
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxide-bridged five-coordinate DyIII single-molecule magnet exhibiting the record thermal relaxation barrier of magnetization among lanthanide-only dimers†
Jin
Xiong
a,
Hai-Yan
Ding
b,
Yin-Shan
Meng
a,
Chen
Gao
a,
Xue-Jing
Zhang
c,
Zhao-Sha
Meng
a,
Yi-Quan
Zhang
b,
Wei
Shi
c,
Bing-Wu
Wang
 *a and
Song
Gao
*a
*a and
Song
Gao
*a
aBeijing National Laboratory of Molecular Science, College of Chemistry and Molecular Engineering, State Key Laboratory of Rare Earth Materials Chemistry and Applications, Peking University, Beijing, 100871, P. R. China. E-mail: wangbw@pku.edu.cn; gaosong@pku.edu.cn; Web: http://www.chem.pku.edu.cn/mmm/ Fax: +8610-6275-1708
bJiangsu Key Laboratory for NSLSCS, School of Physical Science and Technology, Nanjing Normal University, Nanjing 210023, P. R. China
cDepartment of Chemistry, Key Laboratory of Advanced Energy Materials Chemistry (MOE), State Key Laboratory of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China
First published on 3rd October 2016
Abstract
A hydroxide-bridged centrosymmetric DyIII dimer with each DyIII being five-coordinated has been synthesized using bulky hindered phenolate ligands. Magnetic studies revealed that this compound exhibits a slow magnetic relaxation of a single-ion origin together with a step-like magnetic hysteresis of the magnetic coupled cluster. The thermal relaxation barrier of magnetization is 721 K in the absence of a static magnetic field, while the intramolecular magnetic interaction is very large among reported 4f-only dimers. CASSCF calculations with a larger active space were performed to understand the electronic structure of the compound. The thermal relaxation regime and the quantum tunneling regime are well separated, representing a good model to study the relaxation mechanism of SMMs with intramolecular Dy–Dy magnetic interactions.
Introduction
Single-molecule magnets (SMMs) are molecules that can exhibit slow magnetic relaxation behaviour. Such slow relaxation occurs between the bistable ground spin states generated by the magnetic anisotropy of molecules. SMMs have experienced fast development due to their importance in theoretical studies as well as their potential applications in molecular spintronics devices1,2 and quantum computations.3,4 The effective energy barrier (Ueff) and blocking temperature (TB) are universal parameters used to evaluate the ability of magnetization blocking. The former is the thermal energy barrier of magnetization reversal. The latter is the highest temperature for SMMs to display magnetic hysteresis.5 High Ueff and TB are pursued by chemists in the field for potential applications in molecular devices. In the early days, maximizing the spin of the ground state via large 3d metal clusters was considered as a preferred method to obtain SMMs with high Ueff. However, such strategies advanced slowly as a result of the difficulty of manipulating the arrangement of the anisotropy axes of each paramagnetic core so as to obtain a large ground state spin and uniaxial anisotropy of the whole molecule simultaneously.6 Therefore, synthesizing SMMs containing only one paramagnetic centre (so-called single-ion magnets, SIMs) with a large magnetic anisotropy became an alternative choice.7–9 Lanthanide ions were thought to be good candidates to build SIMs because of their strong magnetic anisotropy induced by strong spin–orbit coupling.5,10 Since the first example of lanthanide SIMs, the [NH4][LnPc2] series11 reported in 2003, a lot of knowledge regarding the manipulation of the magnetic anisotropy of a single lanthanide ion has been accumulated. Theoretically, several models and strategies have been proposed to obtain lanthanide SMMs with good performances.12–14 Experimentally, the effective energy barrier (Ueff) and blocking temperature (TB) have increased up to 1025 K (ref. 15) and 20 K (ref. 16) respectively in two DyIII-based SIMs. Hence, the next step would be to question how to manipulate the 4f–4f interaction to build SMMs with both a large ground state spin and strong uniaxial magnetic anisotropy. However, the exchange interaction between the J multiplets of lanthanide ions are complicated to understand17 and are generally very weak, which hindered the development of cluster-based lanthanide SMMs. Only a limited number of examples of 4f-only SMMs with intramolecular 4f–4f magnetic interactions have been reported to date.18–25 It is worth mentioning that the quantum tunnelling of magnetization (QTM) is common for lanthanide SIMs under zero field,26–28 which accelerates the magnetic relaxation rate and limits their potential application. Therefore, suppressing the zero field QTM is also important for the blocking of magnetization when designing SMMs. For the design of high performance SIMs, ensuring SIMs have high uniaxial geometrical symmetry is regarded as a good strategy14,16 but it is hard to achieve in lanthanide compounds due to the large ion radius and the weak metal–ligand bond.29,30Rare examples were reported regarding the synthesis and magnetic studies of DyIII based low coordination number (CN) compounds.31–35 Among the low-CN DyIII-based SMMs, {Dy5},36 {Dy4K2}37 and [Dy(BIPMTMS)(BIPMTMSH)]26 molecules (all CN = 6) exhibited high relaxation temperatures and relatively strong DyIII–ligand bonds, suggesting the existence of interesting magneto-structural correlations. One classical method to synthesize low CN Ln compounds is using bulky ligands to increase the steric hindrance.30 In addition, with proper hindered ligands, such compounds could accept small hindered ligands. This makes it possible to use a small ligand to bridge such monomers to introduce magnetic interactions and build 4f-only SMMs.
Herein, we present a hydroxide-bridged symmetric five-coordinate DyIII dimer, [Dy(μ-OH)(DBP)2(THF)]2 (DBP− = 2,6-di-tert-butylphenolate), which was synthesized by the hydration of low-CN DyIII monomers. This compound exhibits slow magnetic relaxation from a single-ion origin as well as step-like hysteresis curves induced by intramolecular antiferromagnetic Dy–Dy coupling. To the best of our knowledge, this compound represents the highest Ueff in lanthanide-only dimers.
Results and discussion
Inspired by reported works,38–40 we designed an efficient method to get the crystals of the target product in one pot. Briefly, an in situ vapour diffusion reaction of LnN*3 (N*− = N(Si(CH3)3)2−), H-DBP and H2O gave qualified crystals in the formula of [Ln(μ-OH)(DBP)2(THF)]2. (Ln = Dy 1, Y 2. See ESI Section I for details†). An yttrium analogue (3) with 5% dysprosium doping (ICP-AES measurements of the wet-digested solution suggested the molar ratio was about Dy![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Y = 1:19) and a gadolinium analogue (4) were also synthesized for the compared study.
:Y = 1:19) and a gadolinium analogue (4) were also synthesized for the compared study.
Single-crystal X-ray diffraction experiments indicate compounds 1, 2 and 4 are almost isostructural, as they are all crystallized in an orthorhombic space group, Pbca, with close cell parameters (Table S1†). Powder X-ray diffraction experiments confirm that the magnetic diluted sample 3 is a pure single-phase which is isostructural with 2 (Fig. S1†). In 1, each molecule is a centrosymmetric dimer bridged by two hydroxide groups. No H-bonding acceptors can be found for these two hydroxide groups. This is consistent with the FT-IR spectrum (O–H stretching, 3678 cm−1, m, sh, Fig. S2†). Five O atoms coordinate to each DyIII ion, two from the DBP− ligands, two from hydroxide and one from THF (Fig. 1a). The intramolecular Dy–Dy distance is 3.71 Å while the shortest intermolecular Dy–Dy distance is 10.76 Å, suggesting that the intermolecular dipole–dipole interaction could be negligible in comparison to the intramolecular interaction. The crystal field for each DyIII ion exhibits strong anisotropy with four short Dy–O bonds and one longer Dy–O bond (Fig. 1b). The two phenolate O atoms are the most electronegative, leading to very short Ln–O bond lengths of 2.09 Å and 2.12 Å respectively. The two Ln–OH bonds are 2.24 Å and 2.28 Å. The longest Ln–O bond in the dimer is 2.38 Å, in which the O atom is from the electroneutral THF ligand. The bond lengths and angles of Ln2O2 parallelograms are listed in Table 1. To evaluate the geometric symmetry of the first coordination sphere, CShM's values (Table S2†) of the DyIII ion in comparison with all of the reference standard 5-coordinate polyhedrons were calculated using the SHAPE 2.0 program.41–43 The smallest value is 1.923 for C4v symmetry (square pyramid), indicating no apparent geometric symmetry. (CShM's value is a kind of index to evaluate the geometric similarity between two coordination polyhedrons. During the calculations, the atom type is not considered. Smaller CShM values mean that the two polyhedrons are closer. A value of zero means that the two polyhedrons are geometrically identical).
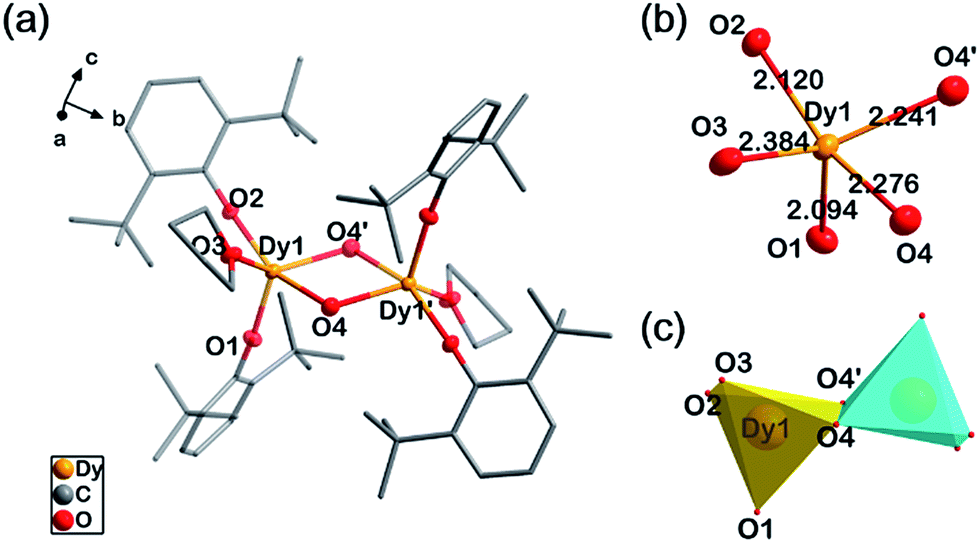 | ||
| Fig. 1 Structure details of 1. (a) The molecular structure of 1 with all of the H atoms omitted. (b) The first coordination shell of DyIII in 1. (c) Polyhedrons of the DyIII ions in 1. | ||
| Compound | Bond length (Å) | Bond angles (°) | ||
|---|---|---|---|---|
| Ln1–O4 | Ln1–O′4 | O4–Ln1–O′4 | Ln1–O4–Ln′1 | |
| 1(DyIII) | 2.28 | 2.24 | 69.5 | 110.5 |
| 2(YIII) | 2.24 | 2.28 | 68.7 | 111.3 |
| 4(GdIII) | 2.31 | 2.25 | 69.5 | 110.5 |
To probe the static magnetic behaviour, direct current (dc) magnetic measurements were performed on both of the polycrystalline samples of 1, 3 and 4. The χmT value of 1 at 300 K was 27.76 cm3 mol−1 K, which is close to the expected value (28.34 cm3 mol−1 K) for the two isolated DyIII ions. The sharp decline of the χmT values at low temperatures may be attributed to three possible reasons: antiferromagnetic coupling, thermal depopulation of low lying crystal field states and magnetic anisotropy. When the χmT value of 3 was divided by the molar ratio of Dy (ω in eqn (1), 5.7%), the result (χmT(3′), blue circles in Fig. 2) could be considered as the value of the two isolated DyIII ions with the same coordination environment of 1. Hence the difference defined in eqn (1) can be mainly attributed to the contribution of the intramolecular magnetic interactions. As the insert of Fig. 2 shows, ΔχmT values are negative between 10 K and 16 K, implying an antiferromagnetic interaction between the DyIII ions. The apparent difference between the zero-field-cooled (ZFC) and field-cooled (FC) magnetic susceptibility curves (Fig. S5 and S7†) indicates strong anisotropy as well as open magnetic hysteresis curves for both 1 and 3.
| ΔχmT = χmT(1) − χmT(3′) = χmT(1) − χmT(3)/ω | (1) |
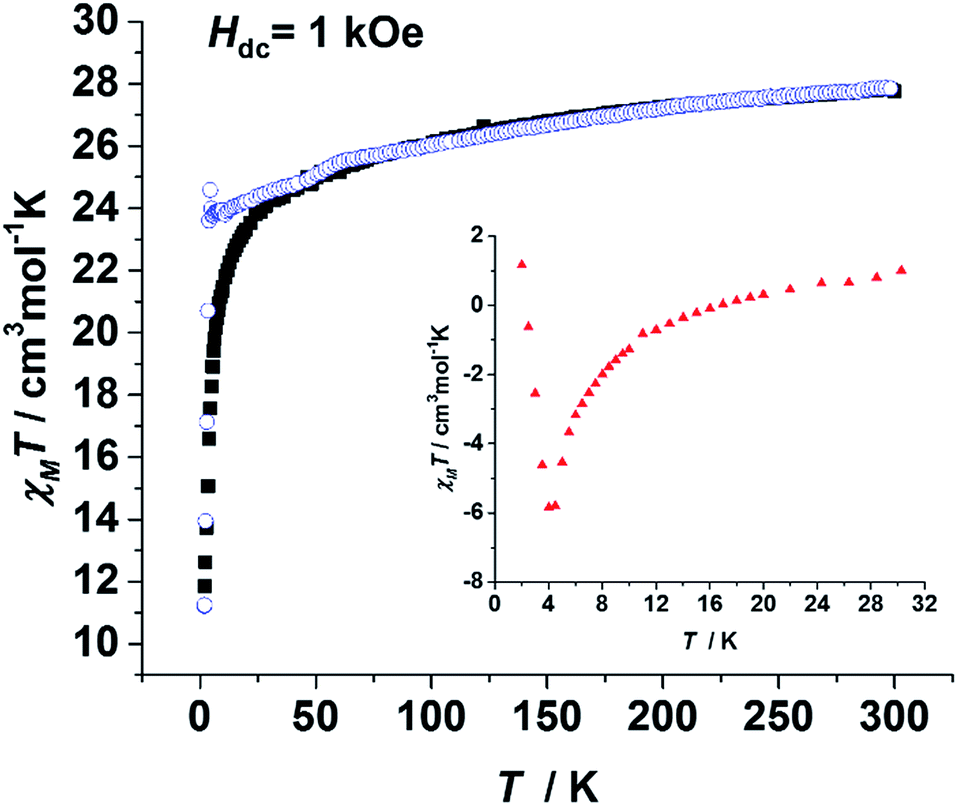 | ||
| Fig. 2 Plots of the temperature dependence of χmT(1) (black square) and χmT(3′) (blue circle). Insert: an enlarged version of the ΔχmT as calculated from eqn (1). All measurements were performed on polycrystalline samples. | ||
As expected, in the magnetic hysteresis measurements of 1, step-like hysteresis curves were observed up until 8 K under a scanning rate of 200 Oe s−1 (Fig. 3). Such steps have been observed on other coupled DyIII dimers, with higher scanning rates or at lower temperatures, before.21–23 The loop was kept open near to zero field with a coercive field (Hc) of about 2500 Oe at 2 K. Steps appeared at the field near to ±2500 Oe, then the loop was enlarged at higher field until it was finally closed at around ±20000 Oe. These steps could be attributed to a level-crossing between the low lying states of the dimer.21–23 Without intramolecular coupling, the hysteresis curve of 3 (Fig. 3 insert) was a typical butterfly-like loop for SIMs with a QTM process near to zero field.
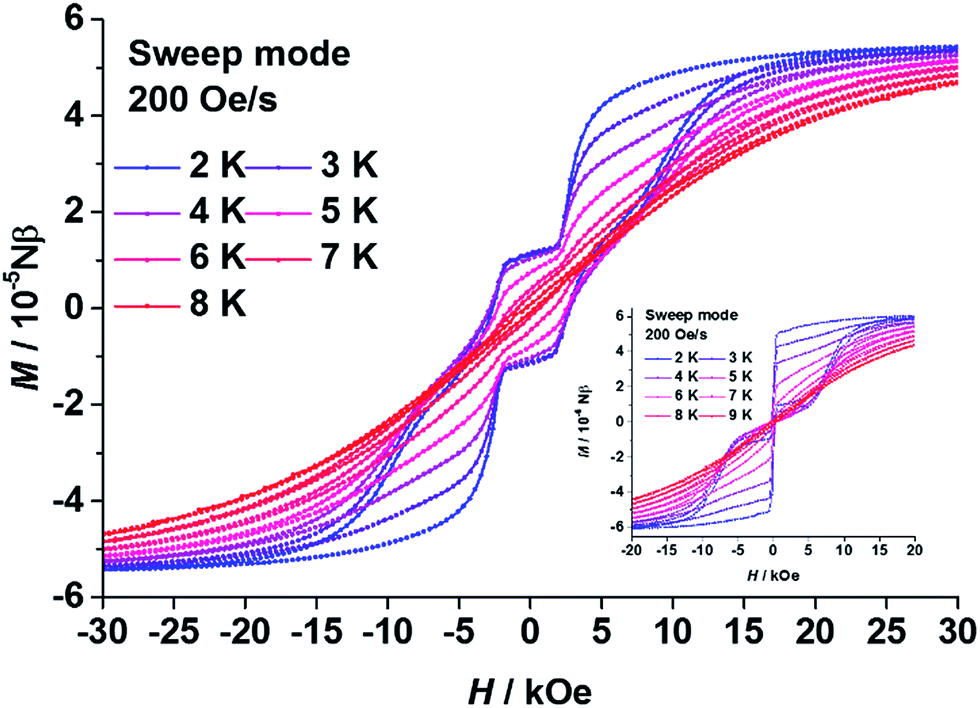 | ||
| Fig. 3 Plots of the magnetic hysteresis of polycrystalline samples of 1 (main) and 3 (insert). The lines are guides for the eyes. | ||
Alternative current (ac) magnetic measurements show the dynamic characteristics of SMMs. When 1 was subjected to an ac field from 100 Hz to 10000 Hz under zero static field, the maxima of both the in-phase (χ′m) and out-of-phase (χ′′m) components showed a clear frequency dependence, revealing the typical slow magnetic relaxation behaviour of SMMs (Fig. 4 top and Fig. S14–S16†). Resonance peaks of χ′′m could be observed between 35 K (100 Hz) and 49 K (10000 Hz). The ac susceptibility measurements as a function of the dc field were also carried out. At 2 K, both χ′m and χ′′m remained very small under a dc field smaller than 2000 Oe, then sharply increased to a maximum around the field of 2500 Oe (Fig. S13†). This behaviour coincides with the level-crossing signal detected in the hysteresis measurement. The raised signal in the low temperature regime should be the consequence of the level-crossing process. The slow thermal relaxation behaviour remained the same under a static field of 2500 Oe (Fig. S22–S24†).
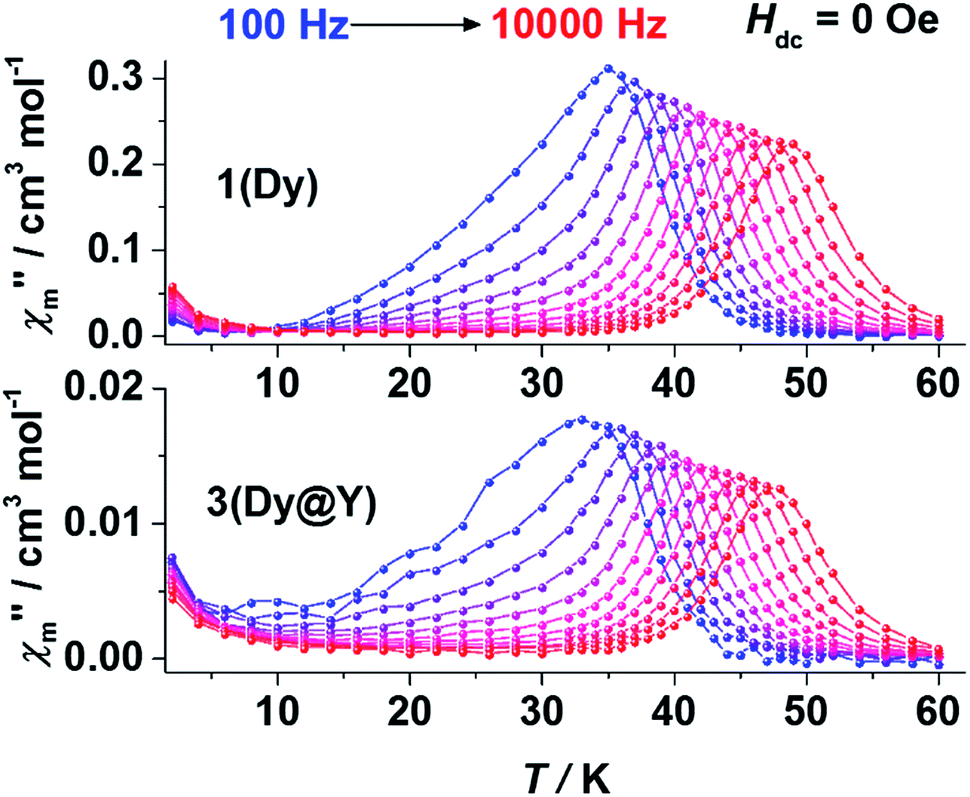 | ||
| Fig. 4 Plots of the temperature dependent out-of-phase ac susceptibility (χ′′m) of 1 (top) and 3 (bottom) at zero static field. The lines are guides for the eyes. | ||
For comparison, ac magnetic measurements under zero (Fig. 4 bottom and Fig. S30–S32†) and a 2500 Oe dc field (Fig. S38–S40†) were also performed on the polycrystalline samples of 3. As expected, two well-separated relaxation regimes were observed under zero static field (Fig. 4 bottom). In the high temperature regime, frequency-dependent relaxation peaks of χ′′m could be observed between 36 K (100 Hz) and 48 K (10000 Hz) under both zero field and a 2500 Oe dc field. The resonance temperature changes are small after magnetic dilution, implying that the thermal relaxation in the high temperature regime may be a single-ion behaviour. In the low temperature regime, the χ′′m peaks are raised when under zero field and disappear at a 2500 Oe field. This is a typical behaviour of the zero-field QTM process. The QTM process appeared under zero field due to the absence of a bias field that is generated by the intramolecular interaction, and could be suppressed by an external dc field. No resonance peaks were observed in the low temperature regime, as the QTM process is slower than the time limit of the measurement.
The ac measurements at a lower frequency range (1–1000 Hz) were also performed (Fig. S18–S20, S26–S28, S34–S36 and S42–S44†). Although the first coordination sphere of each DyIII has no obvious geometric symmetry, the results of the low-frequency ac susceptibility measurements indicate that, even for the diluted sample 3, the QTM process is slower than 1 Hz (Fig. S35†). This is not usual for the DyIII species, since broken axial symmetry will induce the mixing of the mJ states, hence accelerating the QTM process.
The resonance temperatures and extracted relaxation times between the overlapped frequency regimes (100–1000 Hz) of the two measurements (100–10000 Hz on PPMS, 1–1000 Hz on MPMS) were well-matched (see Table S3–S6†). As Fig. 5 shows, the plots of lnτ vs. T−1 for the thermal relaxation process are almost the same regardless of magnetic dilution or an applied dc field, confirming that such relaxation is the nature of a single DyIII ion, which is consistent with the conclusions in previously reported work.23 Arrhenius fitting of the data of 1 at zero static field (1@ZF) between 40 K and 49 K gave a Ueff of 721 K (501 cm−1) and a τ0 of 6.6 × 10−12 s. The Arrhenius fitting parameters of the other data (1@HF, 3@ZF and 3@HF, HF = 2500 Oe dc field) are listed in Table S7.† The parameters are very close to that of 1@ZF. However, the non-linear trend of the plots implies that a contribution of the Raman process cannot be omitted. All of the plots in the whole measurement regime could be fitted well when one Raman process term was added into the fitting equation. Dual process fitting parameters are listed in Table S8.† For 1@ZF, the dual process fitting gave a Ueff of 754 K (524 cm−1) and a τ0 of 3.5 × 10−12 s. These two parameters are close to those from the Arrhenius fitting, and C is very small (2.7 × 10−4 s−1 K−4.00), implying that the Orbach process is predominate.
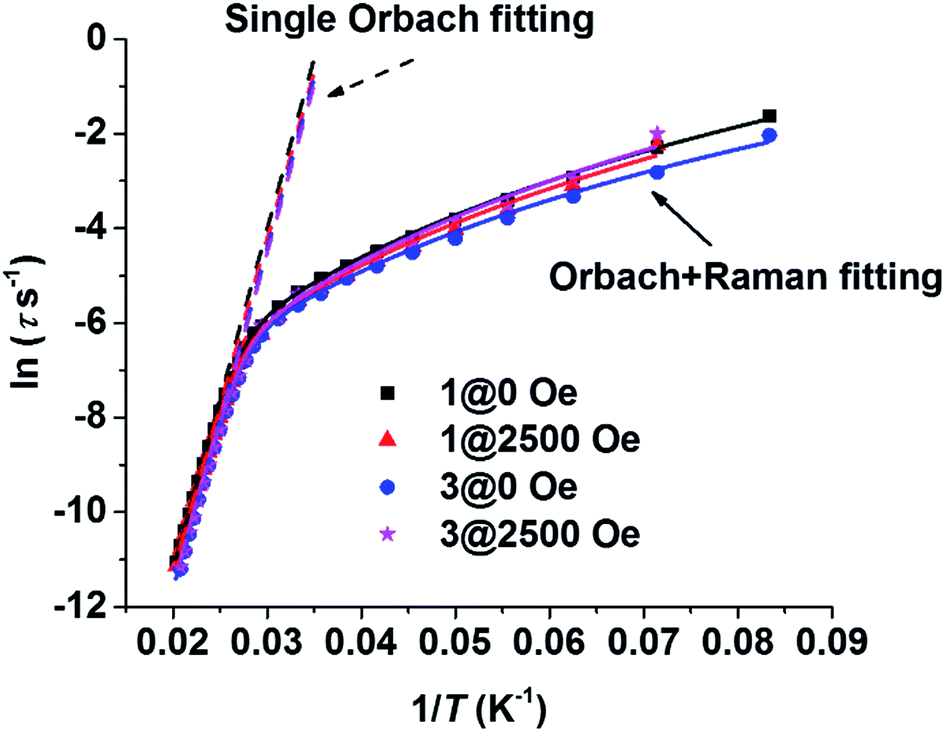 | ||
| Fig. 5 Plots of the logarithm of the relaxation time (ln(τ)) vs. reciprocal temperature (T−1) for 1@0 Oe, 1@2500 Oe, 3@0 Oe and 3@2500 Oe. The dashed lines show the results of the Arrhenius fitting (single Orbach process) for the data in the temperature regime above 40 K (42 K to 49 K for compound 1, 41 K to 48 K for compound 3). The solid lines show the results of the dual process fittings (one Raman and one Orbach process) for the data in the temperature regime of the whole testing range (12 K to 49 K). | ||
Complete-active-space self-consistent field (CASSCF) calculations on individual DyIII fragments of compound 1 (in which the other DyIII is replaced by a diamagnetic LuIII) on the simplified structure of the X-ray determined geometry have been carried out with MOLCAS 7.8 program packs44 (see ESI Section IV for detailed method and results†). However, the results of the conventional method (only choosing the 4f orbitals as the active space, 9 electrons spanning 7 orbitals, CAS(9,7)) could not well explain the effective energy barrier on the basis of a recently reported methodology.45 According to the methodology, the rate of the thermal-assisted tunnelling transition (TAT) through the second-excited Kramers doublet (KD3) is 1.7 times faster than that through the first-excited Kramers doublet (KD2) at 50 K. (Approximately, P ∝ exp(−E/kBT)![[small mu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cd.gif) 2 where P is the transition probability, E is the energy of the doublet relative to the ground state doublet and is the average value of the magnetic moment matrix elements connecting the opposite components of doublets). Hence the TAT process will occur through the second-excited Kramers doublet (KD3 in Table S9†). However, the corresponding energy gap is only 661.8 K (Table S9† and Fig. 6 top), which is smaller than the Ueff obtained by the Arrhenius fitting. This is not reasonable since the Arrhenius Ueff is usually smaller or equal to the corresponding TAT energy gap. One possible reason for the smaller calculated barrier is the improper choice of active space in the computation. Having only seven f orbitals as the active space in the CASSCF part may not be suitable in treating the present compound, considering the existence of short Ln–O bonds, in which the non-electrostatic interaction may be larger. Thus expansion of the active space was attempted in further calculations, considering the possible contribution of the strong coordinated atoms. Limited by our hardware resources, CAS(11,8) calculations were carried out. The corresponding calculated energy gap was increased to 763 K and the relaxation still occurred through the second-excited state (Table S9† and Fig. 6 bottom). These results were consistent with the Orbach energy barrier that was obtained by the dual relaxation process fitting. It seems that the CAS(11,8) calculation may be more appropriate than the conventional CAS(9,7) calculation to understand the electronic structure of the present system, where the ligands strongly coordinate to DyIII.
2 where P is the transition probability, E is the energy of the doublet relative to the ground state doublet and is the average value of the magnetic moment matrix elements connecting the opposite components of doublets). Hence the TAT process will occur through the second-excited Kramers doublet (KD3 in Table S9†). However, the corresponding energy gap is only 661.8 K (Table S9† and Fig. 6 top), which is smaller than the Ueff obtained by the Arrhenius fitting. This is not reasonable since the Arrhenius Ueff is usually smaller or equal to the corresponding TAT energy gap. One possible reason for the smaller calculated barrier is the improper choice of active space in the computation. Having only seven f orbitals as the active space in the CASSCF part may not be suitable in treating the present compound, considering the existence of short Ln–O bonds, in which the non-electrostatic interaction may be larger. Thus expansion of the active space was attempted in further calculations, considering the possible contribution of the strong coordinated atoms. Limited by our hardware resources, CAS(11,8) calculations were carried out. The corresponding calculated energy gap was increased to 763 K and the relaxation still occurred through the second-excited state (Table S9† and Fig. 6 bottom). These results were consistent with the Orbach energy barrier that was obtained by the dual relaxation process fitting. It seems that the CAS(11,8) calculation may be more appropriate than the conventional CAS(9,7) calculation to understand the electronic structure of the present system, where the ligands strongly coordinate to DyIII.
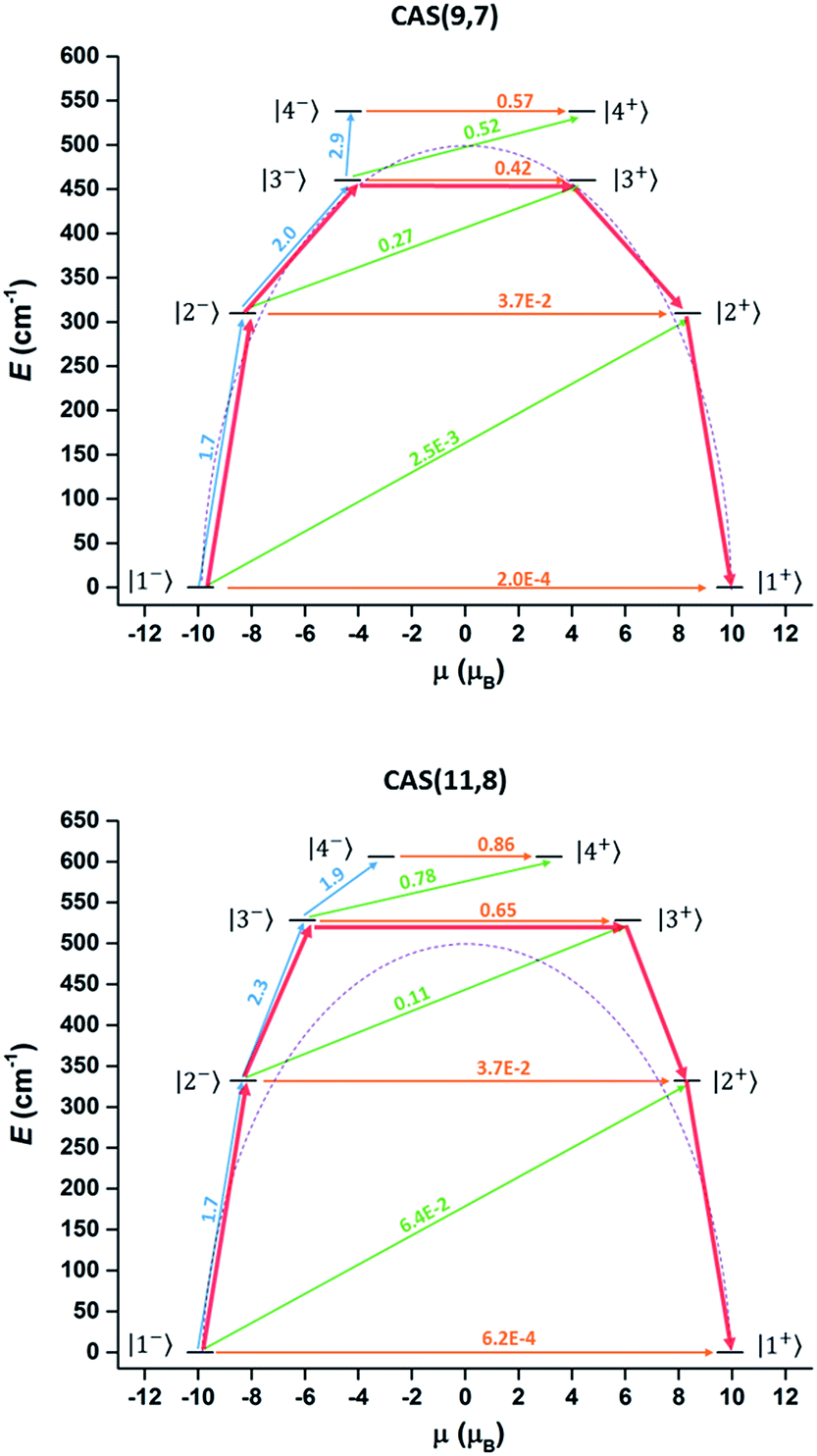 | ||
| Fig. 6 Magnetization blocking barrier in the DyIII fragment in the CAS(9,7) (top) and CAS(11,8) (bottom) cases. The KDs are arranged according to the values of their magnetic moments and noted with the same number in Table S9.† The values and corresponding arrows were marked with the same colours (orange for |i−〉 → |i+〉, green for |i−〉 → |i + 1+〉 and blue for |i−〉 → |i + 1−〉). The red arrows show the relaxation pathway. The purple dashed line is a guide curve of the Ueff (501 cm−1). | ||
Both calculations gave a ground spin state very close to the Ising limit state (Table S10†). The easy axis is close to the perpendicular direction relative to the Dy–O3 (O3 is the O atom of THF, Fig. 1a) bond (Fig. S46†). O3 is the atom which has the least negative Mulliken charge distribution among the five O atoms of the first coordination sphere (Fig. S48†). This demonstrates that when the other ligands are negatively charged, the only one neutral ligand has a decisive role in whether to ‘pin’ the easy axis in the perpendicular direction.46
The intramolecular DyIII–DyIII interaction was also analysed using theoretical and experimental methods (Table 2). The 4f–4f interaction comprises two components, the dipole–dipole interaction and the exchange interaction (coupling constants are represented as Jdip and Jexch). Ab initio calculations (CAS(11,8)) gave gz = 19.86, gx = 0.002 and gy = 0.002, which are very close to the values of the Ising limit state of DyIII with a pseudospin of ±1/2. Thus an Ising approximation in the Lines model47 is reasonable. The total coupling constant can be described as shown in eqn (2) using the Hamiltonian description as shown in eqn (3). The magnetic dipole–dipole contribution to the coupling constant was calculated as Jdip = 1.8 cm−1, ferromagnetically (see ESI Section V for the detailed process†).
| J = Jdip + Jexch | (2) |
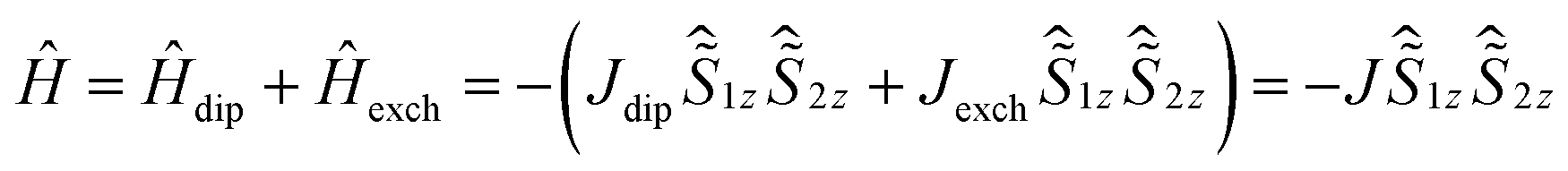 | (3) |
| Method | J dip | J exch | J |
|---|---|---|---|
| Level-crossing | 1.8 | −6.4 | −4.6 |
| POLY_ANISO | 1.8 | −5.4 | −3.6 |
Two methods were applied to obtain the magnetic coupling interaction constant. One method is based on the Zeeman effect. Considering that the steps on hysteresis curves are the consequence of level-crossing between the ground state and the first-excited state of the dimer, eqn (4) could be applied to calculate the J value.21 The first derivative of the magnetization of 1 reveals that the crossing field could be estimated as being 2.5 kOe (Fig. S10 and S11†). Thus J = −4.6 cm−1 was obtained when g = gz = 19.85, hence Jexch = −6.4 cm−1. The other method involves fitting the dc magnetic susceptibility of compound 1 during 2 K to 300 K using the program POLY_ANISO,48–50 in which all the spin orbitals of each DyIII were considered using the Lines model (see Section V in ESI for details†). The fitting gave a result of J = −3.6 cm−1 and Jexch = −5.4 cm−1. Both results indicate that the exchange coupling transferred through the two hydroxide bridges is strong among the bridged lanthanide coordination compounds, leading to an apparent antiferromagnetic interaction although the dipolar component is ferromagnetic.
| Hcross = −J/2gβ | (4) |
For comparison studies, the intramolecular GdIII–GdIII interaction in 4 was also examined using both an analytical method and numerical method (Fig. S50, see ESI Section V for details†). After applying the van Vleck equation with an isotropic spin Hamiltonian, we obtained Jiso(GdIII–GdIII) = −0.231 ± 0.000 cm−1, g = 2.01 ± 0.00 (S = 7/2 and R2 = 1.000). Numerically, we used the PHI 2.1.6 program51 to fit the magnetic susceptibility. The fitting gave Jiso(Gd–Gd) = −0.23 cm−1 and g = 2.01, revealing the large coupling constants among the reported GdIII-only dimers.21,23,35,52 Therefore, the hydroxide-bridged structure is indeed a good candidate for transferring a strong magnetic exchange interaction.
Conclusions
In conclusion, we used the hydration method to introduce two hydroxide bridges between two low-CN DyIII ions to obtain 4f-only dimer SMMs, which exhibited a significant magnetic coercive field and high effective energy barrier. The intramolecular magnetic interaction was analyzed with the help of magnetic dilutions and GdIII analogues. Relatively large antiferromagnetic interaction constants among the Dy-only systems were observed due to the strong superexchange interactions transferred by the hydroxide bridges, even though the dipole–dipole interaction is ferromagnetic. The GdIII analogue also displays a large antiferromagnetic exchange coupling constant. This shows that the hydroxide-bridge is a good bridge ligand for transferring the exchange coupling interaction. The results of the ab initio calculation imply that a larger active space in the CASSCF calculation may be needed to understand the relaxation mechanism of such systems with short Dy–O bonds. In addition, the well-separated thermal relaxation and QTM regime, the high resonance temperature for slow relaxation and flexible chemical substitution sites make it a good model compound for studies on the magneto-structural relationship of 4f-only systems. Further work will focus on any other possible methods to obtain the energy levels precisely and will focus on the study of any substituted derivatives, in order to fully understand the key factors that are involved in enhancing the performance of the magnetic dynamics of SMMs.Acknowledgements
This work was supported by the NSFC (21290171, 21321001, 91422302 and 21571008) and the National Basic Research Program of China (2013CB933401). J. X. gratefully thanks Mr Cong Lin, Mr Jian Li and Prof. Jun-Liang Sun for assistance with the PXRD measurements, Ms Hai-Xia Rao, Mr Yong-Liang Zhang and Dr Wei Pan for help with the FT-IR measurements, and Ms Zhi-Xian Wang for the general assistance with EA measurements.Notes and references
- T. Komeda, H. Isshiki, J. Liu, Y. F. Zhang, N. Lorente, K. Katoh, B. K. Breedlove and M. Yamashita, Nat. Commun., 2011, 2, 217 CrossRef PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed.
- C. Schlegel, J. van Slageren, M. Manoli, E. K. Brechin and M. Dressel, Phys. Rev. Lett., 2008, 101, 147203 CrossRef CAS PubMed.
- D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clerac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 4926–4929 CrossRef CAS PubMed.
- G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- S. Gómez-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289–290, 379–392 CrossRef.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- M. Nakano and H. Oshio, Chem. Soc. Rev., 2011, 40, 3239–3248 RSC.
- N. Ishikawa, M. Sugita, T. Okubo, N. Tanaka, T. Iino and Y. Kaizu, Inorg. Chem., 2003, 42, 2440–2446 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS.
- L. Ungur and L. F. Chibotaru, Inorg. Chem., 2016 DOI:10.1021/acs.inorgchem.6b01353.
- J. Liu, Y. C. Chen, J. H. Jia, J. L. Liu, V. Vieru, L. Ungur, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- Y. C. Chen, J. L. Liu, L. Ungur, J. Liu, Q. W. Li, L. F. Wang, Z. P. Ni, L. F. Chibotaru, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed.
- N. Iwahara and L. F. Chibotaru, Phys. Rev. B, 2015, 91, 174438 CrossRef.
- H. Wang, K. Qian, K. Wang, Y. Bian, J. Jiang and S. Gao, Chem. Commun., 2011, 47, 9624–9626 RSC.
- K. Katoh, T. Kajiwara, M. Nakano, Y. Nakazawa, W. Wernsdorfer, N. Ishikawa, B. K. Breedlove and M. Yamashita, Chem.–Eur. J., 2011, 17, 117–122 CrossRef CAS PubMed.
- Y. Peng, V. Mereacre, A. Baniodeh, Y. Lan, M. Schlageter, G. E. Kostakis and A. K. Powell, Inorg. Chem., 2016, 55, 68–74 CrossRef CAS PubMed.
- X. Yi, K. Bernot, F. Pointillart, G. Poneti, G. Calvez, C. Daiguebonne, O. Guillou and R. Sessoli, Chem.–Eur. J., 2012, 18, 11379–11387 CrossRef CAS PubMed.
- Y. N. Guo, G. F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. Tang, H. J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948–11951 CrossRef CAS PubMed.
- J. Long, F. Habib, P. H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed.
- F. Tuna, C. A. Smith, M. Bodensteiner, L. Ungur, L. F. Chibotaru, E. J. McInnes, R. E. Winpenny, D. Collison and R. A. Layfield, Angew. Chem., Int. Ed., 2012, 51, 6976–6980 CrossRef CAS PubMed.
- J. J. Le Roy, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2014, 136, 8003–8010 CrossRef CAS PubMed.
- M. Gregson, N. F. Chilton, A.-M. Ariciu, F. Tuna, I. Crowe, W. Lewis, A. J. Blake, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and S. Liddle, Chem. Sci., 2016, 7, 155–165 RSC.
- S. D. Jiang, B. W. Wang, H. L. Sun, Z. M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730–4733 CrossRef CAS PubMed.
- S. D. Jiang, B. W. Wang, G. Su, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448–7451 CrossRef CAS PubMed.
- S. A. Cotton, C. R. Chim., 2005, 8, 129–145 CrossRef CAS.
- K. Dehnicke and A. Greiner, Angew. Chem., Int. Ed., 2003, 42, 1340–1354 CrossRef CAS PubMed.
- P. Zhang, J. Jung, L. Zhang, J. Tang and B. Le Guennic, Inorg. Chem., 2016, 55, 1905–1911 CrossRef CAS PubMed.
- B. Na, X. J. Zhang, W. Shi, Y. Q. Zhang, B. W. Wang, C. Gao, S. Gao and P. Cheng, Chem.–Eur. J., 2014, 20, 15975–15980 CrossRef CAS PubMed.
- D. N. Woodruff, F. Tuna, M. Bodensteiner, R. E. P. Winpenny and R. A. Layfield, Organometallics, 2013, 32, 1224–1229 CrossRef CAS.
- S. N. Konig, N. F. Chilton, C. Maichle-Mossmer, E. M. Pineda, T. Pugh, R. Anwander and R. A. Layfield, Dalton Trans., 2014, 43, 3035–3038 RSC.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed.
- R. J. Blagg, C. A. Muryn, E. J. McInnes, F. Tuna and R. E. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530–6533 CrossRef CAS PubMed.
- R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. McInnes, L. F. Chibotaru and R. E. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- T. J. Boyle, S. D. Bunge, P. G. Clem, J. Richardson, J. T. Dawley, L. A. Ottley, M. A. Rodriguez, B. A. Tuttle, G. R. Avilucea and R. G. Tissot, Inorg. Chem., 2005, 44, 1588–1600 CrossRef CAS PubMed.
- G. B. Deacon, G. D. Fallon, C. M. Forsyth, S. C. Harris, P. C. Junk, B. W. Skelton and A. H. White, Dalton Trans., 2006, 802–812, 10.1039/b511609k.
- H.-T. Sheng, H. Zhou, H.-D. Guo, H.-M. Sun, Y.-M. Yao, J.-F. Wang, Y. Zhang and Q. Shen, J. Organomet. Chem., 2007, 692, 1118–1124 CrossRef CAS.
- J. Cirera, E. Ruiz and S. Alvarez, Chem.–Eur. J., 2006, 12, 3162–3167 CrossRef CAS PubMed.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed.
- D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, J. Am. Chem. Soc., 2004, 126, 1755–1763 CrossRef CAS PubMed.
- G. Karlström, R. Lindh, P.-Å. Malmqvist, B. O. Roos, U. Ryde, V. Veryazov, P.-O. Widmark, M. Cossi, B. Schimmelpfennig, P. Neogrady and L. Seijo, Comput. Mater. Sci., 2003, 28, 222–239 CrossRef.
- L. Ungur, M. Thewissen, J. P. Costes, W. Wernsdorfer and L. F. Chibotaru, Inorg. Chem., 2013, 52, 6328–6337 CrossRef CAS PubMed.
- D. Aravena and E. Ruiz, Inorg. Chem., 2013, 52, 13770–13778 CrossRef CAS PubMed.
- M. E. Lines, J. Chem. Phys., 1971, 55, 2977 CrossRef CAS.
- L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126–4129 CrossRef CAS PubMed.
- L. F. Chibotaru, L. Ungur, C. Aronica, H. Elmoll, G. Pilet and D. Luneau, J. Am. Chem. Soc., 2008, 130, 12445–12455 CrossRef CAS PubMed.
- L. Ungur, W. Van den Heuvel and L. F. Chibotaru, New J. Chem., 2009, 33, 1224 RSC.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- F. Pointillart, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Chem.–Eur. J., 2011, 17, 10397–10404 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, structure, magnetism, ab initio calculation, magnetic interaction analysis, Fig. S1–S50, Table S1–S10, the crystal information files of compounds 1, 2 and 4. CCDC 1418657 for 1, 1418658 for 2 and 1506174 for 4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03621j |
| This journal is © The Royal Society of Chemistry 2017 |