has been discussed on the basis of direct EET mechanisms such as the Dexter and Förster mechanisms,24,25 and this can occur only at relatively short distances. For instance, Malta reported that the EET rate between two Ln3+ (Ln = Yb) ions with 10 Å distance took place mainly through the quadrupole–quadrupole coupling, however, it is an order of magnitude smaller than that of Ln3+ f–f emission.24 In the case of the chameleon thermometer,18 the distance between two Ln3+ ions is 13.6 Å; this means that the direct EET from Tb3+ to Eu3+ does not affect the quenching of Tb3+.
To design luminescence materials and sensors that comprise more than two Ln compounds, an understanding of their emission and quenching mechanisms is indispensable. In this study, we discuss the reason for the difference in the thermosensitivities of Tb3+ and Eu3+ and the mechanism of the EET from Tb3+ to Eu3+ using computational calculations. On the basis of this theoretical investigation, a new idea to control the thermosensitivity of chameleon thermometers is proposed. This proposed method is then validated via experimental measurements.
Theoretical methods
Describing the potential energy surfaces (PESs) of Ln3+ complexes
Theoretical molecular design of luminescent materials on the basis of the information on crossing points (minimal seams of crossing or conical intersections) has been reported for some organic molecules.26–30 For Ln3+ compounds, however, no study has computed and discussed the crossing points because of the difficulty of ab initio calculations for the excited states of Ln3+ compounds. To overcome this problem, we applied a reasonable approximation—the energy shift method.31 In this section, the approximation is explained on the basis of the Jablonski diagram shown in Fig. 2(a), which shows the possible emission and quenching mechanisms of Ln3+ (=Tb3+ and Eu3+) with hfa ligands. The emission process starts with ligand-centered excitation (1) from the singlet ground state (S0) to a singlet excited state (Sn), followed by an intersystem crossing (ISC) (2) from Sn to the lowest triplet state (T1). Next, spin-allowed EET (3) from the ligand T1 to the Ln3+-centered 4fN excited state (5DJ) occurs, and then Ln3+ emits light via an f–f transition (4). The mechanism of quenching of Ln3+ can also be understood from Fig. 2(a). The lifetime of the 5DJ state is long because of the parity-forbidden f–f transition. Thus, the quenching process, which starts with a backward EET (5) from the Ln3+-centered 4fN excited state (5DJ) to the ligand T1 and is followed by an ISC from ligand T1 to S0 (6), can take place when the reaction barriers of these two steps are sufficiently low. To evaluate the reaction barrier for the quenching process, the energy levels of the minimal crossing points between two potential energy surfaces (PESs) for both (5) and (6) need to be evaluated.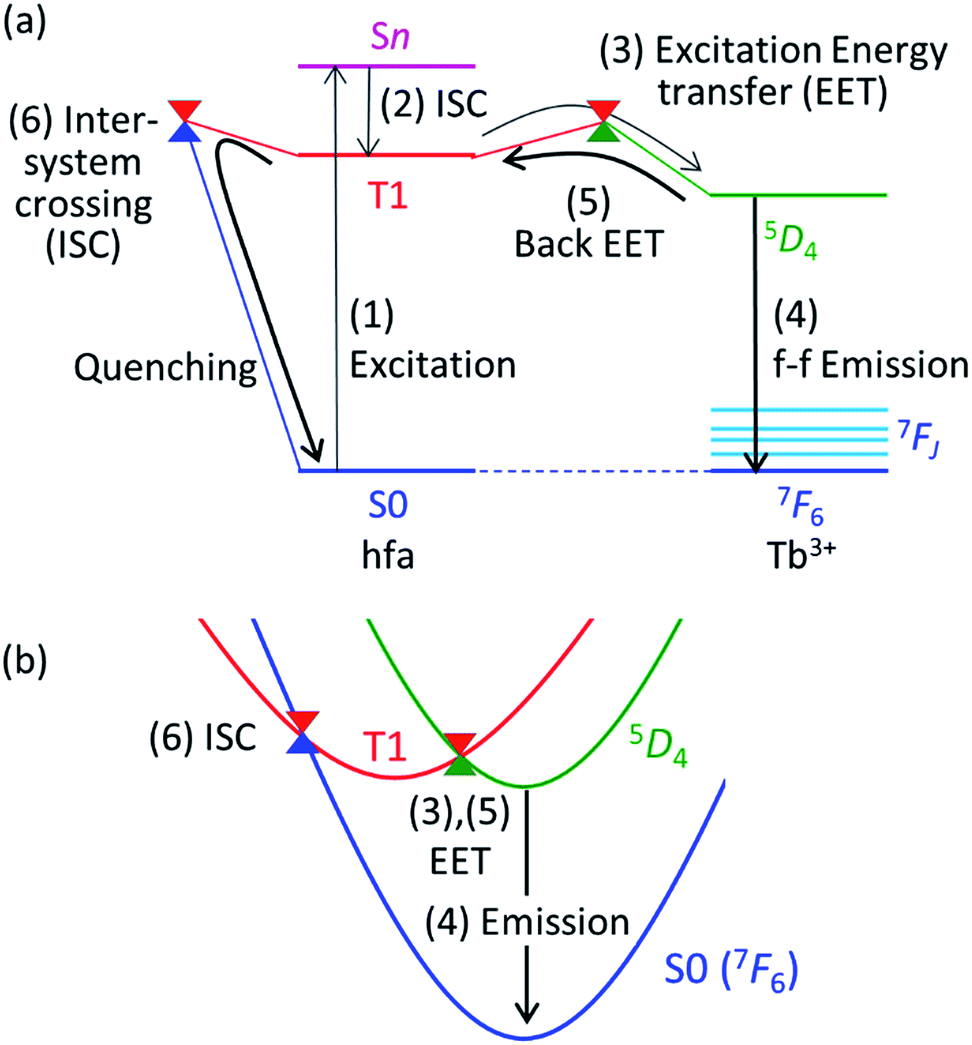 | ||
| Fig. 2 Emission and quenching mechanisms of Tb3+ coordinated by hfa ligands shown in the Jablonski diagram (a) and the schematic potential energy surfaces (PESs) (b). Sn (or T1) and 2S+1LJ represent the electronic states of hfa and Tb3+, respectively. | ||
Thus, we need to describe the three PESs shown in Fig. 2(b)—the ground state, the ligand-centered T1 state, and the Ln3+-centered excited (5DJ) state. To compute these PESs (with a reasonable computational cost), we focus on the character of the intra-4fN transition states. The PES of the Ln3+-centered excited state (5DJ) has a shape nearly identical to that of the ground state, and the excitation energy is independent of the environment because the 4f electrons are shielded from outside by the closed-shell 5s and 5p electrons. Therefore, the PES of 5DJ is described by that of the ground state, corrected by an “energy shift” (for details of the energy shift approximation, see ref. 31). For determining the energy-shift parameters of the 5DJ state, we used the experimental excitation energies of aqueous Ln3+, i.e., 580 nm and 490 nm for 5D0 of Eu3+ and 5D4 of Tb3+, respectively.3 Using this scheme, the electronic states of Ln3+ become identical (7FJ) for all the three states. Thus, Ln3+ can be described using the Stuttgart–Dresden large-core relativistic effective core potentials (RECP),36 in which the 5s, 5p, 5d, and 6s electrons are explicitly considered and the 4f electrons are included in the RECP. Therefore, for the three states shown in Fig. 2(b), only the lowest singlet (S0) and the lowest triplet (T1) states need to be computed explicitly using a conventional ground-state energy calculation method. This approximation is applicable to model complexes for chameleon polymers that comprise Tb3+ and Eu3+ (details are given in ESI†).
Computational details
All the PESs in this study were calculated using the ONIOM method,32 in which the high- and low-level regions were described using the density functional theory (DFT) with the ωB97XD functional,33 and the molecular mechanics (MM) with the UFF force field parameters,34 and the QEq method.35 The basis set for the high-level region was the (8s7p6d5f2g)/[6s5p5d3f2g] RECP basis set36,37 for Ln3+ and cc-pVDZ38 for others. For Ln(hfa)3(tppo)2 (Ln = Eu, Tb; tppo: triphenylphosphine oxide) complexes, we carried out full geometry optimizations of local minima (LMs) and minimum structures on the seams of crossing (MSXs) on and between the ground state, hfa-centered triplet state, and Ln3+-centered excited state. For the chameleon-model complexes, the geometry optimizations, with freezing of the position of the surroundings, were applied on the basis of the crystal structures18,39 (details are given in ESI†). The energy levels of LMs and MSXs of the chameleon-model complexes were also calculated using different DFT functionals as shown in Tables S2 and S3.† Though their energy levels depended slightly on the functional, the magnitude relation was independent of the functional. All optimizations were performed via the Global Reaction Route Mapping (GRRM) program,40,41 using the energies and energy derivatives computed using the Gaussian09 program.42Experimental methods
The linker ligands dpbp, dpb, dppcz, and dpbt (dpb: 1,4-bis(diphenylphosphoryl)benzene, dppcz: 3,6-bis(diphenylphosphoryl)-9-phenylcarbazole, dpbt: 4,4′-bis(diphenylphosphoryl)bithiophene) were prepared as described in ref. 39. The ligands (dpbp: 0.44 g, dpb: 0.38 g, dppcz: 0.51 g, dpbt: 0.45 g, 1 eq.) were dissolved in methanol (30 mL). Tb(hfa)3(H2O)2 (0.65 g, 0.99 eq.) and Eu(hfa)3(H2O)2 (6.5 mg, 0.01 eq.) were dissolved in methanol (30 mL) and were added to each ligand/methanol solution. The mixtures were heated to reflux while stirring for 3 h to give white precipitates of [Tb0.99Eu0.01(hfa)3(dpbp)]n, [Tb0.99Eu0.01(hfa)3(dpb)]n, [Tb0.99Eu0.01(hfa)3(dppcz)]n, and [Tb0.99Eu0.01(hfa)3(dpbt)]n. The precipitates were filtered, washed with methanol and chloroform several times, and dried in vacuo (details are given in ESI.†)Results and discussion
Reason for the difference in the thermosensitivities of the emission intensities of Tb3+ and Eu3+
To understand the difference in the thermosensitivities of the emission intensities of Tb3+ and Eu3+, we computed the LMs and MSXs of the model complexes Ln(hfa)3(tppo)2. Fig. 3 and 4 show the structures and the energy levels of the LMs and MSXs optimized using the ONIOM method. The electronic and Gibbs free-energy levels shown in Fig. 4 are quite similar; therefore, we will discuss the energy levels mainly on the basis of the electronic energies. As shown in Fig. 3(b) and 4(a), the geometry and electronic energy level of the ligand (hfa)-centered T1 state (61.9 kcal mol−1) are similar to those of the Tb3+-centered 5D4 state (58.6 kcal mol−1). This T1 energy level is consistent with the experimental energy level of the hfa-centered T1 state of Gd(hfa)3(H2O)2 (63.5 kcal mol−1).23 The MSX between the T1 and the 5D4 states, where the EET between hfa and Tb3+ occurs, also has a geometry and energy level similar to those of the abovementioned two states. Thus, forward and backward EET can occur almost without a barrier. Conversely, the electronic energy of the MSX for the ISC from the ligand-centered T1 to S0 is 14.3 kcal mol−1 higher than that of the Tb3+-centered 5D4. Thus, the ISC of the ligand via the T1/S0 MSX is the rate-determining step for the quenching process. Comparing the structures, one of the hfa ligands (bold-faced in Fig. 3(b)) has a bent structure at the T1/S0 MSX for the ISC, whereas all the hfa ligands are planar at all other LMs and MSX. This means that the excitation is localized on an hfa ligand and that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bending motion of this hfa ligand induces vibrational relaxation.
O bending motion of this hfa ligand induces vibrational relaxation.
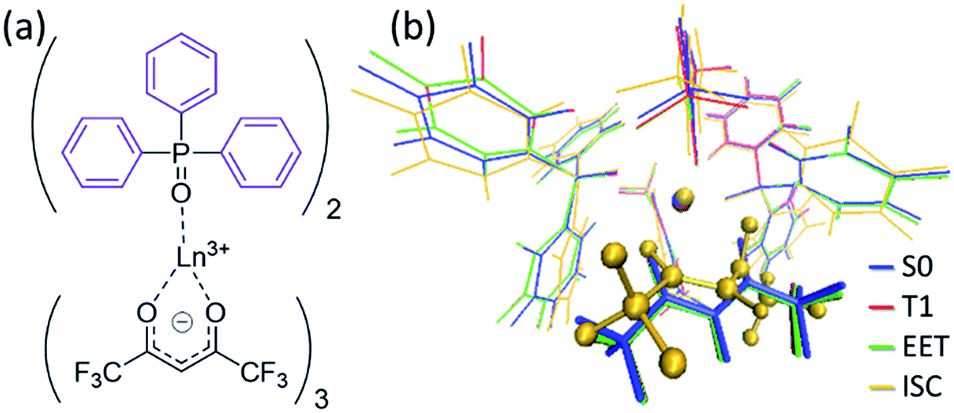 | ||
| Fig. 3 (a) Chemical structure and (b) the optimized geometries of the Tb(hfa)3(tppo)2 complex. Blue, red, green, and yellow in (b) are the LMs on S0, T1, and the MSXs for the EET and ISC processes, respectively. Geometrical optimizations were carried out using the ONIOM(ωB97XD:UFF) method, in which the phenyl groups of tppo (purple in (a)) and others (black in (a)) were treated as the low- and high-level regions, respectively. | ||
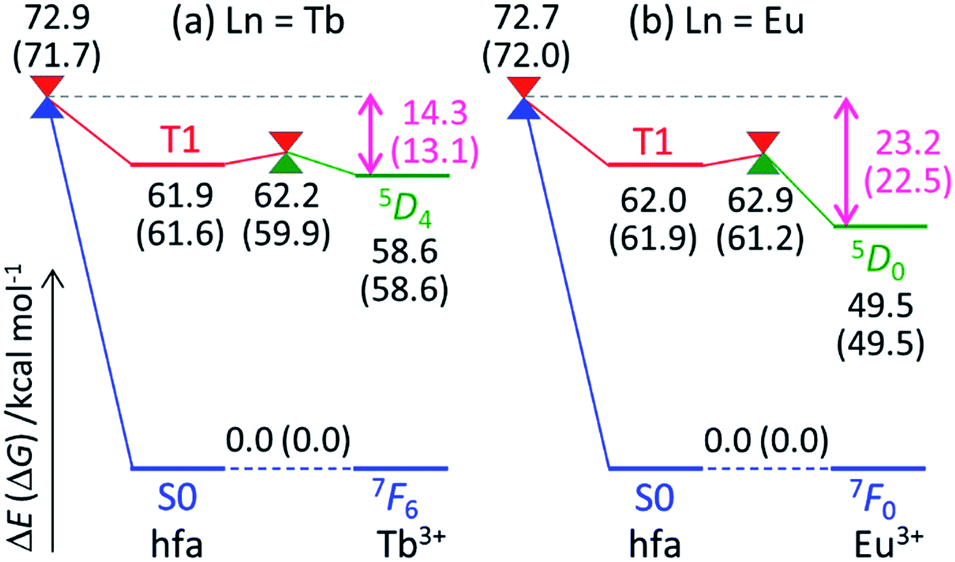 | ||
| Fig. 4 Electronic and Gibbs free energies (in parentheses; kcal mol−1) of the LMs and MSXs on and between PESs, respectively, for Ln(hfa)3(tppo)2 (Ln = (a) Tb, (b) Eu) calculated using the ONIOM(ωB97XD:UFF) level of theory. | ||
Next, we focus on the differences between the Tb3+ and Eu3+ complexes. The geometries of the LMs and MSXs of the Eu3+ complex are similar to those of the Tb3+ complex, as shown in Fig. S1.† Their energy profiles are also quite similar, except for the energy levels of the Ln3+-centered excited states (5DJ), as shown in Fig. 4. Thus, the barrier for the quenching process of the Eu3+ complex is 8.9 kcal mol−1 higher than that of the Tb3+ complex; this makes quenching the red emission from Eu3+ difficult. To compare the timescales for the emission and quenching processes, their rate constants were estimated. The rate constant of the Tb3+ emission was estimated using the inverse of the experimental emission lifetime, i.e., 0.8 ms, measured for Tb(hfa)3(tppo)2 at 80 K.23 The rate constants of the quenching processes were evaluated via the Gibbs free activation energies (13.1 and 22.5 kcal mol−1 for the Tb3+ and Eu3+ complexes, respectively) using transition state theory.43–45 The rate constants of emission are comparable to those of quenching for the Tb3+ and Eu3+ complexes at 300 K and 500 K, respectively (see Table S1†). These values are consistent with the experimental facts that emission intensity decreases for the Tb3+ complex and does not change for the Eu3+ complex as the temperature increases in the range of 200–300 K.18,21–23
Mechanism of the EET from Tb3+ to Eu3+ in the chameleon thermometer
Next, we focus on the thermosensitivity of the luminescence of the chameleon emitter.18 As mentioned above, direct EET from Tb3+ to Eu3+ is negligible.24 Thus, in order to consider an alternative pathway for the EET from Tb3+ to Eu3+, we focus on the phosphine oxide “linker” (dpbp in Fig. 1). Fig. 5 shows the electronic energy levels of the LMs and MSXs of the model complex, which were constructed on the basis of the crystal structure and were optimized (see the computational details in ESI†). The early stage of the emission process is the same as that of Tb3+ and Eu3+ complexes with hfa ligands; the hfa ligand absorbs the light and the EET from hfa-centered T1 state to Tb3+- or Eu3+-centered 5DJ excited state takes place as shown in Fig. 5. Compared with the Tb3+-centered 5D4 reference, the LMs on the linker-centered T1 state is 11.8 kcal mol−1 higher, and the MSXs between linker-centered T1 and Tb3+- and Eu3+-centered 5DJ are 12.2 and 12.0 kcal mol−1 higher, respectively. Thus, the stepwise EET from Tb3+ to Eu3+via the linker-centered T1 state can occur with a reasonable reaction barrier, which is lower than the barrier (at 14.3 kcal mol−1) for the quenching via an ISC on the hfa ligand. Additionally, we obtained a MSX for the ISC between the linker-centered T1 state to the ground state, which is 28.3 kcal mol−1 higher than the Tb3+-centered 5D4 state. Toward the ISC, one of the C–H bonds in the phenyl group bends, as shown in Fig. S5.† The effect of the ISC via linker T1 is almost negligible due to the too high barrier. Thus, there exist two comparable quenching pathways from Tb3+—the ISC on hfa and the EET to Eu3+.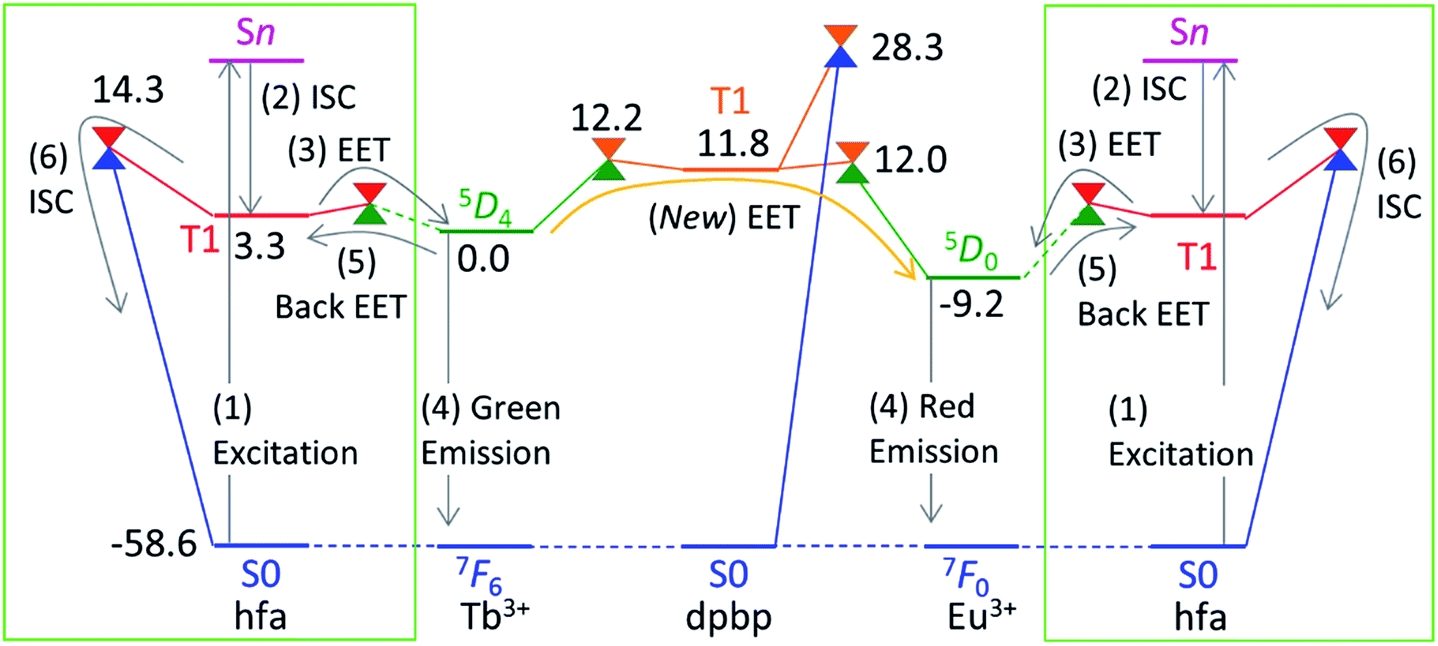 | ||
| Fig. 5 Electronic energy levels (in kcal mol−1) of the LMs and MSXs for the model complex of the chameleon thermometer comprising the dpbp linker calculated at the ONIOM(ωB97XD:UFF) level of theory. The energy levels of the hfa-centered T1 and MSXs (in the green box) are obtained from Fig. 4. For convenience, the energy zero (5D4) is shifted from zero (the ground state) in Fig. 4. | ||
Computational prediction of the thermosensitivity of chameleon thermometers comprising different linker molecules
As shown in Fig. 5, the linker-centered T1 state is thoroughly involved in the quenching of Tb3+. This suggests that the barrier for the quenching of Tb3+, i.e., the thermosensitivity of the emission from Tb3+, could be controlled by changing the linker molecule. In other words, we may be able to design a thermometer with the desired thermosensitivity by optimizing the energy level of the linker-centered T1 state and the ISC on the linker.To confirm this new quenching mechanism, other polymers that comprise three different linker molecules, i.e., [Ln(hfa)3(dpb)]n, [Ln(hfa)3(dppcz)]n, and [Ln(hfa)3(dpbt)]n, were examined. Fig. 6 shows the energy levels of the LMs and MSXs of the model complexes, each involving a linker (dpb, dppcz, or dpbt), Eu(hfa)3, Tb(hfa)3, and surroundings, whose structures were constructed on the basis of crystal structures and were optimized (as shown in Fig. S6†).39 The energy level of the linker-centered T1 state is the highest for dpb, followed by dppcz, dpbp, and dpbt, and they are 23.0, 19.1, 11.8, and −7.9 kcal mol−1 higher, respectively, than the energy level of the Tb3+-centered 5D4 state. The energy levels and geometries of the MSXs of the EET between Ln3+ and the linker are similar to those of the linker-centered T1 states, especially when the linker-centered T1 state is less stable than 5D4. Thus, the barrier for the EET from Tb3+ to Eu3+ is essentially determined by the linker-centered T1 state. In the case of the dpbt model complex, the energy levels of the linker-centered T1 state and the MSXs for the EET between Ln3+ and dpbt are lower than that of 5D4. Thus, the EET from Tb3+ to Eu3+ should be an almost barrierless process.
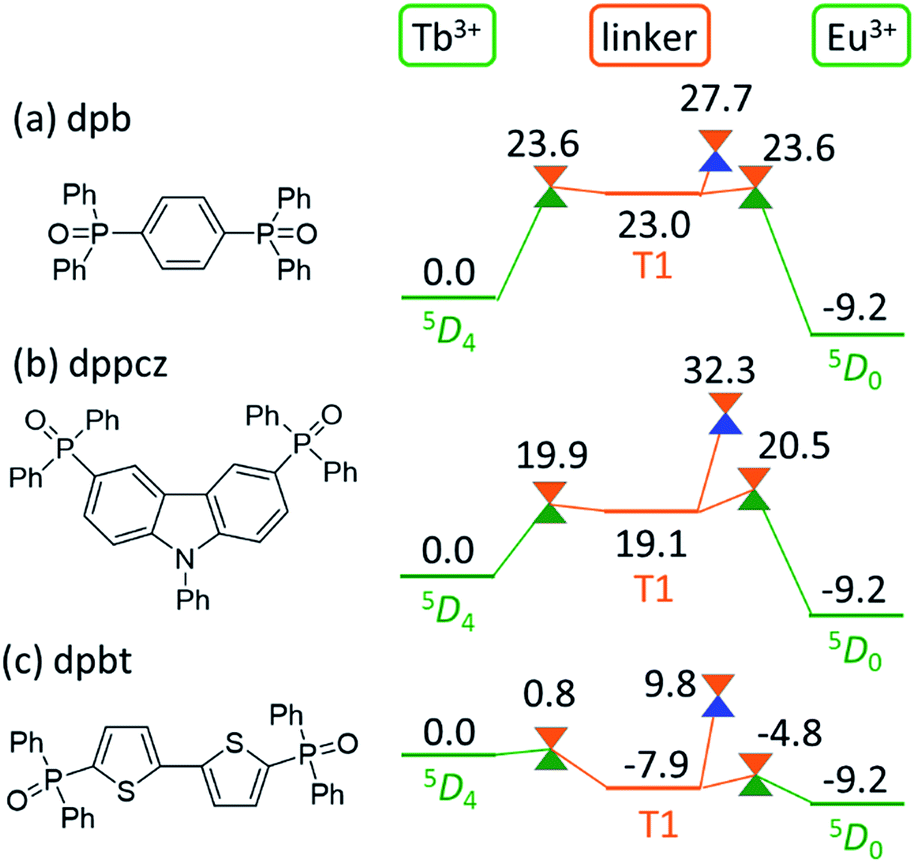 | ||
| Fig. 6 Chemical structures of the linker molecules and electronic energy levels (in kcal mol−1) of the LMs and MSXs for the model complexes involving (a) dpb, (b) dppcz, and (c) dpbt. Ln3+-centered excited 5DJ states, the linker-centered T1 states, and the ground states are shown in green, orange, and blue, respectively. | ||
Next, we focus on the MSXs for the ISC from the linker-centered T1 state to the ground state. The order of their energy levels is different from that of the T1 states. The energy level of the MSX on the linker is the highest for dppcz, followed by dpbp, dpb, and dpbt, and they are 32.3, 28.3, 27.7, and 9.8 kcal mol−1 higher, respectively, than the energy level of the Tb3+-centered 5D4 state. These barriers are too high to affect the quenching rate of Tb at room temperature, except for dpbt. The MSX for the ISC on dpbt is lower than that for the ISC on hfa (14.3 kcal mol−1). Thus, the quenching of Eu3+ and Tb3+ in this polymer should occur much faster than in other polymers. Note that the structural changes from the linker-centered T1 minimum to the ISC are similar for the model complexes comprising dppcz, dpbp, and dpb; one of the C–H bonds in the phenyl group bends (shown in Fig. S7†). The reason for the instability of the ISC in dppcz can be attributed to the packing effect from the surroundings, because the stability of the ISC for isolated linker molecules is similar (see Fig. S8†).
Experimental confirmation of the EET mechanism via the linker-centered triplet state
Finally, we compared the computational predictions with the experimental thermosensitivities. The three polymers, [Tb0.99Eu0.01(hfa)3(X)]n (X = dpb, dppcz, and dpbt), were synthesized and their temperature-dependent emission spectra in the temperature range 100–450 K in the solid state were observed. Fig. 7 shows these spectra for the polymers along with that of the original chameleon thermometer, [Tb0.99Eu0.01(hfa)3(dpbp)]n. The emission spectrum of [Tb0.99Eu0.01(hfa)3(dpbt)]n is not shown in Fig. 7 because no emission was observed. This is consistent with the computational result, which showed barrierless quenching for Tb and a low barrier for the quenching of Eu3+via dpbt. The other polymers exhibited different temperature sensitivities. The sensitivities in the temperature range of 200–300 K were 0.83% K−1, 0.82% K−1, and 0.45% K−1 for [Tb0.99Eu0.01(hfa)3(X)]n, in which X = dpbp, dpb, and dppcz, respectively. Compared to the original chameleon thermometer [Tb0.99Eu0.01(hfa)3(dpbp)]n, [Tb0.99Eu0.01(hfa)3(dppcz)]n showed an inferior temperature sensitivity; this can be attributed to the higher barrier for the EET from Tb3+ to Eu3+via the linker-centered T1 state. [Tb0.99Eu0.01(hfa)3(dpb)]n had a slightly inferior temperature sensitivity than the original chameleon thermometer even though the barrier for the EET from Tb3+ to Eu3+ was 11.3 kcal mol−1 higher. The reason of the discrepancy between experiment and theoretical prediction (i.e. the high sensitivity of [Tb0.99Eu0.01(hfa)3(dpb)]n) could be attributed by the mixing of charge transfer to the hfa-centered triplet states. To examine the characters of the triplet excited states, the excited energies of the Franc–Condon region of the model complexes comprising dpbp, dpb and dppcz were calculated using the full-QM time-dependent DFT (TDDFT) method as shown in Fig. S9 and Table S4.† The six lower and the seventh triplet states are the hfa-centered and the linker-centered excited states, respectively. The linker-centered triplet states of the model complexes comprising dpbp, dpb and dppcz are well localized on the linker as shown in Fig. S10,† which confirms the adequacy of our ONIOM calculation scheme for Fig. 5 and 6. Moreover, the hfa-centered triplet states of the model complex comprising dpb are localized on each hfa and the excitation energies are close to the results in Fig. 4. On the other hand, the six lower excited states of the model complexes comprising dpbp and dppcz are about 2–4 kcal mol−1 destabilized due to the mixing of charge transfer component from hfa to the linker. Therefore, the high thermosensitivity of [Tb0.99Eu0.01(hfa)3(dpb)]n could be attributed to the faster quenching via the ISC on hfa ligands.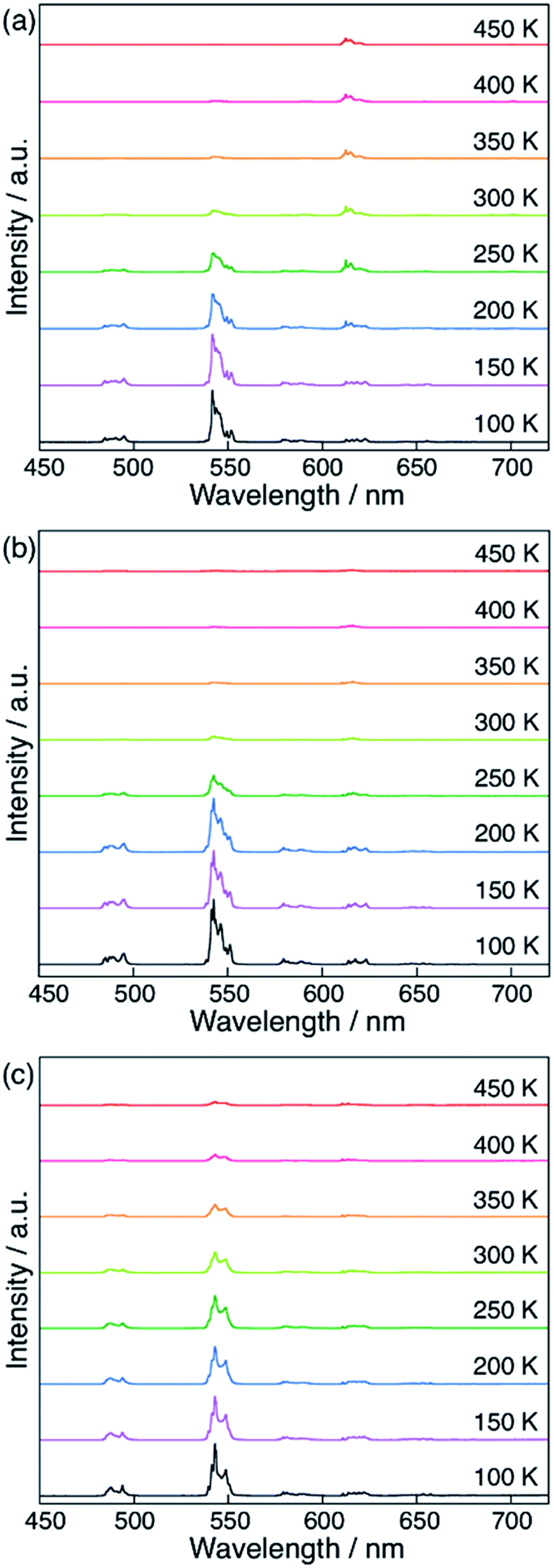 | ||
| Fig. 7 Temperature-dependent emission spectra of (a) [Tb0.99Eu0.01(hfa)3(dpbp)]n, (b) [Tb0.99Eu0.01(hfa)3(dpb)]n, and (c) [Tb0.99Eu0.01(hfa)3(dppcz)]n in solid state (λex = 380 nm). The spectra for [Tb0.99Eu0.01(hfa)3(dpbt)]n are not shown because they have very weak emission intensities. | ||
As shown above, it was confirmed that thermosensitivity depends on the linker molecules. This phenomenon was mainly explained by the difference in the energy levels of the linker-centered T1 state. Although the numerical estimation of the temperature sensitivities is difficult by our calculation scheme due to the mixing of the charge transfer to the hfa-centered excited states, we can accelerate the development of the chameleon thermometers by focusing on the linkers whose T1 energies are higher than the energy level of the emissive state of Tb3+ (5D4). The effect of the Tb/Eu ratio was also not considered in our computational models shown in Fig. 5 and 6, however, we expect that the effect of the EET from Tb3+ to Eu3+ could increase as the amount of Eu3+ increases. In fact, the thermosensitivity of [Tb(1−n)Eun(hfa)3(dpbp)]n with a different Tb3+/Eu3+ ratio was observed in a previous study.46 The effect of the EET from Tb3+ to Eu3+ increased as the amount of Eu3+ increased.
Conclusions
In this study, we observed that there are three quenching pathways from the Tb3+-centered excited state in chameleon-type polymers. Moreover, we found that the thermosensitivity of the emission from Tb3+ can be controlled by changing the reaction barriers for the three quenching pathways. The first quenching pathway is through an ISC from the β-diketone ligand-centered T1 state to the ground state.31 The second is through the stepwise EET from the excited state in Tb3+ to that in Eu3+; this pathway is determined mainly by the linker-centered T1 state. The third is through an ISC from the linker-centered T1 state to the ground state. The experimental linker-dependence of thermosensitivity confirms the contributions of the newly proposed linker-centered T1 quenching pathway. Our proposed strategy to use linker T1 for designing a chameleon thermometer should be applicable not only to polymers but also to other materials, such as metal–organic frameworks and nanoclusters.11–14Acknowledgements
The authors are grateful to Prof. Satoshi Maeda of Hokkaido University for the Global Reaction Route Mapping (GRRM) code and discussions on the GRRM method. This work was in part supported by the JST through PRESTO program to MH and by JSPS KAKENHI Grant Numbers JP15KT0142, JP16H00853 (to MH), JP24245005, JP26105733 and JP15H02158 (to KM). MH acknowledges the Fukui Fellowship of Kyoto University and the Collaborative Research Program for Young Scientists of ACCMS and IIMC, Kyoto University. We also acknowledge the computer resources provided by the Academic Center for Computing and Media Studies (ACCMS) at the Kyoto University and by the Research Center of Computer Science (RCCS) at the Institute for Molecular Science.Notes and references
- J. C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- J. C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- S. V. Eliseeva and J. C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed.
- X. H. Wang, H. J. Chang, J. Xie, B. Z. Zhao, B. T. Liu, S. L. Xu, W. B. Pei, N. Ren, L. Huang and W. Huang, Coord. Chem. Rev., 2014, 273, 201–212 CrossRef.
- Y. J. Cui, B. L. Chen and G. D. Qian, Coord. Chem. Rev., 2014, 273, 76–86 CrossRef.
- P. K. Chen, Q. C. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590–11593 CrossRef CAS PubMed.
- H. B. Zhang, X. C. Shan, Z. J. Ma, L. J. Zhou, M. J. Zhang, P. Lin, S. M. Hu, E. Ma, R. F. Li and S. W. Du, J. Mater. Chem. C, 2014, 2, 1367–1371 RSC.
- N. Kerbellec, D. Kustaryono, V. Haquin, M. Etienne, C. Daiguebonne and O. Guillou, Inorg. Chem., 2009, 48, 2837–2843 CrossRef CAS PubMed.
- Y. J. Cui, F. L. Zhu, B. L. Chen and G. D. Qian, Chem. Commun., 2015, 51, 7420–7431 RSC.
- C. D. S. Brites, P. P. Lima, N. J. O. Silva, A. Millan, V. S. Amaral, F. Palacio and L. D. Carlos, New J. Chem., 2011, 35, 1177–1183 RSC.
- D. Ananias, F. A. A. Paz, D. S. Yufit, L. D. Carlos and J. Rocha, J. Am. Chem. Soc., 2015, 137, 3051–3058 CrossRef CAS PubMed.
- X. T. Rao, T. Song, J. K. Gao, Y. J. Cui, Y. Yang, C. D. Wu, B. L. Chen and G. D. Qian, J. Am. Chem. Soc., 2013, 135, 15559–15564 CrossRef CAS PubMed.
- H. Dong, L. D. Sun and C. H. Yan, Chem. Soc. Rev., 2015, 44, 1608–1634 RSC.
- P. Ramasamy, P. Manivasakan and J. Kim, RSC Adv., 2014, 4, 34873–34895 RSC.
- M. V. DaCosta, S. Doughan, Y. Han and U. J. Krull, Anal. Chim. Acta, 2014, 832, 1–33 CrossRef CAS PubMed.
- K. Miyata, Y. Konno, T. Nakanishi, A. Kobayashi, M. Kato, K. Fushimi and Y. Hasegawa, Angew. Chem., Int. Ed. Engl., 2013, 52, 6413–6416 CrossRef CAS PubMed.
- Y. Hasegawa, Y. Kimura, K. Murakoshi, Y. Wada, J. H. Kim, N. Nakashima, T. Yamanaka and S. Yanagida, J. Phys. Chem., 1996, 100, 10201–10205 CrossRef CAS.
- Y. Hasegawa, Y. Wada and S. Yanagida, J. Photochem. Photobiol., C, 2004, 5, 183–202 CrossRef CAS.
- S. Katagiri, Y. Hasegawa, Y. Wada and S. Yanagida, Chem. Lett., 2004, 33, 1438–1439 CrossRef.
- S. Katagiri, Y. Hasegawa, Y. Wada, K. Mitsuo and S. Yanagida, J. Alloys Compd., 2006, 408–412, 809–812 CrossRef CAS.
- S. Katagiri, Y. Tsukahara, Y. Hasegawa and Y. Wada, Bull. Chem. Soc. Jpn., 2007, 80, 1492–1503 CrossRef CAS.
- O. L. Malta, J. Non-Cryst. Solids, 2008, 354, 4770–4776 CrossRef CAS.
- V. S. Mironov, Spectrochim. Acta, Part A, 1998, 54, 1607–1614 CrossRef.
- M. Filatov and M. Olivucci, J. Org. Chem., 2014, 79, 3587–3600 CrossRef CAS PubMed.
- S. Maeda, Y. Harabuchi, T. Taketsugu and K. Morokuma, J. Phys. Chem. A, 2014, 118, 12050–12058 CrossRef CAS PubMed.
- S. Suzuki, S. Maeda and K. Morokuma, J. Phys. Chem. A, 2015, 119, 11479–11487 CrossRef CAS PubMed.
- S. Maeda, T. Taketsugu, K. Ohno and K. Morokuma, J. Am. Chem. Soc., 2015, 137, 3433–3445 CrossRef CAS PubMed.
- Y. Harabuchi, T. Taketsugu and S. Maeda, Phys. Chem. Chem. Phys., 2015, 17, 22561–22565 RSC.
- M. Hatanaka and K. Morokuma, J. Chem. Theory Comput., 2014, 10, 4184–4188 CrossRef CAS PubMed.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. F. Ke, F. Y. Liu, H. B. Li, L. N. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- A. K. Rappé and W. A. Goddard, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef.
- M. Dolg, H. Stoll, A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173–194 CrossRef CAS.
- M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1993, 85, 441–450 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- K. Miyata, T. Ohba, A. Kobayashi, M. Kato, T. Nakanishi, K. Fushimi and Y. Hasegawa, ChemPlusChem, 2012, 77, 277–280 CrossRef CAS.
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683–3701 RSC.
- S. Maeda, Y. Osada, T. Taketsugu, K. Morokuma and K. Ohno, GRRM14, http://www.grrm.chem.tohoku.ac.jp/GRRM/index_e.html.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, A. Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09, Rev. D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H. Eyring and A. E. Stearn, Chem. Rev., 1939, 24, 253–270 CrossRef CAS.
- J. Z. Pu, J. L. Gao and D. G. Truhlar, Chem. Rev., 2006, 106, 3140–3169 CrossRef CAS PubMed.
- T. Ziegler and J. Autschbach, Chem. Rev., 2005, 105, 2695–2722 CrossRef CAS PubMed.
- Y. Hirai, T. Nakanishi, K. Miyata, K. Fushimi and Y. Hasegawa, Mater. Lett., 2014, 130, 91–93 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational and experimental details, and the Cartesian coordinates. See DOI: 10.1039/c6sc03006h |
| This journal is © The Royal Society of Chemistry 2017 |