
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reagent-controlled enantioselectivity switch for the asymmetric fluorination of β-ketocarbonyls by chiral primary amine catalysis†
Yang'en
You
ab,
Long
Zhang
abc and
Sanzhong
Luo
*abc
aBeijing National Laboratory for Molecule Sciences (BNLMS), Key Laboratory for Molecular Recognition and Function, Institute of Chemistry, The Chinese Academy of Sciences, Beijing 100190, China. E-mail: luosz@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cCollaborative Innovation Center of Chemical Science and Engineering, Tianjin, 300071, China
First published on 26th August 2016
Abstract
A reagent-controlled enantioselectivity switch was uncovered in the asymmetric α-fluorination of β-ketocarbonyls by a chiral primary amine catalyst. By a simple swap of fluorination reagents, both enantiomers of the quaternary fluorination adducts could be obtained with good yields and high enantioselectivity. Mechanistic studies disclosed dual H-bonding and electrostatic stereocontrolling modes for the catalysis.
Asymmetric catalysis is arguably the most effective and atom-economic approach for access to optically pure compounds. Using a single chiral catalyst to get two enantiomeric products is attractive as it eliminates the need for the synthesis of catalyst enantiomers, which is not a trivial effort in most cases.1 Recently, a switch of enantioselectivity has been noted in a number of metal catalyzed reactions by simply varying the solvent, temperature and additives without structural modifications of the chiral ligands.2 However, such an external switch with good enantioselectivity for both enantiomers is still very rare for organocatalytic reactions.3 Nagasawa successfully achieved a solvent-switch of enantioselection in an asymmetric Mannich reaction with middle to high ee. Maruoka developed asymmetric aldol and Mannich reactions by an achiral-acid-additive induced switch. Matsubara reported a procedure-controlled enantioselectivity switch with moderate ee. Herein, we reported an enantioselective switch for synthetically important asymmetric fluorination reactions by two different stereocontrolling modes.
Catalytic enantioselective construction of carbon–fluorine bonds is of significant synthetic interest due to the prevalence of fluorinated drugs and agricultural agents.4d,h In this regard, enamine catalysis has appeared as a prominent strategy for the fluorination of aldehydes and ketones. Pioneering works by the groups7 of Jørgensen, Barbas and MacMillan have achieved asymmetric α-fluorination of linear aldehydes with high enantioselectivity. The reaction has recently been extended to α-branched aldehydes by primary amine catalysis.8 In contrast, the asymmetric fluorination of ketones by amine catalysis has remained far less developed, particularly for branched ketones. Two elegant contributions along this line have been recently reported for the reactions of cyclic ketones by the research groups of MacMillan9 and Toste.10 However, the asymmetric α-fluorination of acyclic ketones remains largely undeveloped not only in aminocatalysis, but in general in asymmetric catalysis. Previously, Lewis acid catalysis (Scheme 1, I) has been frequently attempted in the asymmetric fluorination of acyclic ketones,4–6 but successes have only been seen with β-ketoesters bearing bulky (e.g. t-butyl) ester groups.5 We have pursued the fluorination reaction of acyclic β-ketocarbonyls using our primary amine catalysts12 and during this process we observed an unprecedented reagent controlled switch of enantioselectivity. Thus, by simply choosing two readily available fluorination reagents, one single chiral primary amine catalyst would be able to provide two enantiomers, respectively, bearing fluorinated quaternary centers with good enantioselectivity in both cases (Scheme 1, II).
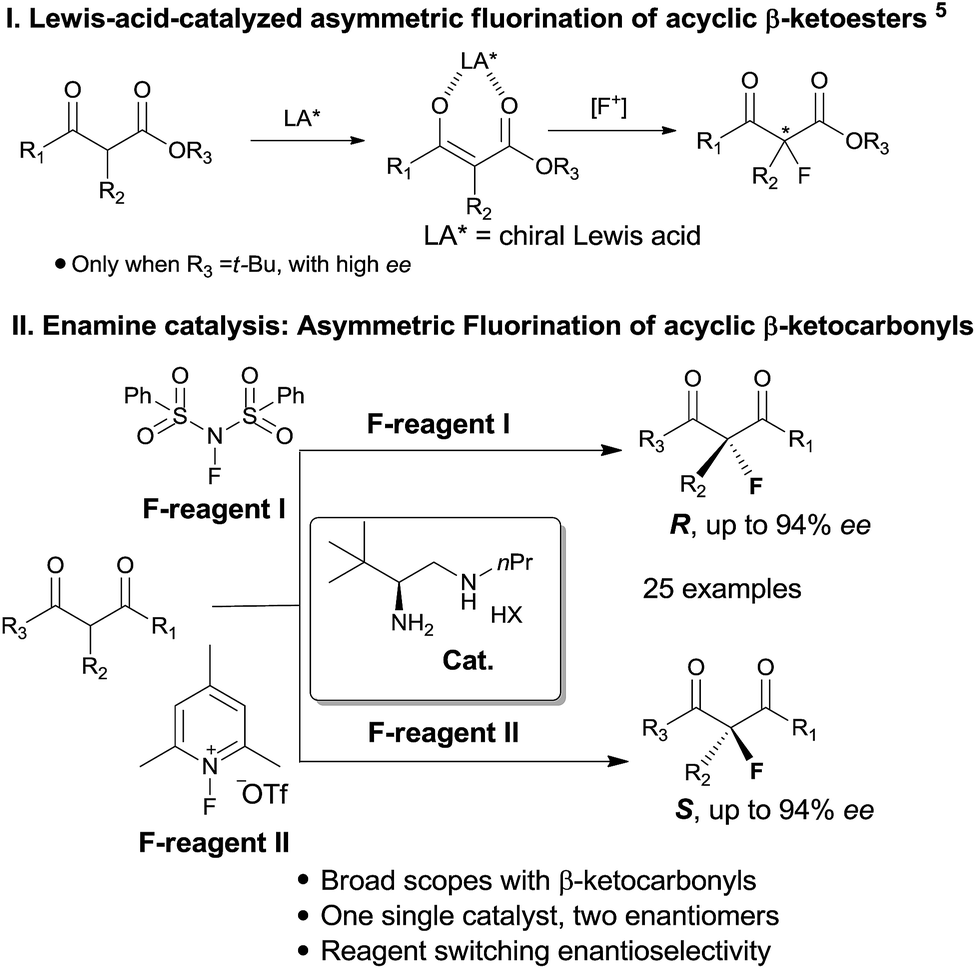 | ||
| Scheme 1 Asymmetric α-fluorination of cyclic and acyclic ketones. | ||
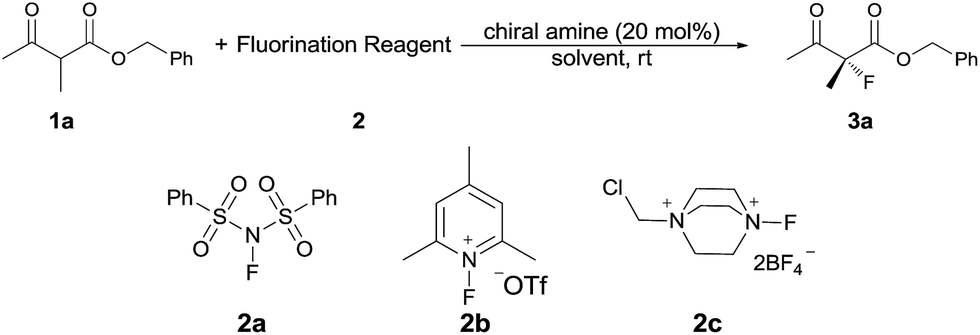
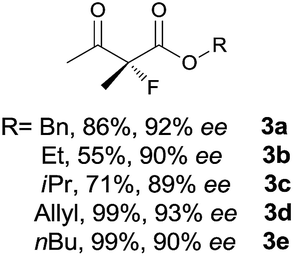
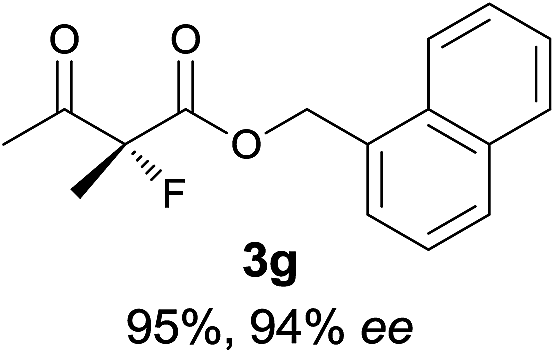
Our initial investigation was performed with acetoacetate 1a in the presence of our typical primary amine catalyst I/TfOH. A quick survey of different fluorination reagents was conducted. The reaction proceeded smoothly in all the tests and a switch of chiral induction was clearly noted among different fluorination reagents. While the reaction with NFSI (2a) gave 51% ee (Table 1, entry 1), the use of 2b and 2c led to −71% ee and −38% ee, respectively, with opposite chiral induction (Table 1, entries 2 and 3). A switch of chiral induction was generally observed with other primary–tertiary diamine catalysts such as IV and V (Table 1, entries 7–10). Though enantioselectivity varied in different solvents (see the ESI for details†), the switching phenomenon was uniformly observed (Table 1, entries 4–6 vs. 1–3). In all the cases examined, fluorination reagent 2b showed better enantioselectivity than 2c. The reagents 2a and 2b were then selected for subsequent optimization with the aim to find a generally applied enantio-switching catalytic system (see the ESI for details†). In this regard, we were delighted to identify a simple primary–secondary diamine II that gave good enantioselectivity in both of the switching reactions (Table 1, entries 11 and 12). Thus, the best reaction conditions are a combination of II/(DNBA I) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 2a as the fluorination reagent in chloroform at room temperature, providing the product with 86% yield and 92% ee, and the configuration of the product is R-configuration. On the other hand, when 2b was used as the fluorination reagent, by the same primary amine II/(DNBA II) in methanol, the desired product was obtained with 95% yield and −90% ee, and the product was S-configuration. At this stage, alteration of the acidic additive from TfOH to dinitrobenzoic acid was found to give a small but noticeable improvement in enantioselectivity (Table 1, entries 13 and 14 vs. 11 and 12). An additional benefit of the use of dinitrobenzoic acid is the ease in handling and manipulation as the resulting salts are crystal solids and bench stable. Adamantyl primary amine III, a close analogue of II, was also found to deliver an excellent −93% ee for the S-selective process, however, this catalyst performed rather poorly in the R-selective process with only 72% ee (Table 1, entries 15 and 16). This observation indicated a critical balance of steric hindrance for both the two enantio-switching processes.
:1) with 2a as the fluorination reagent in chloroform at room temperature, providing the product with 86% yield and 92% ee, and the configuration of the product is R-configuration. On the other hand, when 2b was used as the fluorination reagent, by the same primary amine II/(DNBA II) in methanol, the desired product was obtained with 95% yield and −90% ee, and the product was S-configuration. At this stage, alteration of the acidic additive from TfOH to dinitrobenzoic acid was found to give a small but noticeable improvement in enantioselectivity (Table 1, entries 13 and 14 vs. 11 and 12). An additional benefit of the use of dinitrobenzoic acid is the ease in handling and manipulation as the resulting salts are crystal solids and bench stable. Adamantyl primary amine III, a close analogue of II, was also found to deliver an excellent −93% ee for the S-selective process, however, this catalyst performed rather poorly in the R-selective process with only 72% ee (Table 1, entries 15 and 16). This observation indicated a critical balance of steric hindrance for both the two enantio-switching processes.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Amine catalyst | Fluorination reagent | Solvent | Yieldb (%) | eec (%) |
| a General conditions: 1a (0.075 mmol), 2 (0.05 mmol), amine catalyst (20 mol%) in solvent at r.t. for 24 h. b Isolated yield. c Determined by HPLC on a chiral stationary phase. d DNBA-I: 2,4-(NO2)2PhCO2H; DNBA-II: 3,4-(NO2)2PhCO2H. | |||||
| 1 | I/TfOH | 2a | CHCl3 | 41 | 51 |
| 2 | I/TfOH | 2b | CHCl3 | 75 | −71 |
| 3 | I/TfOH | 2c | CHCl3 | 71 | −38 |
| 4 | I/TfOH | 2a | CH3OH | 54 | 21 |
| 5 | I/TfOH | 2b | CH3OH | 75 | −83 |
| 6 | I/HOTf | 2c | CH3OH | 94 | −69 |
| 7 | IV/TfOH | 2a | CHCl3 | 50 | 42 |
| 8 | IV/TfOH | 2b | CH3OH | 88 | −60 |
| 9 | V/TfOH | 2a | CHCl3 | 45 | 33 |
| 10 | V/TfOH | 2b | CH3OH | 75 | −93 |
| 11 | II/TfOH | 2a | CHCl3 | 72 | 81 |
| 12 | II/TfOH | 2b | CH3OH | 90 | −89 |
| 13 | II/DNBA-I | 2a | CHCl 3 | 85 | 92 |
| 14 | II/DNBA-II | 2b | CH 3 OH | 95 | −90 |
| 15 | III/TfOH | 2a | CHCl3 | 42 | 72 |
| 16 | III/TfOH | 2b | CH3OH | 82 | −93 |
| Entryb,c | R product | S product | Entryb,c | R product | S product |
|---|---|---|---|---|---|
| a General conditions: 1 (0.075 mmol), 2a (0.05 mmol), and II/DNBA-I (0.01 mmol, 20 mol%) in CHCl3 (0.25 mL) at r.t. for 24–36 h; 1 (0.075 mmol), 2b (0.05 mmol), and II/DNBA-II (0.01 mmol, 20 mol%) in CH3OH (0.4 mL) at r.t. for 24–36 h. b Yields shown are of isolated products. c The ee was determined by GC or HPLC on a chiral stationary phase. d Catalyst III/TfOH was used instead of II/DNBA-II at r.t. for 36 h. | |||||
| 1–5 |
|
|
16 |
|
|
| 6 |
|
|
17 |
|
|
| 7 |
|
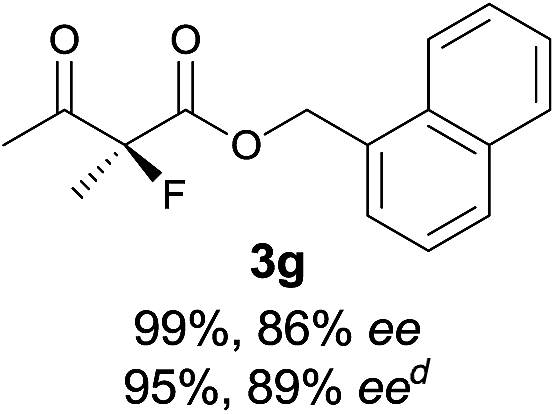
|
18 |
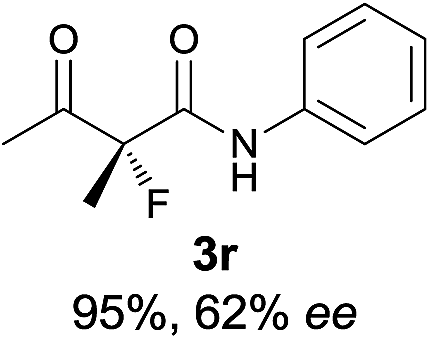
|
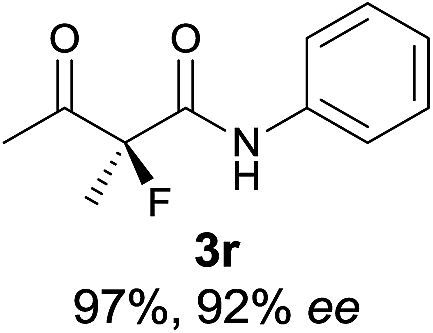
|
| 8 |
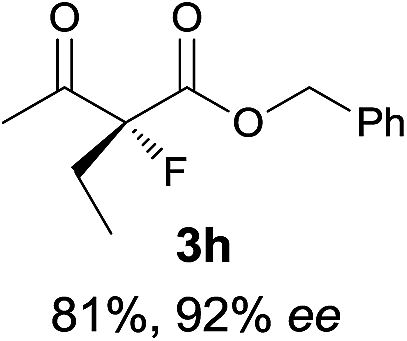
|
|
19 |
|
|
| 9 |
|
|
20 |
|
|
| 10 |
|
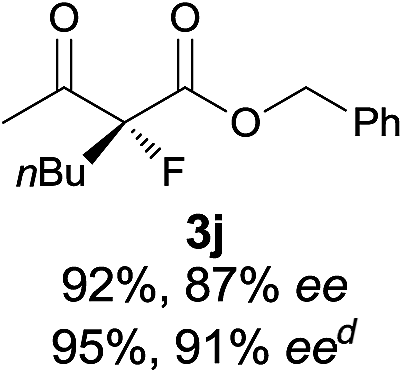
|
21 |
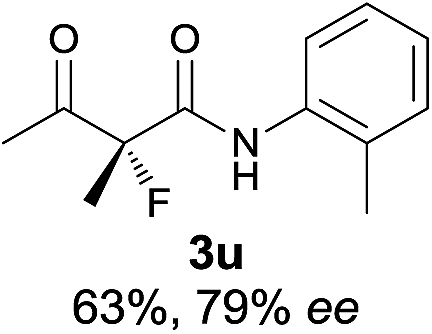
|
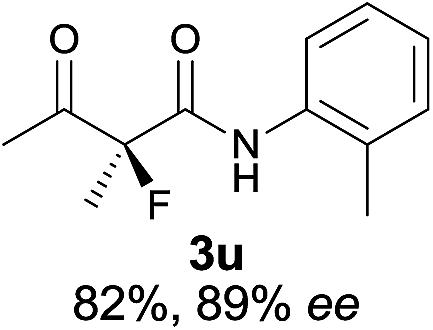
|
| 11 |
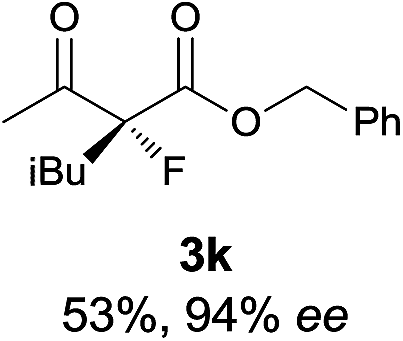
|
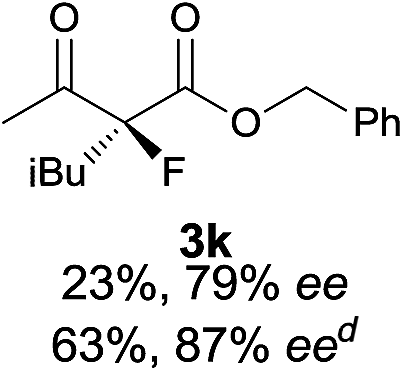
|
22 |
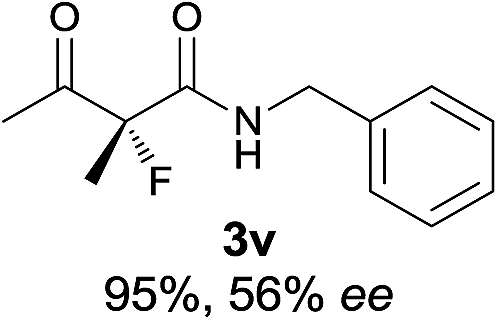
|
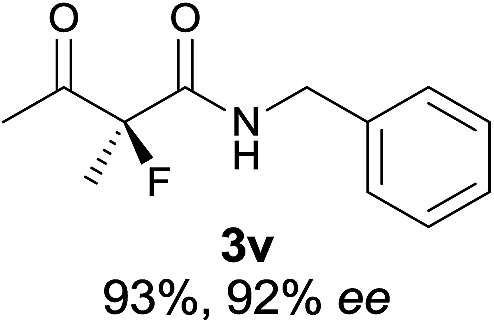
|
| 12 |
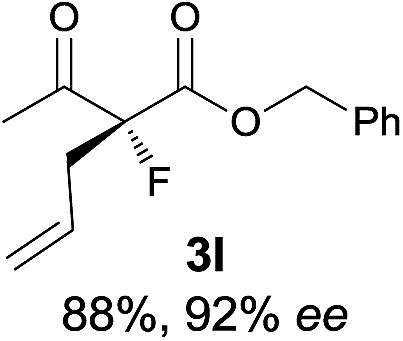
|
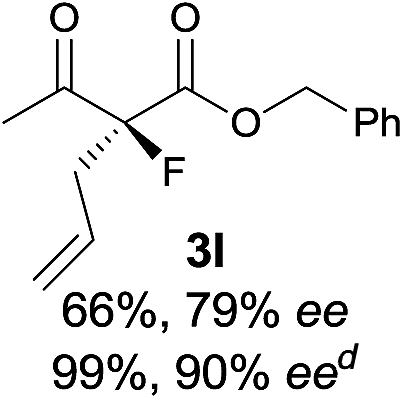
|
23 |
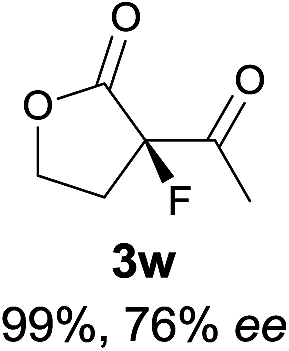
|
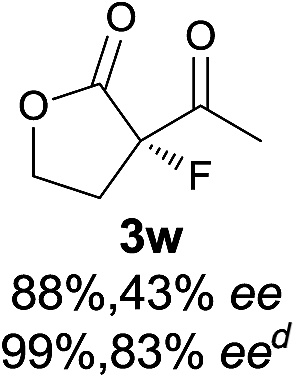
|
| 13 |
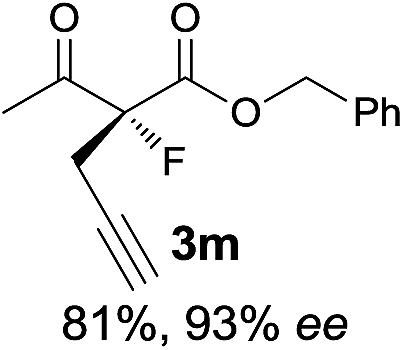
|
|
24 |
|
|
| 14 |
|
|
25 |
|
|
| 15 |
|
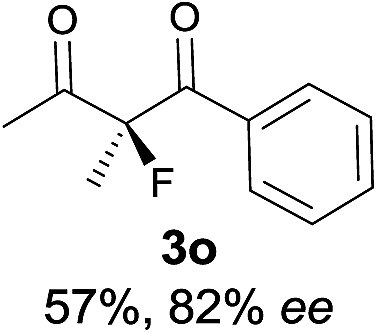
|
|||
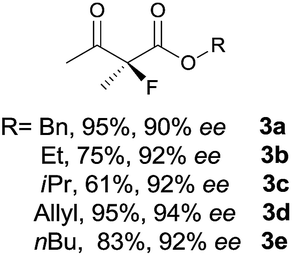
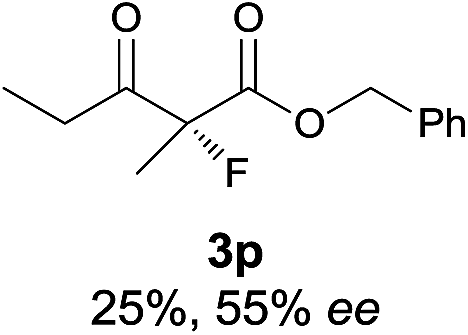
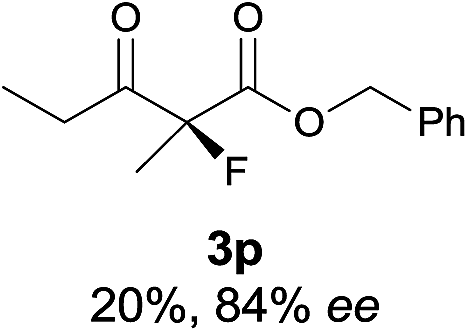
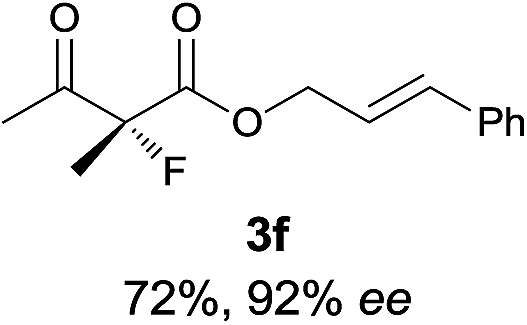
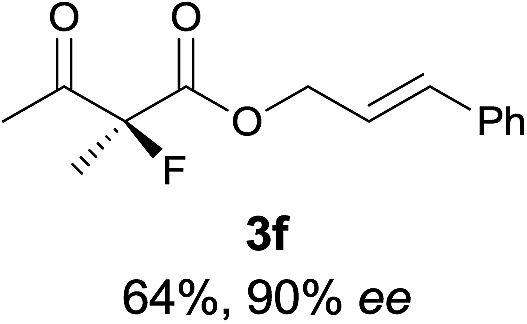
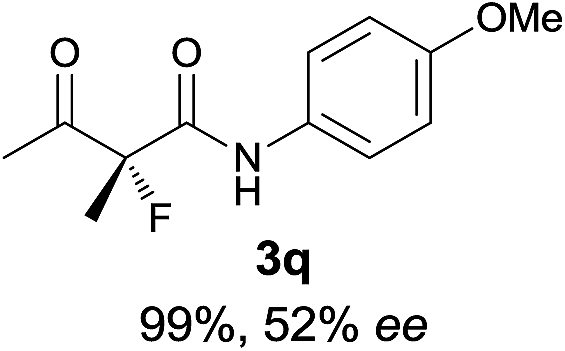
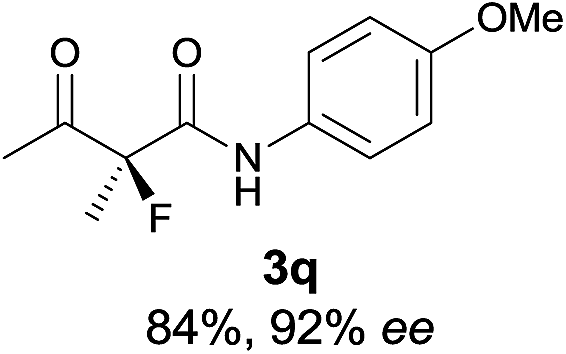
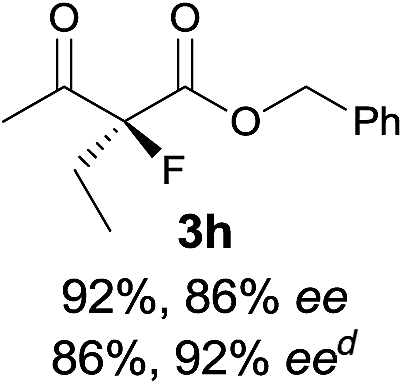
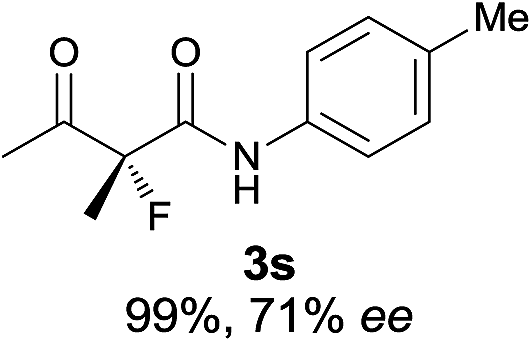
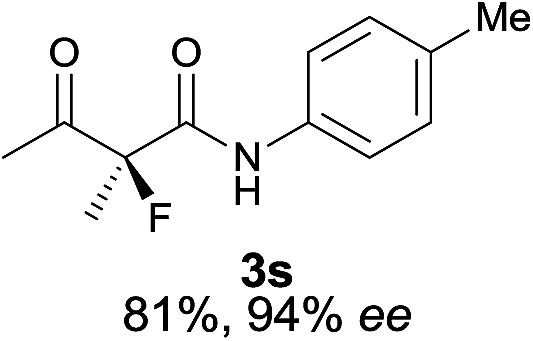
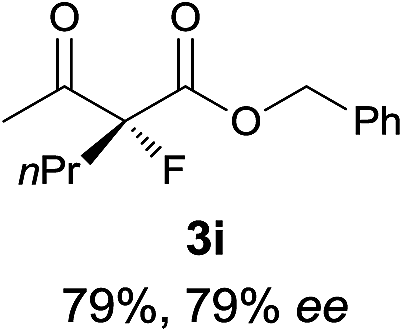
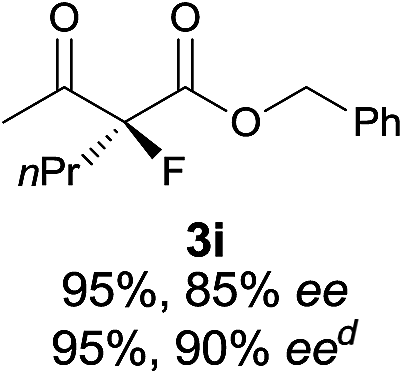
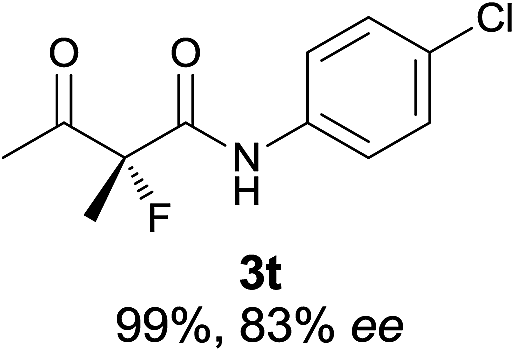
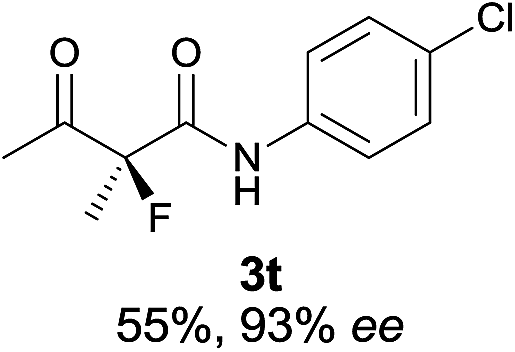
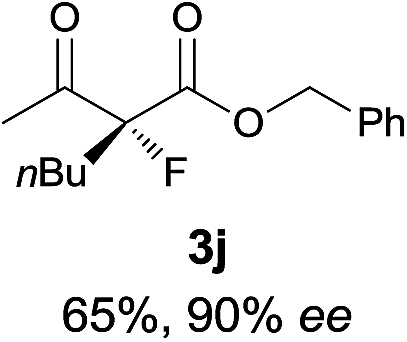
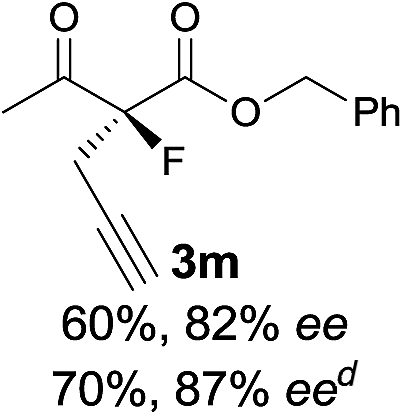
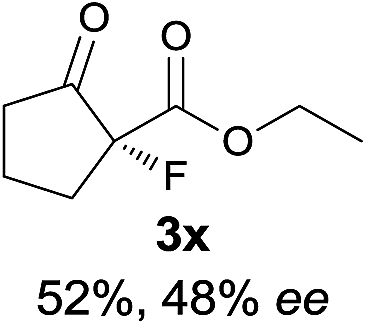
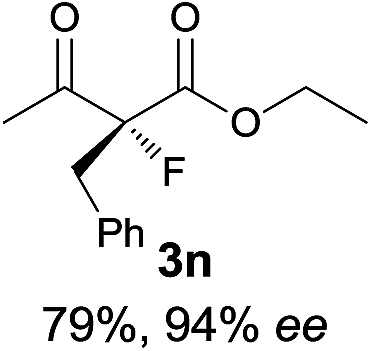
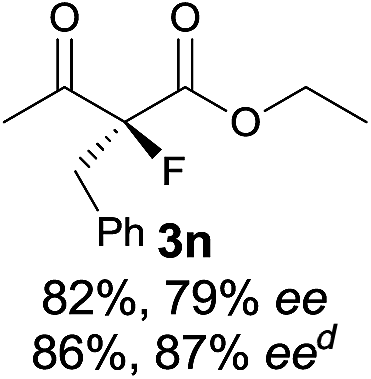
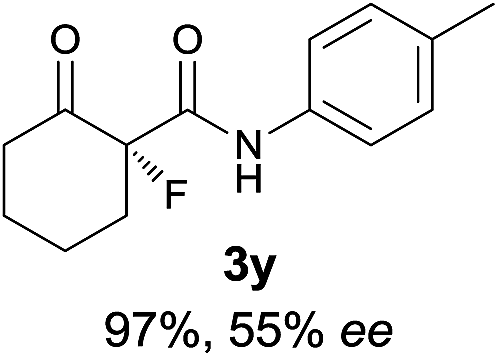
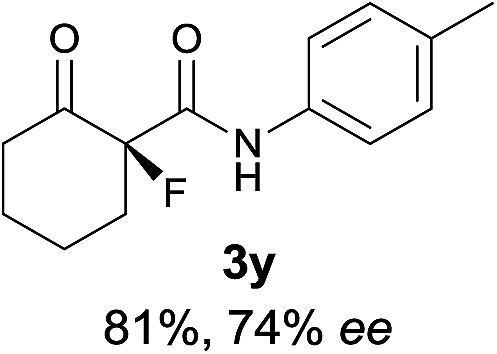
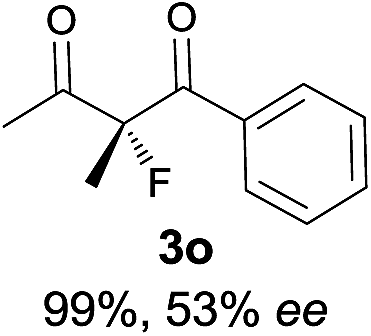
 The substrate scope could be further extended to β-ketoamides.11 Both N-aryl and N-aliphatic amides worked well in the reactions with good yields and moderate to good enantioselectivities (Table 2, entries 17–22 and 25). It seems that the free N–H moiety has a marginal effect on the reactions. In particular, N-aryl amides bearing either electron-donating or electron-withdrawing groups were equally applied, with the latter giving slightly better enantioselectivity (Table 2, entry 20 vs. 19). A lactone-type substrate 3w could also be incorporated in the current catalysis with 76% ee and 99% yield.6c Cyclic ketocarbonyls have also been examined, showing good reactivity but moderate enantioselectivity (Table 2, entries 24 and 25).
The substrate scope could be further extended to β-ketoamides.11 Both N-aryl and N-aliphatic amides worked well in the reactions with good yields and moderate to good enantioselectivities (Table 2, entries 17–22 and 25). It seems that the free N–H moiety has a marginal effect on the reactions. In particular, N-aryl amides bearing either electron-donating or electron-withdrawing groups were equally applied, with the latter giving slightly better enantioselectivity (Table 2, entry 20 vs. 19). A lactone-type substrate 3w could also be incorporated in the current catalysis with 76% ee and 99% yield.6c Cyclic ketocarbonyls have also been examined, showing good reactivity but moderate enantioselectivity (Table 2, entries 24 and 25).
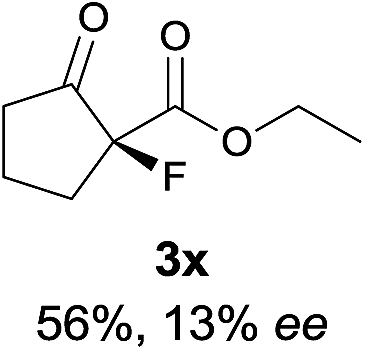
The same substrate scope was also tested for the S-selective reactions. In these cases, the reaction was examined with 2b as the fluorination reagent, and 20 mol% II/DNBA-II or III/HOTf as the catalyst in CH3OH. In most cases, the reactivity and enantioselectivity were comparable to those obtained in the R-selective processes. One particular exception was with the reactions of β-ketoamides, wherein the S-selective process with primary amine catalyst II worked extremely well to afford the desired adducts in good yields and high enantioselectivities (Table 2, entries 17–22 and 25). In comparison, the enantioselectivity with the R-process was moderate. The use of primary amine catalyst III/TfOH has been found to deliver improved enantioselectivity in the S-selective reactions (Table 2, entries 7–14 and 23).
To probe the utility of our fluorination reaction in preparative synthesis, a gram-scale reaction of β-ketoester 1a (7.5 mmol, 1.545 g) was performed with NFSI (5 mmol, 1.576 g) as the fluorination reagent in CHCl3 for 48 h delivering the desired product 3a with a good yield (0.952 g, 85% yield) and excellent enantioselectivity (94% ee). The chiral primary amine catalyst could be quantitatively recovered by concentrating the aqueous solution after work up.
Though an enol-type could not be completely ruled out, the current experimental observations as well as our previous studies strongly favour an enamine mechanism (see the ESI for details†). To account for the switch of enantioselectivity, we have proposed two plausible enamine-based intermolecular F-attack transition states.13 For the NFSI (2a) based R-selective process, an H-bonding mode I between the sulfonyl moiety and the protonated ammonium N–H was invoked in the stereocontrol. In this model, the Si-facial fluorination was largely disfavoured due to geometrically unfavourable H-bonding with the ammonium N–H. On the other hand, an electrostatic mode II was proposed for the N-fluoro-pyridinium (2b) based S-selective process, wherein the electrostatic repulsion between the cationic charged ammonium and pyridinium species plays a dominant role. Steric effects would also contribute in this model, however, the impact should be minor, as we note that primary amine II performed equally as well as its more bulky counterparts such as catalysts III and V. The observed solvent effect (e.g. CH3OH vs. CHCl3, Table 1, entries 1–6) is also in line with the electrostatic model since ionic species would become highly dissociated in polar protic CH3OH, a feature favourable for the electrostatic repulsion interaction.
The stereocontrolling modes (I and II) could be further verified by DFT calculations at the B3LYP/6-31G* level of approximation (see the ESI for details†). For mode II, the S-selective TS-S was favoured over the R-selective TS-R by 3.0 kcal mol−1, which is consistent with the experimental observations. We further calculated the electrostatic surface potential (ESP) of the two located TSs. As revealed in Fig. 1b, the ammonium and pyridinium moieties of TS-R, both bearing positively charged surfaces, seem not to have any noticeable steric interaction from being close together. Thus, TS-R would be mainly disfavoured by electrostatic repulsion not by steric effects. By AIM analysis, we could also identify an attractive C–H⋯F interaction between the tert-butyl group of the amine catalyst and the fluorination reagent 2b, as shown in Fig. 1b, which may also contribute in facilitating the Si-facial attack in TS-S.14
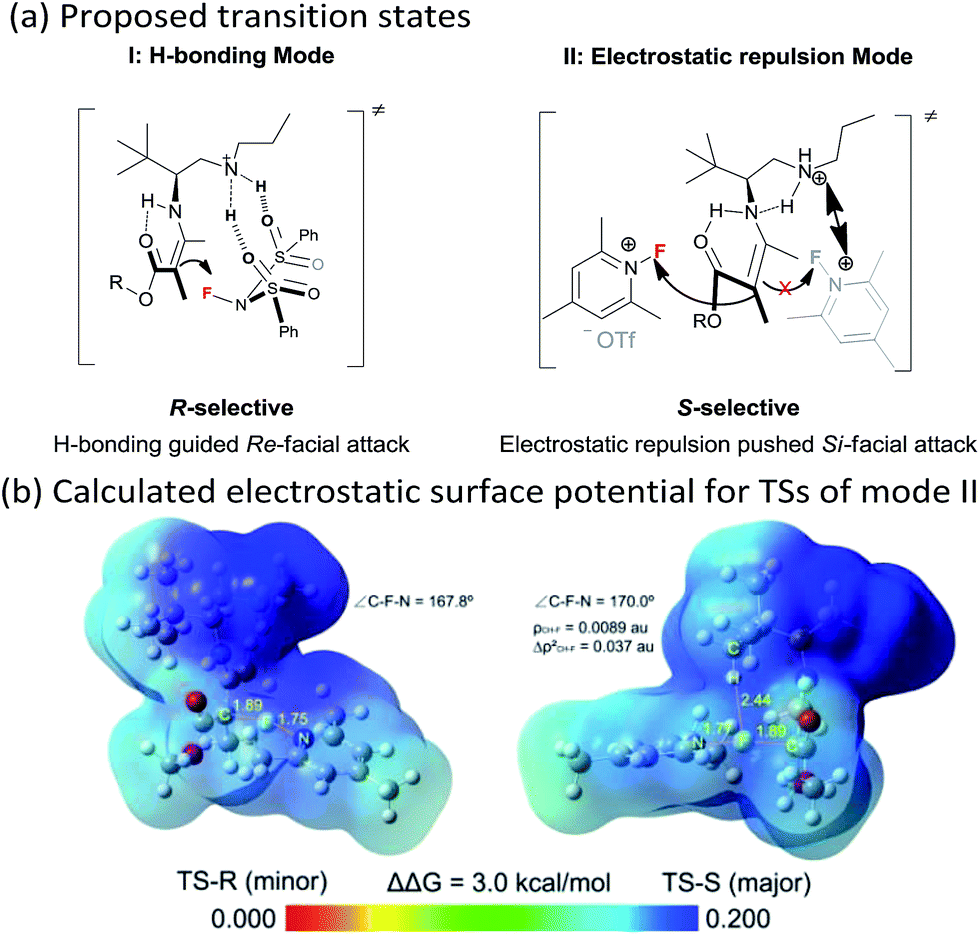 | ||
| Fig. 1 (a) Proposed transition states (I and II) for the two enantioselectivity switch fluorination reactions; (b) calculated electrostatic surface potential for TSs of the electrostatic mode II. | ||
Conclusions
In summary, we have presented herein a reagent-controlled enantioselectivity switch for the organocatalytic asymmetric fluorination of β-ketocarbonyls. A simple swap of the fluorination reagent switched the enantioselectivity with good reactivity and enantiomeric excess in both cases. Mechanistic studies revealed dual H-bonding and electrostatic stereocontrolling modes for a single chiral primary amine catalyst. Further explorations of switchable enantioselectivity in other reactions are currently underway in our laboratory.Acknowledgements
The project was supported by Ministry of Science and Technology (2012CB821600), the Natural Science Foundation of China, and the Chinese Academy of Sciences.Notes and references
- For recent reviews on switching selectivity, see: (a) G. Zanoni, F. Castronovo, M. Franzin, G. Vidari and E. Giannini, Chem. Soc. Rev., 2003, 32, 115 RSC; (b) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999 Search PubMed; (c) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2000 Search PubMed; (d) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341 RSC; (e) X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515 CrossRef CAS PubMed; (f) T. Tanaka and M. Hayashi, Synthesis, 2008, 3361 CAS; (g) M. Bartók, Chem. Rev., 2010, 110, 1663 CrossRef PubMed; (h) J. Escorihuela, M. I. Burguete and S. V. Luis, Chem. Soc. Rev., 2013, 42, 5595 RSC.
- For recent examples, see: (a) J. Wang and B. L. Feringa, Science, 2011, 331, 1429 CrossRef CAS PubMed; (b) S. Mortezaei, N. R. Catarineu and J. W. Canary, J. Am. Chem. Soc., 2012, 134, 8054 CrossRef CAS PubMed; (c) S. A. Moteki, J. Han, S. Arimitsu, M. Akakura, K. Nakayama and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1187 CrossRef CAS PubMed; (d) J. Lv, Y. Qin, J. Cheng and S. Luo, Acta Chim. Sin., 2014, 72, 809 CrossRef; (e) Y. Zhang, N. Yang, X. Liu, J. Guo, X. Zhang, L. Lin, C. Hu and X. Feng, Chem. Commun., 2015, 51, 8432 RSC; (f) N. Shibata, T. Ishimaru, T. Nagai, J. Kohno and T. Toru, Synlett, 2004, 10, 1703 CrossRef.
- For organocatalytic enantioselectivity switches, see: (a) Y. Sohtome, S. Tanaka, K. Takada, T. Yamaguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 9254 CrossRef CAS PubMed; (b) A. Garzan, A. Jaganathan, N. Salehi Marzijarani, R. Yousefi, D. C. Whitehead, J. E. Jackson and B. Borhan, Chem.–Eur. J., 2013, 19, 9015 CrossRef CAS PubMed; (c) S. A. Moteki, J. Han, S. Arimitsu, M. Akakura, K. Nakayama and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1187 CrossRef CAS PubMed; (d) Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2013, 135, 12160 CrossRef CAS PubMed.
- For recent reviews on asymmetric fluorination reactions, see: (a) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (c) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (d) R. Britton and B. Kang, Nat. Prod. Rep., 2013, 30, 227 RSC; (e) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637 CrossRef CAS PubMed; (f) S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708 CrossRef CAS; (g) J.-H. Lin and J.-C. Xiao, Tetrahedron Lett., 2014, 55, 6147 CrossRef CAS; (h) J. Wang, M. Sánchez-Rosollo, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (i) V. Bizet, T. Besset, J.-A. Ma and D. Cahard, Curr. Top. Med. Chem., 2014, 14, 901 CrossRef CAS PubMed.
- For leading examples on metal Lewis acid catalysis, see: (a) L. Hintermann and A. Togni, Angew. Chem., Int. Ed., 2000, 39, 4359 CrossRef CAS; (b) Y. Hamashima, K. Yagi, H. Takano, L. Tamás and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 14530 CrossRef CAS PubMed; (c) N. Shibata, J. Kohno, K. Takai, T. Ishimaru, S. Nakamura, T. Toru and S. Kanemasa, Angew. Chem., Int. Ed., 2005, 44, 4204 CrossRef CAS PubMed; (d) A. Narayama, K. Shibatomi, Y. Soga, T. Muto and S. Iwasa, Synlett, 2013, 24, 375 CrossRef CAS.
- For organocatalytic fluorination of β-ketocarbonyls, see: (a) D. Y. Kim and E. J. Park, Org. Lett., 2002, 4, 545 CrossRef CAS PubMed; (b) X. Wang, Q. Lan, S. Shirakawa and K. Maruoka, Chem. Commun., 2010, 46, 321 RSC; (c) J. Xu, Y. Hu, D. Huang, K.-H. Wang, C. Xu and T. Niu, Adv. Synth. Catal., 2012, 354, 515 CrossRef CAS.
- (a) M. Marigo, D. Fielenbach, A. Braunton, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 3703 CrossRef CAS PubMed; (b) D. D. Steiner, N. Mase and C. F. Barbas, Angew. Chem., Int. Ed., 2005, 44, 3706 CrossRef CAS PubMed; (c) T. D. Beeson and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 8826 CrossRef CAS PubMed.
- (a) S. Brandes, B. Niess, M. Bella, A. Prieto, J. Overgaard and K. A. Jørgensen, Chem.–Eur. J., 2006, 12, 6039 CrossRef CAS PubMed; (b) M. R. Witten and E. N. Jacobsen, Org. Lett., 2015, 17, 2772 CrossRef CAS PubMed; (c) K. Shibatomi, K. Kitahara, T. Okimi, Y. Abe and S. Iwasa, Chem. Sci., 2016, 7, 1388–1392 RSC.
- (a) P. Kwiatkowski, T. D. Beeson, J. C. Conrad and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 1738 CrossRef CAS PubMed; (b) Y. Lam and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 9556 CrossRef CAS PubMed.
- X. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225 CrossRef CAS PubMed.
- (a) L.-S. Zheng, Y.-L. Wei, K.-Z. Jiang, Y. Deng, Z.-J. Zheng and L.-W. Xu, Adv. Synth. Catal., 2014, 356, 3769 CrossRef CAS; (b) S. J. Kwon and D. Y. Kim, J. Fluorine Chem., 2015, 180, 201 CrossRef CAS; (c) K. Hayamizu, N. Terayama, D. Hashizume, K. Dodo and M. Sodeoka, Tetrahedron, 2015, 71, 6594 CrossRef CAS.
- L. Zhang, N. Fu and S. Luo, Acc. Chem. Res., 2015, 48, 986 CrossRef CAS PubMed.
- The intramolecular F-attack with the secondary NH–F species (see ref. 9b), in situ transferred from the fluorination reagent to the aminocatalyst, though not completely excluded, may not be applicable in our case. First, there is no such precedence on fluorination with NH–F species. In addition, even if an F-transfer did occur, facile proton release instead of F-attack might be the dominant pathway, leading to an unreactive neutral-N–F species or even poisoning of the catalyst. Experimentally, we did not detect such species by in situ ESI-MS in both cases.
- In TS-S, a short C–H⋯F distance was found of 2.44 Å. Atoms in Molecules (AIM) analysis at the bond critical point suggested a weak interaction between H and F where the electron density (ρ) is 0.0089 au and the Laplacian value (Δρ2) is 0.037 au. These values indicate that a weak C–H⋯F interaction is present. For a review on C–H⋯F–C interaction, see: C.-C. Liu and M. C. W. Chan, Acc. Chem. Res., 2015, 48, 1580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including characterization date, copies of 1H, 13C, and 19F NMR and HPLC traces. See DOI: 10.1039/c6sc03109a |
| This journal is © The Royal Society of Chemistry 2017 |