
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering catalytic coordination space in a chemically stable Ir-porphyrin MOF with a confinement effect inverting conventional Si–H insertion chemoselectivity†
Yingxia
Wang
,
Hao
Cui
,
Zhang-Wen
Wei
,
Hai-Ping
Wang
,
Li
Zhang
* and
Cheng-Yong
Su
 *
*
MOE Laboratory of Bioinorganic and Synthetic Chemistry, Lehn Institute of Functional Materials, School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou 510275, China. E-mail: zhli99@mail.sysu.edu.cn; cesscy@mail.sysu.edu.cn
First published on 1st September 2016
Abstract
An iridium-porphyrin ligand, Ir(TCPP)Cl (TCPP = tetrakis(4-carboxyphenyl)porphyrin), has been utilized to react with HfCl4 to generate a stable Ir(III)-porphyrin metal–organic framework of the formula [(Hf6(μ3-O)8(OH)2(H2O)10)2(Ir(TCPP)Cl)3]·solvents (Ir-PMOF-1(Hf)), which possesses two types of open cavities (1.9 × 1.9 × 1.9 and 3.0 × 3.0 × 3.0 nm3) crosslinked through orthogonal channels (1.9 × 1.9 nm2) in three directions. The smaller cavity is surrounded by four catalytic Ir(TCPP)Cl walls to form a confined coordination space as a molecular nanoreactor, while the larger one facilitates reactant/product feeding and release. Therefore, the porous Ir-PMOF-1(Hf) can act as a multi-channel crystalline molecular flask to promote the carbenoid insertion reaction into Si–H bonds, featuring high chemoselectivity towards primary silanes among primary, secondary and tertiary silanes under heterogeneous conditions that are inaccessible by conventional homogeneous catalysts.
Introduction
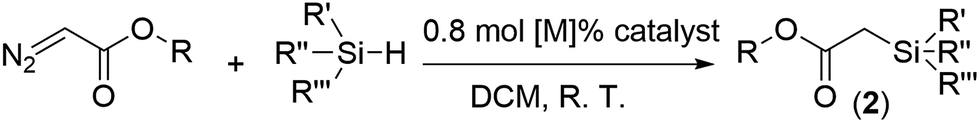
Crystal engineering of metal–organic frameworks (MOFs) provides a powerful platform to explore their versatile potential correlating to designable porosity and complexity.1 In this regard, functionalization of a MOF may be realized through coordination space engineering (CSE) in MOF pores via one-pot synthesis or post-modification.2 Thanks to the plentiful reticular chemistry of MOFs, engineering of coordination spaces (CSs) as the basic functional units for a specific application becomes feasible by virtue of deliberate selection of metal ion, organic linker and framework topology.3 For example, engineering catalytic CSs in a MOF can start from the design of active sites with the selectivity controllable by a confinement effect from the framework cavities and pore windows.Transition-metal-catalyzed carbenoid insertion into the Si–H bond is an attractive method to prepare α-silyl carbonyl compounds. Besides dirhodium(II)4 and copper5 complexes, which are generally superior catalysts for carbene transfer reactions, only a few examples involving Zn,6 Ag,7 Au8 and Ir9 have been reported to catalyze Si–H insertion reactions. The majority of these catalytic reactions prefer tertiary silanes. Pérez has studied the relative reactivity of substituted silanes in the presence of a Cu or Ag-based catalyst, and found that these catalysts display activity in the order of tertiary > secondary > primary for ethyl substituted silanes, but an inverted order of secondary > tertiary ≈ primary for phenyl substituted silanes.7 Nevertheless, none of the reported catalysts display the highest chemoselectivity for primary silanes. The bond dissociation energy (BDE) of the Si–H bond increases in the order of tertiary, secondary and primary silanes, and thus primary Si–H bonds are the most inert towards the insertion reaction with carbenoids.10 However, it was noted that a considerable steric effect could be aroused by multiple substituents in secondary and tertiary silanes. Therefore, potential selectivity for primary silanes may be achieved if the reaction takes place in a confined space, where the active sites become more inaccessible to bulky substrates. In this context, CSE of confined and catalytic nanoreactors in MOF pores may offer an ideal and easy approach to such a purpose, owing to additional selectivity endowed by the channel size, making it possible to convert less active primary silanes into more active secondary silanes with plenty of practical uses.11
Herein, we test this speculation by designing a robust and self-supported iridium-porphyrin MOF catalyst, [(Hf6(μ3-O)8(OH)2(H2O)10)2(Ir(TCPP)Cl)3]·solvents (Ir-PMOF-1(Hf), TCPP = tetrakis(4-carboxyphenyl)porphyrin), through incorporating Ir(III)-porphyrin active sites on the pore surface to generate catalytic CSs. Owing to its continuous and permeable channels crosslinking the catalytic CSs, Ir-PMOF-1(Hf) behaves as a multi-channel crystalline molecular flask12 featuring an effective confinement effect and easy transportation of reactant/product into/from inner reactive vessels.13–24 The high selectivity and reactivity for the primary Si–H insertion reaction (Scheme 1) has been successfully established, supplementing alternative chemoselective choices in contrast to conventional homogeneous catalysts. To the best of our knowledge, this is the first report of an inverted selectivity order, i.e., primary > secondary > tertiary, for Si–H insertion induced by a metal catalyst.
 | ||
| Scheme 1 Heterogeneous chemoselectivity of primary Si–H insertion, induced by confined coordination spaces in Ir-PMOF-1(Hf), showing the opposite reactivity order to conventional homogeneous catalysis. | ||
Experimental
General information
All the reagents in the present work were obtained from commercial sources and used directly without further purification. The metalloporphyrin ligands Ir(TCPPCO2Me)(CO)Cl and Ir(TCPP)Cl were synthesized according to our recent work.14 The elemental analyses were performed with a Perkin-Elmer 240 elemental analyzer. HRESI-MS was performed by using a Bruker Daltonics ESI-Q-TOF maXis4G. Infrared spectra on KBr pellets were collected with a Nicolet/Nexus-670 FT-IR spectrometer in the region of 4000–400 cm−1. UV-vis spectra were tested on a Shimadzu/UV-3600 spectrophotometer. 1H and 13C NMR spectra were recorded on a Bruker AVANCE III 400 MHz. PXRD patterns were recorded on a SmartLab X-ray powder diffractometer (Rigaku Co.) at 40 kV and 30 mA with a Cu target tube. Thermogravimetric (TG) analyses were performed under an air atmosphere at a heating rate of 2 °C min−1 by using a NETZSCH TG 209 system. X-ray photoelectron spectroscopy (XPS) was performed on a ULVAC PHI Quantera microprobe. Binding energies (BE) were calibrated by setting the measured BE of C 1s to 284.65 eV. The sorption isotherms for N2 (77 K) gas were measured with an Autosorb-iQ2-MP gas sorption analyzer (Quantachrome, USA).Caution! Although we have not experienced any problems in the handling of diazo compounds, extreme care should be taken when manipulating them due to their explosive nature.
Synthesis of (Hf6(μ3-O)8(OH)2(H2O)10)2(Ir(TCPP)Cl)3·solvents (Ir-PMOF-1(Hf))
Ir(TCPP)Cl (5 mg, 4.9 × 10−3 mmol), HfCl4 (15 mg, 4.7 × 10−2 mmol), benzoic acid (400 mg, 3.27 mmol) and dimethylformamide (DMF, 1 mL) were placed in a glass vial, which was then sealed and heated to 120 °C in an oven. After 48 h, red block crystals were obtained (7 mg, 77% yield based on Ir(TCPP)Cl) and air-dried. Anal. calcd for (Hf6(μ3-O)8(OH)2(H2O)10)2(Ir(TCPP)Cl)3·9(C6H5COOH)·2H2O (C207H176Cl3Ir3N12O85Hf12): C, 35.43; H, 2.53; N, 2.40. Found: C, 35.01; H, 3.01; N, 2.28%. FT-IR (KBr) ν 3402 (br), 1658 (s), 1601 (s), 1552 (s), 1418 (s), 1075 (m), 1015 (m), 718 (m), 664 (m) cm−1.Single crystal X-ray crystallography
The X-ray diffraction data was collected with an Agilent Technologies SuperNova X-ray diffractometer system equipped with Cu-Kα radiation (λ = 1.54178 Å). The crystal was kept at 150(10) K during data collection. The structure was solved with the ShelXS structure solution program integrated in Olex2 using Direct Methods, and refined with the ShelXL refinement package using CGLS minimisation.25a The refinement was restricted to one set of molecules by using DFIX and FLAT to constrain the porphyrin ligand. ISOR/SIMU was applied to all non-hydrogen atoms to simulate isotropic behaviors. All solvent molecules have been removed by the SQUEEZE program.25b The positions of the hydrogen atoms are generated geometrically. A summary of the crystal structure refinement data and selected bond angles and distances are listed in Tables S1 and S2. CCDC 1491233.†Typical procedure for Si–H insertion
A solution of ethyl-2-diazoacetate (EDA, 45.6 mg, 0.4 mmol, 1.0 eq.) in DCM (1.0 mL) was added slowly to the mixture of phenylsilane (PhSiH3, 216.4 mg, 2.0 mmol, 5.0 eq.) and activated Ir-PMOF-1(Hf) (6.4 mg, 0.0032 mmol, 0.8 mol [Ir]%) in dichloromethane (DCM, 1 mL). The resulting suspension was stirred at room temperature for 3 min until EDA was completely consumed. The undissolved catalyst was removed through centrifugation, and washed with DCM (3 × 8 mL). The combined supernatant was evaporated to dryness, and the residue was dissolved in CDCl3 and analyzed by 1H NMR to determine the conversion of EDA to ethyl-2-(phenylsilyl)acetate (2a, 93%).Results and discussion
The solvothermal reaction of Ir(TCPP)Cl and HfCl4 in the presence of benzoic acid in DMF at 120 °C for 48 h yielded red block crystals of Ir-PMOF-1(Hf). Single-crystal X-ray diffraction studies reveal that Ir-PMOF-1(Hf) crystallizes in the cubic space group Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m as an isostructure of Ir-PMOF-1(Zr)14 and PCN-224,24 albeit based on Hf6-clusters (Fig. 1). The basic Hf-oxide node may consist of a core Hf6(μ3-O)8 octahedron and 12 terminal H2O/OH− groups, in which each vertex of the Hf6-octahedron is occupied by a Hf(IV) atom and each face is capped by one μ3-oxygen atom. Six edges of the Hf6-octahedron are bridged by carboxylates from six different 4-connecting Ir(TCPP)Cl ligands, thus leading to the formation of a three-dimensional (3D) framework of (4,6)-connected she net with the short vertex symbol of 4.4.4.4.8(26).8(26).26 There are two types of cavities with open windows in such a topological framework. One is bigger, surrounded by 8 Hf6(μ3-O)8 clusters (3.0 × 3.0 × 3.0 nm3), while the other is smaller, enclosed by 4 Ir(TCPP)Cl (1.9 × 1.9 × 1.9 nm3) units which interconnect with each other to form square-like pores (1.9 × 1.9 nm2) intercrossing in three orthogonal directions. Therefore, Ir-PMOF-1(Hf) can be regarded as a multi-channel platform for heterogeneous catalysis with self-supporting parallel nanoreactors and transportation channels, where catalytic Ir-porphyrin sites are confined in CSs with open windows for convenient reactant/product feeding and release.
m as an isostructure of Ir-PMOF-1(Zr)14 and PCN-224,24 albeit based on Hf6-clusters (Fig. 1). The basic Hf-oxide node may consist of a core Hf6(μ3-O)8 octahedron and 12 terminal H2O/OH− groups, in which each vertex of the Hf6-octahedron is occupied by a Hf(IV) atom and each face is capped by one μ3-oxygen atom. Six edges of the Hf6-octahedron are bridged by carboxylates from six different 4-connecting Ir(TCPP)Cl ligands, thus leading to the formation of a three-dimensional (3D) framework of (4,6)-connected she net with the short vertex symbol of 4.4.4.4.8(26).8(26).26 There are two types of cavities with open windows in such a topological framework. One is bigger, surrounded by 8 Hf6(μ3-O)8 clusters (3.0 × 3.0 × 3.0 nm3), while the other is smaller, enclosed by 4 Ir(TCPP)Cl (1.9 × 1.9 × 1.9 nm3) units which interconnect with each other to form square-like pores (1.9 × 1.9 nm2) intercrossing in three orthogonal directions. Therefore, Ir-PMOF-1(Hf) can be regarded as a multi-channel platform for heterogeneous catalysis with self-supporting parallel nanoreactors and transportation channels, where catalytic Ir-porphyrin sites are confined in CSs with open windows for convenient reactant/product feeding and release.
 | ||
| Fig. 1 Assembly of 3D Ir-PMOF-1(Hf) based on 6-connected Hf6(μ3-O)8(OH)2(H2O)10 clusters and 4-connected Ir(TCPP)Cl ligands, showing two types of cavities interconnected to generate open channels in three directions. | ||
The powder X-ray diffraction (PXRD) patterns of bulk samples closely match with the simulated one from single-crystal data, indicative of satisfactory phase purity (Fig. S1†). As the crystal structure of Ir-PMOF-1(Hf) shows, the atomic ratio of Hf![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ir:Cl is 4:1:1, which is further confirmed by energy-dispersive X-ray spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) analyses (Fig. S2 and S3; Tables S3 and S4†). Two intense peaks around 65.1 and 62.2 eV appear in the XPS spectrum, which are assignable to Ir 4f5/2 and Ir 4f7/2, respectively.27
:Ir:Cl is 4:1:1, which is further confirmed by energy-dispersive X-ray spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) analyses (Fig. S2 and S3; Tables S3 and S4†). Two intense peaks around 65.1 and 62.2 eV appear in the XPS spectrum, which are assignable to Ir 4f5/2 and Ir 4f7/2, respectively.27
The thermogravimetric (TG) curve of the desolvated Ir-PMOF-1(Hf) crystallites displays no obvious weight loss up to 340 °C, where the framework starts to decompose (Fig. S4†). The variable temperature PXRD experiments disclose that the framework crystallinity can sustain heating up to 220 °C (Fig. S5†). Ir-PMOF-1(Hf) exhibits excellent chemical stability. After immersing the fresh crystals in a wide range of solvents, such as dichloromethane (DCM), ethyl acetate (EA), diethyl ether (Et2O), acetonitrile (MeCN), tetrahydrofuran (THF), methanol (MeOH) and water for 3–5 days, the PXRD patterns of the recycled samples still retain the prominent peak profiles and crystallinity (Fig. 2), verifying that the porous Ir-PMOF-1(Hf) crystals are capable of heterogeneous catalysis in these solvents. After soaking samples of Ir-PMOF-1(Hf) in pH = 0–11 and aqueous solutions of inorganic salts for 24 h, the PXRD patterns show comparable diffraction profiles to the as-synthesized sample (Fig. S6†).
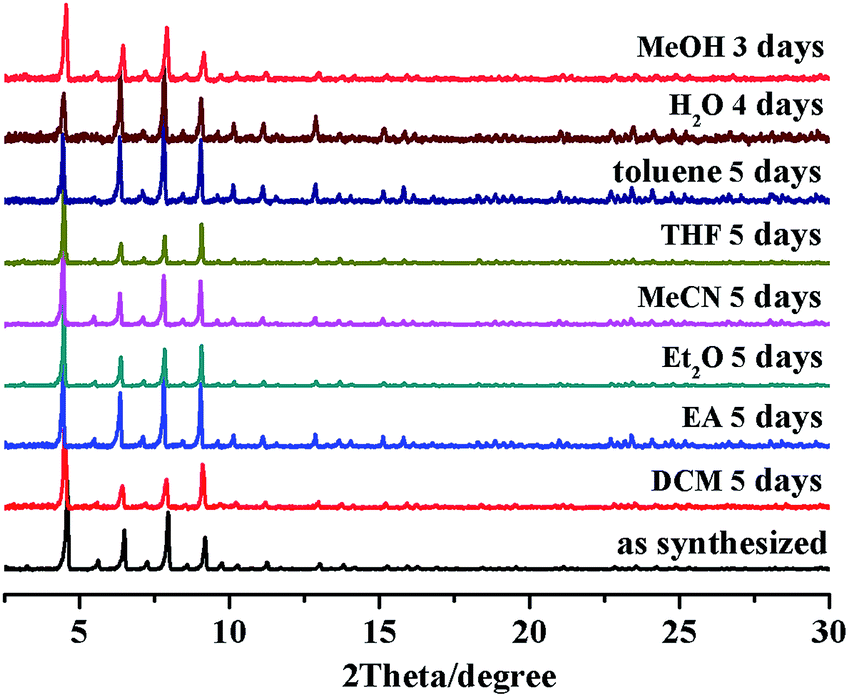 | ||
| Fig. 2 Stability test of Ir-PMOF-1(Hf) in different solvents. | ||
The porosity of Ir-PMOF-1(Hf) has been evaluated by N2 adsorption isotherms at 77 K (Fig. S7†). Prior to sorption measurements, the samples were subjected to solvent exchange with acetone for 10 h and then activated by heating at 120 °C under vacuum for 12 h. The N2 adsorption of Ir-PMOF-1(Hf) shows a typical type-I isotherm, indicative of micro-porosity. A N2 uptake of 535 cm3 g−1 was obtained, and the Brunauer–Emmett–Teller (BET) surface area was calculated to be 1758 m2 g−1. According to the Density Functional Theory (DFT) pore distribution plot of Ir-PMOF-1(Hf), the pore diameter of the sample is approximately 1.6 nm (Fig. S8†), which approximates to the single-crystal analysis (1.9 nm). As calculated by PLATON analysis, the effective solvent accessible void in the crystal lattice is 78.4% of the cell volume.
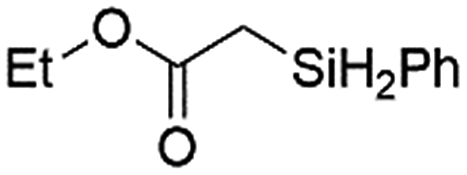
The superiority of Ir-PMOF-1(Hf) in Si–H insertion reactions has been examined by the model reaction of ethyl-2-diazoacetate (EDA) and phenylsilane (PhSiH3). In the presence of 0.8 mol [M]% of the metal catalysts, including Ir-PMOF-1(Hf), Ir(TPP)(CO)Cl (TPP = 5,10,15,20-tetraphenylporphyrin) and Rh2(OAc)4, the reaction of a 1:5 molar ratio mixture of EDA and PhSiH3 in DCM after 3 min produced the Si–H insertion product of ethyl-2-(phenylsilyl)acetate (2a) in 93, 86 and 83% yields, respectively (Table 1, entries 1–3). In each reaction, EDA was completely consumed. In addition to the Si–H insertion products, the self-coupling reaction products of EDA, i.e. diethyl fumarate and maleate, account for all initial EDA. The similar reaction catalyzed by Cu(OTf)2 is found to be ineffective, and 2a is formed in only 41% yield after 48 h (entry 4). When decreasing the molar percent of [Ir] to 0.04%, the reaction in the presence of Ir-PMOF-1(Hf) still achieves a 76% yield, although a much longer reaction time of 12 h is required (entry 5). Based on these catalysis results, the turnover frequency (TOF) and the turnover number (TON) of Ir-PMOF-1(Hf) can be estimated to be up to 2325 h−1 and 1900, respectively.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time | Product | Yield (%) | |
| a Reaction conditions: 0.8 mol [M]% of the catalyst. The yields are based on the conversions of diazoacetates to the Si–H insertion products. In each reaction, EDA has been completely consumed. b Catalyst amount: 0.04 mol [Ir]%. | |||||
| 1 | Ir-PMOF-1(Hf) | 3 min | 2a |
|
93 |
| 2 | Ir(TPP)(CO)Cl | 3 min | 2a | 86 | |
| 3 | Rh2(OAc)4 | 3 min | 2a | 83 | |
| 4 | Cu(OTf)2 | 48 h | 2a | 41 | |
| 5b | Ir-PMOF-1(Hf) | 12 h | 2a | 76 | |
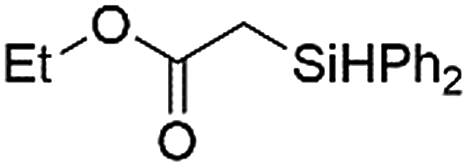
| 6 | Ir-PMOF-1(Hf) | 6 min | 2b |
|
82 |
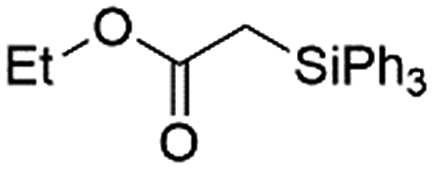
| 7 | Ir-PMOF-1(Hf) | 7 min | 2c |
|
33 |
| 8 | Ir-PMOF-1(Hf) | 5 min | 2d |
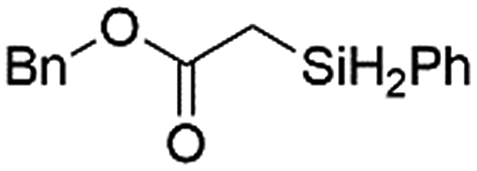
|
73 |
| 9 | Ir-PMOF-1(Hf) | 20 min | 2e |
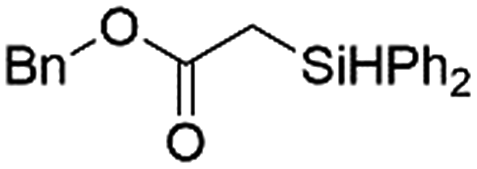
|
51 |
| 10 | Ir-PMOF-1(Hf) | 1 h | 2f |
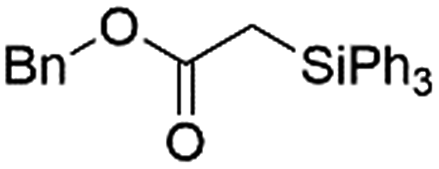
|
28 |
| 11 | Ir-PMOF-1(Hf) | 3 min | 2g |
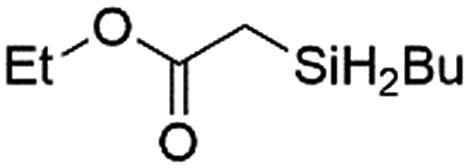
|
91 |
| 12 | Ir-PMOF-1(Hf) | 3 min | 2h |
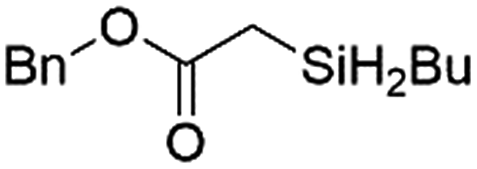
|
84 |
Ir-PMOF-1(Hf) is also efficient in inducing the reaction of a secondary silane. The reaction of EDA and diphenylsilane (Ph2SiH2) in the presence of 0.8 mol [Ir]% of Ir-PMOF-1(Hf) leads to the formation of ethyl-2-(diphenylsilyl)acetate (2b) in 82% yield (entry 6). In comparison, the Si–H insertion reaction of a tertiary silane such as triphenylsilane (Ph3SiH) is sluggish, and the product, ethyl-2-(triphenylsilyl)acetate (2c), is generated in only 33% yield (entry 7). In changing the diazo compound to benzyl 2-diazoacetate (BDA), the reactions with PhSiH3, Ph2SiH2 and Ph3SiH give the Si–H insertion products 2d–f in yields of 73, 51 and 28%, respectively, and the reaction time has been lengthened from 5 min to 1 h (entries 8–10). Besides phenylsilane, other primary silanes like n-butylsilane (BuSiH3) can also react with both EDA and BDA in the presence of Ir-PMOF-1(Hf) to generate Si–H insertion products 2g and 2h in high yields (entries 11 and 12). After the above catalytic reactions, the PXRD patterns of the recycled catalyst samples remain almost intact, confirming that the porous framework of Ir-PMOF-1(Hf) can well tolerate the catalytic process (Fig. S9†).
Comparison among the above catalytic activities of differently substituted silanes obviously discloses a reactivity order of primary > secondary > tertiary silanes (entries 1, 6–10). This reactivity order induced by Ir-PMOF-1(Hf) is strikingly different from the known order of Si–H insertion when homogeneously catalyzed by dirhodium(II) tetracarboxylates,4 metalloporphyrin complexes,9 Cu(I/II)5 and Ag(I)7 salts, under whose catalysis the secondary and tertiary silanes display higher activity than primary silanes.
To further evaluate the chemoselectivity of Ir-PMOF-1(Hf) towards primary silanes, a competition experiment has been carried out, employing a 0.9:1.35:2.7 molar ratio mixture of PhSiH3, Ph2SiH2 and Ph3SiH as the Si–H sources, and thus the amounts of the primary, secondary and tertiary Si–H bonds are equal (Fig. S10†).7 One equivalent of EDA reacts with this mixture to give rise to Si–H products 2a–c in a molar ratio of 1:0.7:0.3 in the presence of Ir-PMOF-1(Hf). In a similar way, BDA reacts with this mixture to produce 2d–f in a molar ratio of 1:0.7:0.3. In comparison, the similar reactions induced by Ir(TPP)(CO)Cl and Rh2(OAc)4 generate 2a–c in the ratios of 1:1:0.4 and 1:1.5:1.2, respectively. Competition experiments between two of the three silanes under the catalysis of Ir-PMOF-1(Hf) have also been carried out, and the molar ratios of 2a:2b and 2a:2c are 1:0.7 and 1:0.2, respectively. Considering that the Si–H insertion products 2a–c do not undergo further Si–H insertion in the presence of Ir-PMOF-1(Hf), we have also carried out a similar competition experiment but employed a 1:1:1 molar ratio mixture of PhSiH3, Ph2SiH2 and Ph3SiH instead. This catalysis result showed that 2a, 2b and 2c were formed in a molar ratio of 1:0.5:0.1 (Fig. 3).
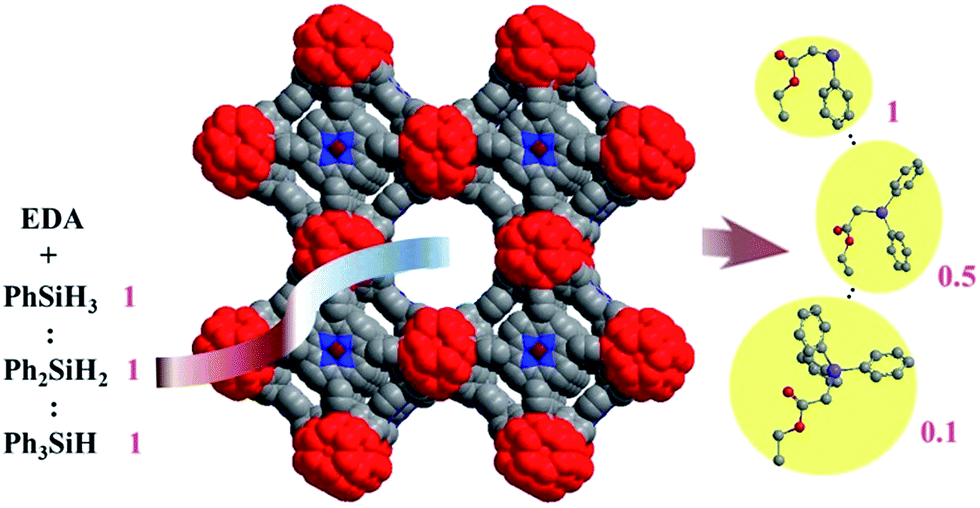 | ||
| Fig. 3 Chemoselective Si–H insertion heterogeneously catalysed by the Ir-PMOF-1(Hf) framework. Numbers show the molar ratio. | ||
The inverted chemoselectivity for the carbenoid insertion into primary Si–H bonds induced by Ir-PMOF-1(Hf) may be ascribed to the following two reasons. On one hand, the active sites are located on the axial positions of the metalloporphyrin motifs, which enclose a confined nanospace, imposing more steric effect toward bulky reactants than free metal catalysts such as dirhodium(II) tetracarboxylates. As a result, the sterically unfavored tertiary silanes display poor chemoselectivity in the case of the heterogeneous catalyst Ir-PMOF-1(Hf) compared to the similar metalloporphyrin-based homogeneous catalyst Ir(TPP)(CO)Cl. On the other hand, the porosity of Ir-PMOF-1(Hf) brings additional size selectivity and channel diffusion differences to enforce the confinement effect; therefore, the least bulky primary silane and its corresponding Si–H insertion product can be transported through the multi-channels of the self-supported catalytic Ir-PMOF-1(Hf) more easily than those of secondary and tertiary analogues. The overall result might explain the high chemoselectivity and reactivity towards primary silanes in the Ir-PMOF-1(Hf)-catalyzed heterogeneous Si–H insertion reactions.
Moreover, the crystalline catalysts can be easily isolated by centrifugation, and reused at least ten times for Si–H insertion of PhSiH3 with EDA (Fig. 4; Table S5†). During the ten reaction runs, the yields of 2a were in the range of 74–92%. From the 1st to 6th run, it took less than 15 min to finish the reaction, whereas the 7th, 8th and 9th runs needed 26, 55 and 90 min, respectively, to complete. The reduced activity from 7th run might be due to reactants and products stuck in the pores of the Ir-PMOF-1(Hf) framework. In order to remove the adsorbed organic compounds on the interior and exterior surfaces during the catalytic reactions, the recycled Ir-PMOF-1(Hf) catalyst should be washed with DCM (3 × 8 mL) before successive runs. The PXRD patterns of the recycled catalyst after the 1st, 3rd, 5th and 10th runs show comparable diffraction profiles with the as-synthesized sample (Fig. S11†).
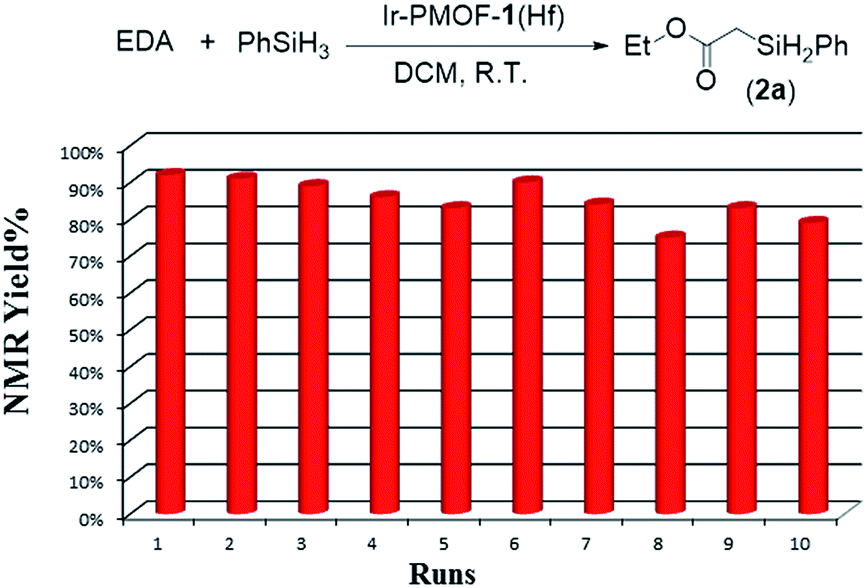 | ||
| Fig. 4 Recycling experiments. | ||
Conclusions
In summary, we have successfully imparted the outstanding carbenoid transfer catalysis of homogeneous Ir-porphyrin to a porous, robust and stable Ir-PMOF-1(Hf), underlining a new field of heterogeneous MOF catalysis via introducing rarely explored noble metal-porphyrins (e.g. Ir, Ru and Rh). Engineering of confined and catalytic CSs in a MOF, in collaboration with its open porosity, presents multi-channel crystalline nanoreactors for selective Si–H insertion, offering higher efficiency than the corresponding homogeneous catalysts with excellent TOF and TON. An inverted reactivity and selectivity order of Si–H insertion reactions, i.e. primary > secondary > tertiary, which is unattainable by conventional metal catalysts, is achieved, and the self-supported catalyst is easy to recycle and separate. These results demonstrate the versatility of MOF catalysis which can not only incorporate the excellent features of homogeneous catalysts, but also endow metal catalysts with a unique property under heterogeneous conditions to accomplish catalytic processes unachievable by homogeneous catalysts.Acknowledgements
This work was supported by the 973 Program (2012CB821701), the NSFC Projects (21373278, 91222201), the STP Project of Guangzhou (15020016), the NSF of Guangdong Province (S2013030013474), the RFDP of Higher Education of China (20120171130006) and the Fundamental Research Funds for the Central Universities.Notes and references
- (a) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC; (b) T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097 RSC; (c) Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234 CrossRef CAS PubMed; (d) A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC; (e) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (f) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed; (g) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC; (h) J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361 CrossRef CAS.
- (a) S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490 CrossRef CAS; (b) R. A. Fischer and C. Wöll, Angew. Chem., Int. Ed., 2008, 47, 8164 CrossRef CAS PubMed; (c) S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef PubMed.
- (a) H. M. L. Davies, T. Hansen, J. Rutberg and P. R. Bruzinski, Tetrahedron Lett., 1997, 38, 1741 CrossRef CAS; (b) S. Kitagaki, M. Kinoshita, M. Takeba, M. Anada and S. Hashimoto, Tetrahedron: Asymmetry, 2000, 11, 3855 CrossRef CAS; (c) R. T. Buck, D. M. Coe, M. J. Drysdale, L. Ferris, D. Haigh, C. J. Moody, N. D. Pearson and J. B. Sanghera, Tetrahedron: Asymmetry, 2003, 14, 791 CrossRef CAS.
- (a) L. A. Dakin, S. E. Schaus, E. N. Jacobsen and J. S. Panek, Tetrahedron Lett., 1998, 39, 8947 CrossRef CAS; (b) Y.-Z. Zhang, S.-F. Zhu, L.-X. Wang and Q. L. Zhou, Angew. Chem., Int. Ed., 2008, 47, 8496 CrossRef CAS PubMed.
- R. Vicente, J. González, L. Riesgo, J. González and L. A. López, Angew. Chem., Int. Ed., 2012, 51, 8063 CrossRef CAS PubMed.
- M. J. Iglesias, M. C. Nicasio, A. Caballero and P. J. Pérez, Dalton Trans., 2013, 42, 1191 RSC.
- J. Ma, H. Jiang and S. Zhu, Org. Lett., 2014, 16, 4472 CrossRef CAS PubMed.
- (a) Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510 CrossRef CAS PubMed; (b) J.-C. Wang, Z.-J. Xu, Z. Guo, Q.-H. Deng, C.-Y. Zhou, X.-L. Wan and C.-M. Che, Chem. Commun., 2012, 48, 4299 RSC.
- Y.-D. Wu and C.-L. Wong, J. Org. Chem., 1995, 60, 821 Search PubMed.
- (a) D. Mukherjee, R. R. Thompson, A. Ellern and A. D. Sadow, ACS Catal., 2011, 1, 698 CrossRef CAS; (b) D. Peng, Y. Zhang, X. Du, L. Zhang, X. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154 CrossRef CAS PubMed; (c) M. Zhao, W. Xie and C. Cui, Chem.–Eur. J., 2014, 20, 9259 CrossRef CAS PubMed.
- Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS PubMed.
- (a) L. Chen, T. Yang, H. Cui, T. Cai, L. Zhang and C.-Y. Su, J. Mater. Chem. A, 2015, 3, 20201 RSC; (b) T. Yang, H. Cui, C. Zhang, L. Zhang and C.-Y. Su, Inorg. Chem., 2013, 52, 9053 CrossRef CAS PubMed; (c) T. Yang, H. Cui, C. Zhang, L. Zhang and C.-Y. Su, ChemCatChem, 2013, 5, 3131 CrossRef CAS.
- H. Cui, Y. Wang, Y. Wang, Y.-Z. Fan, L. Zhang and C.-Y. Su, CrystEngComm, 2016, 18, 2203 RSC.
- B. J. Burnett, P. M. Barron, C. Hu and W. Choe, J. Am. Chem. Soc., 2011, 133, 9984 CrossRef CAS PubMed.
- Y. Zhao, N. Kornienko, Z. Liu, C. Zhu, S. Asahina, T.-R. Kuo, W. Bao, C. Xie, A. Hexemer, O. Terasaki, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2199 CrossRef CAS PubMed.
- S. Takaishi, E. J. DeMarco, M. J. Pellin, O. K. Farha and J. T. Hupp, Chem. Sci., 2013, 4, 1509 RSC.
- X.-L. Yang, M.-H. Xie, C. Zou, Y. He, B. Chen, M. O'Keeffe and C.-D. Wu, J. Am. Chem. Soc., 2012, 134, 10638 CrossRef CAS PubMed.
- L. Meng, Q. Cheng, C. Kim, W.-Y. Gao, L. Wojtas, Y.-S. Chen, M. J. Zaworotko, X. P. Zhang and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 10082 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440 CrossRef CAS PubMed.
- J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283 CrossRef CAS.
- J. Zheng, M. Wu, F. Jiang, W. Su and M. Hong, Chem. Sci., 2015, 6, 3466 RSC.
- Q. Lin, X. Bu, A. Kong, C. Mao, X. Zhao, F. Bu and P. Feng, J. Am. Chem. Soc., 2015, 137, 2235 CrossRef CAS PubMed.
- D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105 CrossRef CAS PubMed.
- (a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, Appl. Crystallogr., 2009, 42, 339 CrossRef CAS; (b) P. V. D. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- O. Delgado-Friedrichs, M. O’Keeffe and O. M. Yaghi, Acta Cryst., 2006, A62, 350–355 CrossRef CAS PubMed.
- T. V. Voskobojnikov, E. S. Shpiro, H. Landmesser, N. I. Jaeger and G. Schulz-Ekloff, J. Mol. Catal. A: Chem., 1996, 104, 299–309 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Physical characterization of metalloporphyrin ligands, crystal structure data, physical characterization, chemical/thermal stability studies, gas adsorption of porphyrinic MOFs, recycling experiments, and NMR data of Si–H insertion products. CCDC 1491233. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03288e |
| This journal is © The Royal Society of Chemistry 2017 |