
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmonic labeling of subcellular compartments in cancer cells: multiplexing with fine-tuned gold and silver nanoshells†
R. G.
Sobral-Filho
a,
A. M.
Brito-Silva
a,
M.
Isabelle‡
b,
A.
Jirasek
c,
J. J.
Lum
bd and
A. G.
Brolo
 *a
*a
aDepartment of Chemistry, University of Victoria, 3800 Finnerty Road, Victoria BC V8P 5C2, Canada. E-mail: agbrolo@uvic.ca
bBritish Columbia Cancer Agency – Vancouver Island Centre, Trev and Joyce Deeley Research Centre, 2410 Lee Ave., Victoria, BC V8R 6V5, Canada
cDepartment of Mathematics, Statistics, Physics and Computer Science, University of British Columbia Okanagan, 3187 University Way, Kelowna BC V1V 1V7, Canada
dDepartment of Biochemistry and Microbiology, University of Victoria, 3800 Finnerty Road, Victoria BC V8P 5C2, Canada
First published on 3rd February 2017
Abstract
Fine-tuned gold and silver nanoshells were produced via an entirely reformulated synthesis. The new method yielded ultramonodisperse samples, with polydispersity indexes (PI) as low as 0.02 and narrow extinction bands suited for multiplex analysis. A library of nanoshell samples with localized surface plasmon resonances (LSPR) spanning across the visible range was synthesized. Hyperspectral analysis revealed that the average scattering spectrum of 100 nanoshells matched closely to the spectrum of a single nanoshell, indicating an unprecedented low level of nanoparticle-to-nanoparticle variation for this type of system. A cell labeling experiment, targeting different subcellular compartments in MCF-7 human breast cancer cells, demonstrated that these monodisperse nanoparticles can be used as a multiplex platform for single cell analysis at the intracellular and extracellular level. Antibody-coated gold nanoshells targeted the plasma membrane, while silver nanoshells coated with a nuclear localization signal (NLS) targeted the nuclear membrane. A fluorescence counterstaining experiment, as well as single cell hyperspectral microscopy showed the excellent selectivity and specificity of each type of nanoparticle for its designed subcellular compartment. A time-lapse photodegradation experiment confirmed the enhanced stability of the nanoshells over fluorescent labeling and their capabilities for long-term live cell imaging.
Introduction
A variety of nanomaterials with diverse chemical and physical properties have emerged in the last two decades, bringing great promise for applications in environmental, energy and health sciences.1–3 Metallic nanoshells appear prominent among those. Composed of a dielectric core and a thin metallic shell, they hold great promise for bioapplications.4 These nanoparticles are strong scatterers, enabling efficient spectroscopic responses. Their LSPR bands can be tuned throughout visible and near infrared (NIR) wavelengths by simply controlling their core size and shell thickness.4 However, despite this spectral tunability, efficient plasmonic multiplexing experiments based on nanoshells have not yet been outlined. The problem of multiplexing with nanoshells can be attributed to the difficulties in obtaining monodisperse colloids on a multi-step fabrication process. One potential application of metallic nanoparticles that requires multiplexing is in the field of cellular imaging.Recent advances have been made in intracellular probing by LSPR sensors based on nanoparticles.5,6 However, such sensors are very susceptible to changes in the nanoparticles characteristics, and their widespread use requires the development of uniform and well characterized colloids.7,8 Significant efforts have revealed progress in understanding how nanoparticles are internalized by cells and some of the physical and biochemical interactions that lead to their intracellular accumulation.9 However, the use of nanoparticles as labelling and tracking agents for intracellular environments still finds itself in the early stages. Developments in this area have the potential to greatly improve our understanding of cellular responses.
Considering the complexity of biological systems and cellular microenvironments, an ideal labelling experiment should allow simultaneous evaluation of several components in the sub-micrometer range (multiplexing). Immunophenotyping of the plasma membrane is the gold standard for distinguishing between cell types and is essential as a diagnostic tool.10 Changes in protein expression, enzymatic activity, nucleic acid production and many other processes take place intracellularly and are concomitant in a cell. This scenario demands the use of an efficient multiplex approach that can probe specific subcellular compartments (membranes, organelles, etc.) and components (receptors and other biomolecules). To that end, most systems investigated to date, are fluorescence-based.11–13 Such cases, while extremely effective for detection, lack the stability required for long-term real-time analysis of cellular processes that happen in the range of several hours, and do not take place in stationary positions inside the cell. Plasmonic-based approaches have recently been demonstrated for intracellular probing. Gold nanorods have been 3-D imaged inside osteosarcoma cells through two-photon luminescence with precision regarding their spatial orientation,14 and surface-enhanced Raman scattering measurements have been made probing the endosomal environment through the use of magnetite–metal composite nanoparticles.15 In both cases the nanoparticles were internalized through nonspecific interactions with the cells.
Metallic nanoshells have fallen short for this type of application due to the broad extinction bands associated with the size disparity of the colloids. Although valiant efforts have been made to incorporate such variations into models to predict the extinction profile of nanoshells suspensions.16,17 Nanoshell fabrication methods comprise a combination of multiple surface chemistry processes and colloids – each of these carrying their own potential barriers to a uniform size distribution. Such limitations have resulted in spectral overlaps that derail the effectiveness of metallic nanoshells as a multiplex platform.18 In a previous work performed by our group, the characterization checkpoints and techniques to track the progress of the nanoshells fabrication process were presented, indicating improvements which led to a faster synthesis and control over the degree of metal coverage in a single core size.19 However, that did not lead to the desired narrow extinction bands for the obtained colloids. An extensive review of the literature and several discussions within our lab, led to the decision to tackle the size distribution problem by making dramatic changes to our previous protocols and reformulating the nanoshell synthesis to a process that would result in ultramonodisperse colloids with narrow extinction bands, enabling their use for multiplexing.
Here, single cell labelling experiments based on the scattering profiles of finely tuned gold and silver nanoshells are reported. Precise control over the two parameters that define the optical properties of these particles – core size and shell thickness – was achieved. A refined streamlined synthesis, realized using only simple labware (see Fig. SI-1†), generated colloids with unprecedented quality. Highly monodisperse samples were produced, resulting in narrow extinction bands that can, in fact, be used for multiplex analysis at the single cell-single particle level. We designed nanoshell colloids that can operate in the visible range. In principle, dark field scattering images of cells labelled with colour-tuned nanoshells can be realized under white light illumination. This is a simple hardware implementation that can be easily achieved in clinical conditions. These nanoshells also show much better photostability when compared with typical fluorescence labels in cellular imaging experiments.
Results and discussion
Synthesis and characterization
The core size is one of the main factors for tuning the LSPR in a nanoshell. Silica (SiO2) nanoparticles were used as cores. By controlling the amount of tetraethyl orthosilicate (TEOS) and reaction times, colloids with four different sizes were prepared (50, 63, 72 and 80 nm). These diameters were specifically chosen to produce nanoshells whose LSPR would operate in the visible range. A reverse microemulsion system (described below – detailed protocol available as ESI†) allowed a one-batch synthesis and functionalization of the SiO2 cores. Scheme 1 presents the synthetic process outlined here.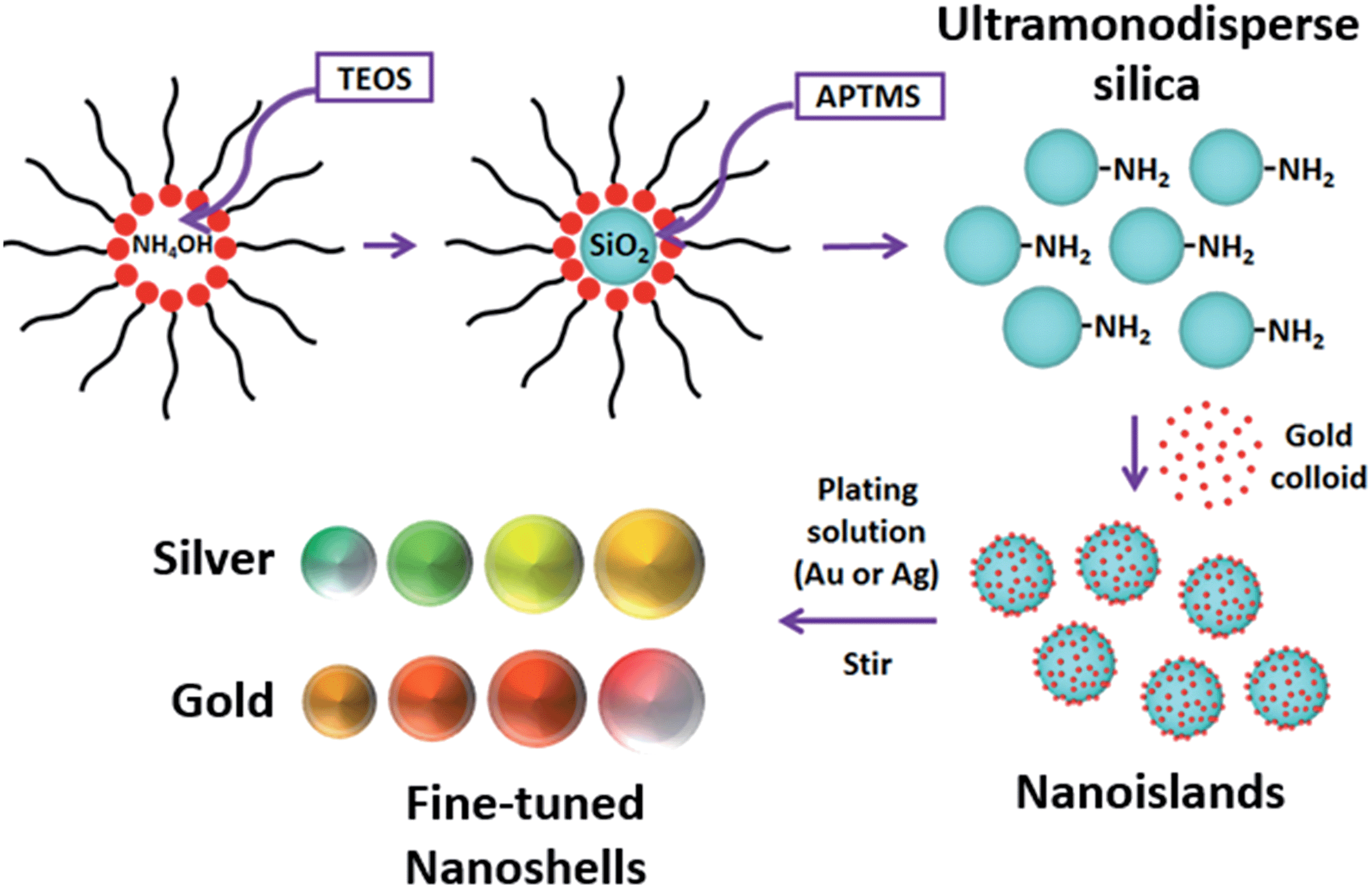 | ||
| Scheme 1 Ultramonodisperse aminated silica particles are produced in a one-batch synthesis via a reverse microemulsion system. Small gold nanoparticles are then attached to the silica and the shell growth takes place under stirring in a plating solution with metal ions at low concentration (150 μM). Different SiO2 sizes and shell thicknesses can be achieved with this method. | ||
Ultramonodisperse samples were obtained with standard deviations ranging from 2.3–2.8 nm and polydispersity indexes (PI) as low as 0.02. These values are one order of magnitude lower than the accepted threshold of PI = 0.2 for monodisperse samples.20 The TEM images in Fig. 1a and b convey the general characteristics of the colloids showing the uniform size and shape distributions of the SiO2 samples. Triton X-100 and n-hexanol were used as surfactants in cyclohexane. Aqueous ammonium hydroxide (NH4OH) was contained to the inner portion of the micelles. TEOS is added to the organic phase and the process of nucleation and growth in the aqueous phase is therefore limited by the TEOS diffusion through the supramolecular structure of the micelles. This grants a slow and controlled growth of the SiO2 particles. External nucleation in the organic phase was prevented by sequential additions of TEOS at every six hours, so that the concentration of the precursor in that phase was kept relatively low throughout the process. For the sizes evaluated, there was no disruption of the reverse microemulsion, even after long reaction times, indicating that the bending rigidity of the micelles21 may also contribute to the high degree of monodispersity attained here.
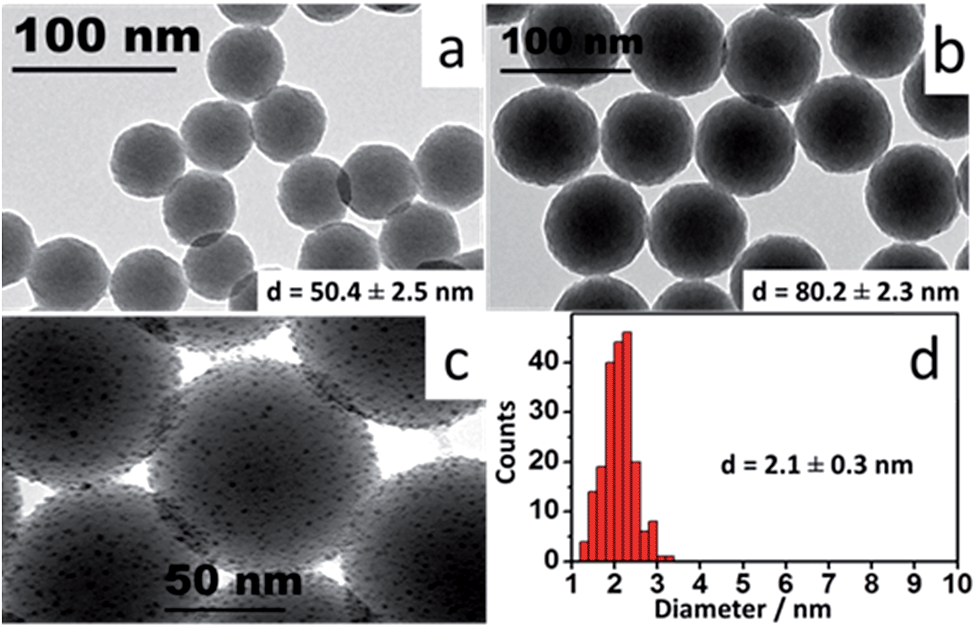 | ||
| Fig. 1 TEM images: (a and b) SiO2 nanoparticles synthesized by reverse microemulsion; (c) nanoislands at high magnification; (d) Au colloid diameter histogram. | ||
After the growth of the SiO2 particles was completed, the pH of the reaction was lowered by simply opening the vial and allowing NH3 to escape. Functionalization of the SiO2 cores was carried out with the addition of an aminosilane to the mixture for 5 minutes before breaking the micelles with ethanol. It is worth mentioning that no complex separation method was required for these samples. Special attention, however, needs to be given to the cleaning process, since several reports in the literature point out that colloids produced by a reverse microemulsion will often generate aggregated samples.22 That issue was solved through a rigorous cleaning process, where the samples were centrifuged, resuspended in ethanol, and kept under vigorous sonication for 15 minutes before each cleaning cycle. Post-cleaning dynamic light scattering (DLS) measurements show that there is no aggregation in the colloids (Fig. SI-2†). These samples can be kept in ethanol at room temperature for several months. Long term stability was assessed by TEM using a 10 month old aminated silica sample. Fig. SI-3† displays the results that show the good colloidal and chemical stability of the samples produced through our method. A variety of SiO2 sizes can be fabricated by controlling reaction parameters such as time and reagent amounts. Table SI-1† shows in detail the parameters utilized for the fabrication of the diameters used in this work.
Coating the functionalized cores with a metallic shell requires the adsorption of small gold particles on the surface of the SiO2. These nanoparticles (2.1 ± 0.3 nm) were prepared as previously described,23 resulting in a colloid with ∼1.36 × 1015 particles per mL, as determined by ICP-MS. A method to calculate the number of particles in the silica samples is outlined in the ESI file.† For the SiO2 samples prepared here, particle concentrations range from ∼2.4–4.1 × 1012 particles per mL. The two colloids were mixed in a small vial at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49 (SiO2:Au) volume ratio and continuously stirred at room temperature for 3 hours in a stirring plate. The small Au particles bind to the amine groups on the silica surface to an estimated coverage of 30%, due to electrostatic repulsion between bound and free gold nanoparticles.24 These composite particles (gold-decorated silica nanoparticles) are referred to as ‘nanoislands’. The sample was cleaned by centrifugation–resuspension cycles in ultrapure water and had its volume reduced to the initial amount of aminated SiO2 colloid. Fig. 1c shows high magnification TEM images of the resulting nanoislands. A narrow size dispersion is important throughout the entire synthesis of the nanoshells, and a diameter histogram for the Au colloid is also shown in Fig. 1d. The uniform size distribution of the Au colloid will determine the homogeneous formation of the metallic shells in the next step of the synthesis.
:49 (SiO2:Au) volume ratio and continuously stirred at room temperature for 3 hours in a stirring plate. The small Au particles bind to the amine groups on the silica surface to an estimated coverage of 30%, due to electrostatic repulsion between bound and free gold nanoparticles.24 These composite particles (gold-decorated silica nanoparticles) are referred to as ‘nanoislands’. The sample was cleaned by centrifugation–resuspension cycles in ultrapure water and had its volume reduced to the initial amount of aminated SiO2 colloid. Fig. 1c shows high magnification TEM images of the resulting nanoislands. A narrow size dispersion is important throughout the entire synthesis of the nanoshells, and a diameter histogram for the Au colloid is also shown in Fig. 1d. The uniform size distribution of the Au colloid will determine the homogeneous formation of the metallic shells in the next step of the synthesis.
Shell growth is the final step of the synthesis and is normally accomplished by adding the nanoislands to a solution containing the ionic precursors of the chosen metal. In the case of Au, a solution of gold hydroxide (Au(OH)3), referred as K-gold,25 is typically prepared. This solution requires aging times on the order of 12–24 h, and is very sensitive to the initial pH of the water. Adjusting its final pH with acids or bases results in the addition of ions to the reaction media that can affect the stability of the colloids and promote aggregation during the growth process. We therefore decided to produce either gold or silver hydroxide (AgOH) plating solutions using a different approach, in order to standardize the coating process for these metals. HAuCl4 (or AgNO3) are dissolved in ultrapure water at low concentrations, in the micromolar range, and ammonium hydroxide is added to the solution in order to generate the designated hydroxide. The NH4OH also sets the pH to an optimal range (9–10) for the shell growth.26 The resulting solution is transferred to a clean vial through a syringe filter. This step will retain impurities and small gold or silver oxide particles that might form in the previous step. The appropriate amount of nanoislands is then added to the plating solution. The formation of the shells proceeded through reduction of Au (or Ag) onto the nanoislands under vigorous stirring by adding ascorbic acid (C6H8O6) (for Au) or formaldehyde (CH2O) (for Ag). The resulting colloid was left undisturbed and the reaction was monitored by measuring the LSPR spectrum, as showed in our previous work.19
The aspect of the colloids evolves with time over the course of 60–90 minutes. Even though the coating process can be stopped at any time by simply cleaning the sample, shell thickness was controlled by the amount of added nanoislands – lower amounts of nanoislands lead to thicker shells and vice versa. It is easy to estimate the number of particles per sample through a simple calculation as shown in the ESI file.† The nanoshell samples were finally cleaned by centrifugation–resuspension in ultrapure water and stored at 10 °C. A combination of Au and Ag nanoshells of varying thicknesses and SiO2 diameters were produced. Fig. 2 illustrates the whole LSPR spectral dataset for the nanoshell samples and highlights the narrow extinction bands for all colloids showed in Fig. 2a. These colloids have extinction maxima ranging from 550–685 nm. The wavelength range can be expanded simply by choosing the appropriate core size and shell thickness. It is clear from Fig. 2 that the narrow LSPR characteristics of these nanoshells are suitable for multiplexing applications based solely on their spectral profiles. This attests to the exceptional high quality of the colloids prepared by the procedure reported here. Low and high magnification TEM images, displayed in Fig. SI-4,† show the good quality of the gold (or silver) coatings for these nanoshells.
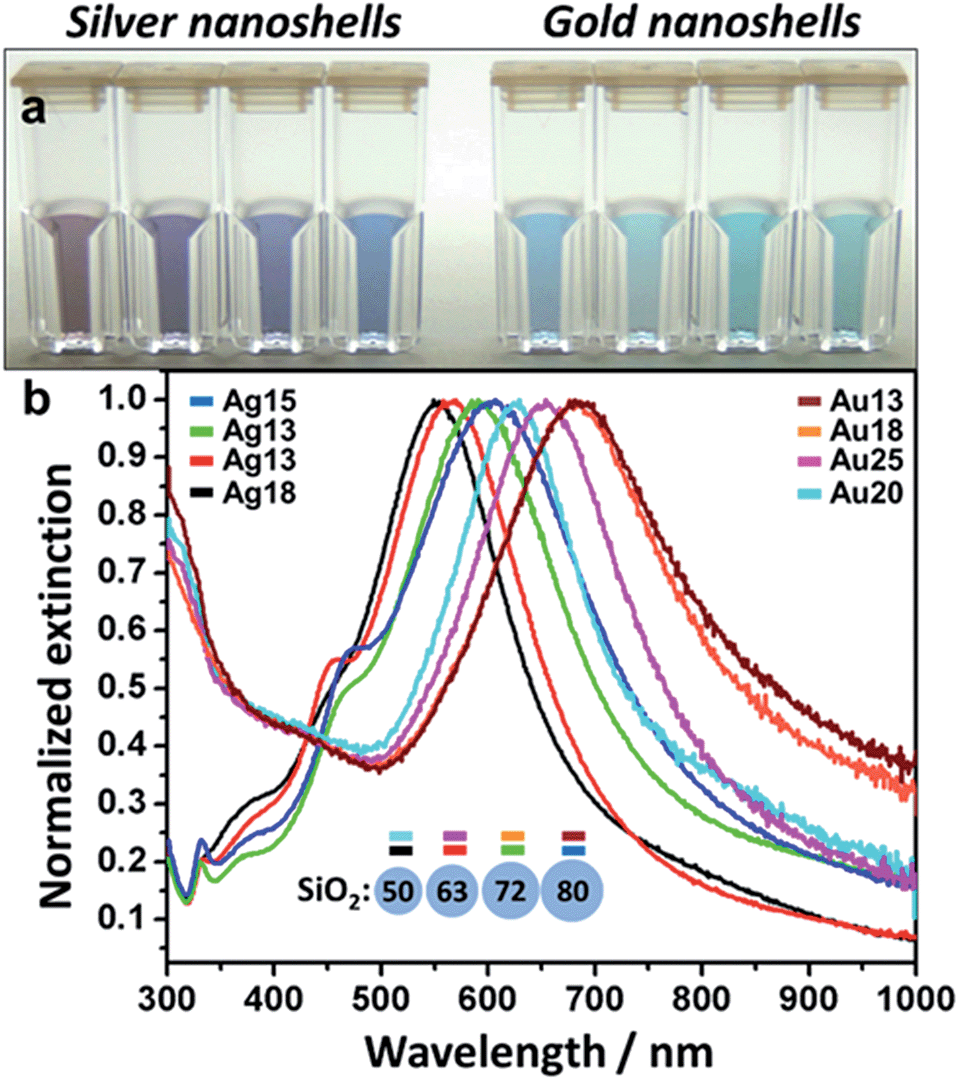 | ||
| Fig. 2 (a) Fine-tuned Ag and Au nanoshells samples – cuvettes match measured spectra in 3b (left to right); (b) extinction spectra for Ag and Au nanoshells – core size and shell thickness are color-coded to the curves (dimensions in nm). | ||
Hyperspectral analysis
It is clear the distinction between the samples displayed in Fig. 2, however, in order to assess the feasibility of effectively using these particles for multiplexing, a more in-depth analysis is required. To that end we performed hyperspectral dark field measurements for several of the nanoshells produced here. Fig. SI-5† displays a hyperspectral image and the scattering spectra for isolated particles from a mixture of 4 nanoshell colloids immobilized onto an aminated glass coverslip (coverslip silanization described in detail in our previous work19). The color distinction and good spectral separation between particles show the potential these colloids hold for multiplex applications. Based on that, two samples, Ag (18 nm)@SiO2 (50 nm) and Au (18 nm)@SiO2 (72 nm), were selected for the single-cell proof of principle experiment. Fig. SI-6† illustrates the dramatic color separation between these particles, indicating the feasibility of achieving similar results even with the use of a regular dark field microscope (without hyperspectral capabilities). An in-depth assessment of Ag (18 nm)@SiO2 (50 nm) and Au (18 nm)@SiO2 (72 nm) was carried out by hyperspectral analysis. The results are presented in Fig. 3, where a comparison between the measured scattering spectra for a single nanoshell and that of 100 averaged particles was performed for each of these two colloids.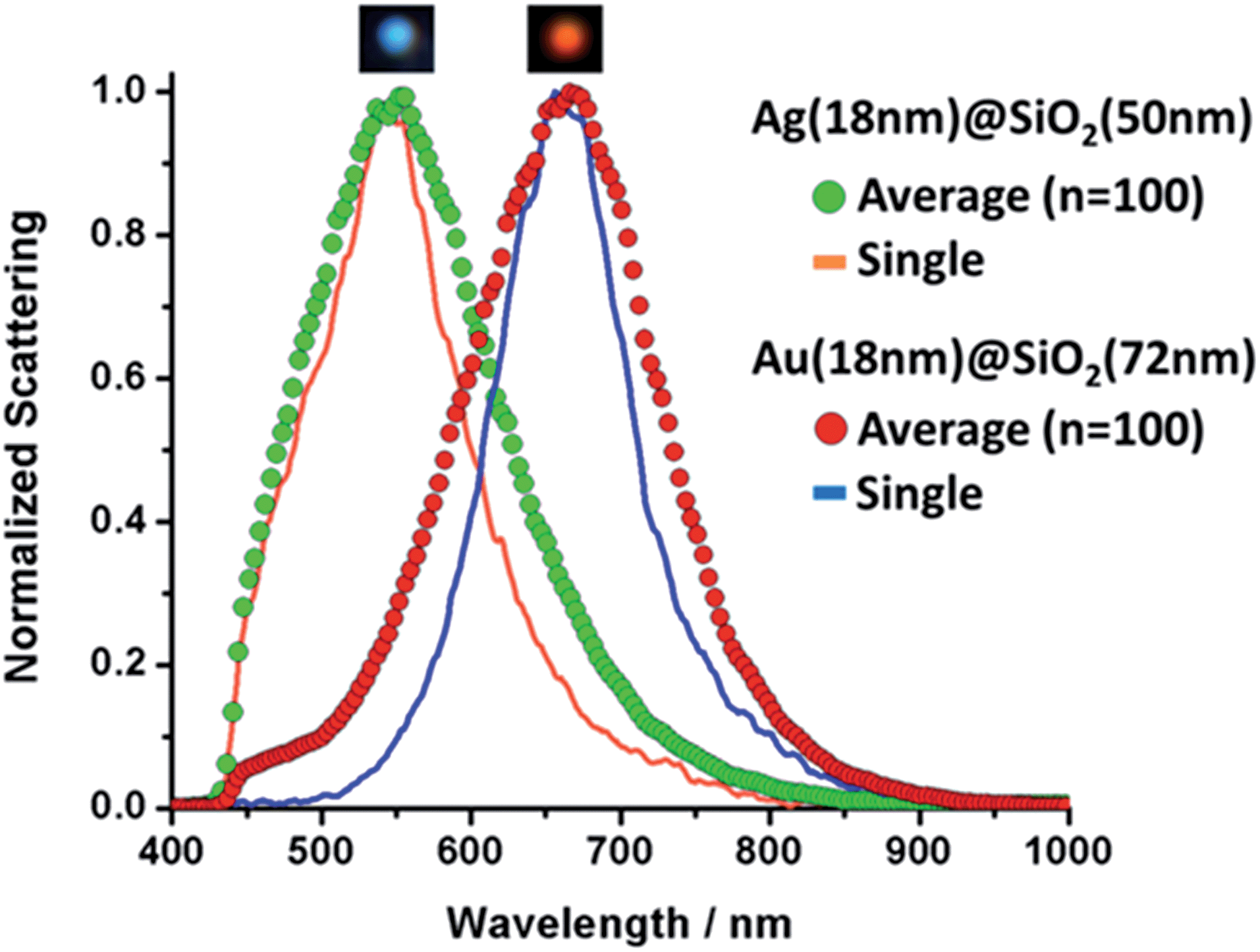 | ||
| Fig. 3 Single and averaged (n = 100) scattering spectra for Ag and Au nanoshells. Inserts above each curve show individual nanoshells as seen under dark field illumination. | ||
Hyperspectral measurements, shown in Fig. 3, were acquired for different particles from each sample. Individual amine-modified glass slides were placed in each of the colloids in order to capture the particles for analysis. Line plots correspond to a single nanoshell and scatter plots show the averaged spectrum. Insets in Fig. 3 show the particles as observed under white light illumination in the dark field configuration. The agreement between the LSPR peak positions is excellent in both cases, and the slightly broader average spectra (compared to that of a single nanoshell) is an indication of the optimal size dispersion of the samples. Fig. 3 indicates the effective level of control over the LSPR bands of metallic nanoshells achieved through the wet-chemistry processes adopted here. We attribute this to the high monodispersity of the colloids produced through this method. Fig. SI-7† shows the individual normalized extinctions for each of the 100 LSPR spectra plotted on the same graph for both samples. Fig. SI-7† provides an indication of the particle-to-particle variations.
Labeling of subcellular compartments
Protein expression patterns on the plasma membrane can provide valuable information about cellular processes such as proliferation, apoptosis and senescence.27 Such patterns often carry prognostic and predictive value in medical diseases and conditions, particularly those related with cancer biology. Insulin-like growth factor receptor (IGFR) expression has recently been associated with increased invasiveness, anti-estrogen resistance and induced epithelial-to-mesenchymal transition in breast cancer cells.28,29 These factors can contribute to increased tumor aggressiveness and reduced sensitivity to chemotherapy, in addition to intensifying the incidence of metastasis and remittance. Moreover, many metabolic processes related to carcinogenesis take place in subcellular compartments and the importance of using nanoparticles to target intracellular processes and compartments recently been outlined.30–33One of the major problems in intracellular and membrane-probing with nanoparticles is to avoid nonspecific interactions between the particles and the cell. Triggering nonspecific endocytic pathways and having particles nonspecifically adsorbing to the membrane are examples of phenomena that can lead to misinterpretation. Endocytosis can happen in a variety of ways and that poses a crucial challenge in intracellular labelling.22 Nonspecific pathways can allow internalized nanoparticles to be allocated to subcellular compartments (e.g. endoplasmic reticulum, Golgi complex).34 In order to demonstrate the feasibility of using nanoshells as a multiplex platform for cell analysis in both domains (intra and extracellular), the nanoparticle surface was functionalized with either a NLS (nuclear localization signal) peptide or an anti-IGFR antibody. The nanoshells modified with NLS peptides are expected to be internalized and localized to the nuclear membrane. NLS are peptides capable of selectively transporting cargoes into the nucleus;35 therefore, they should promote entering through the nuclear pores. However, the nanoparticles used in this work are large enough to preclude nuclear internalization leading to accumulation on the nuclear membrane. The anti-IGFR antibody targets the plasma membrane. It is important to clarify that the use of NLS peptides does not necessarily mean nuclear transport.36 Different cell lines may require different NLS peptides and for applications involving multiple cell lines, a screening step is necessary. The NLS used in this work was specifically screened for MCF-7 cells (see ESI† file for peptide sequence).37 Simpler than that, only cells expressing IGF receptors should be targeted by the Au-IGFR nanoshells, and a negative control was established using SKBR-3 breast cancer cells that express only low basal levels of IGF receptors. With this, we aim not only to demonstrate the capacity to target distinct subcellular compartments, but also (in the case of IGF receptors) the ability to target specific biochemical elements within such compartments. Scheme 2 summarizes and pinpoints the main features of the experiment.
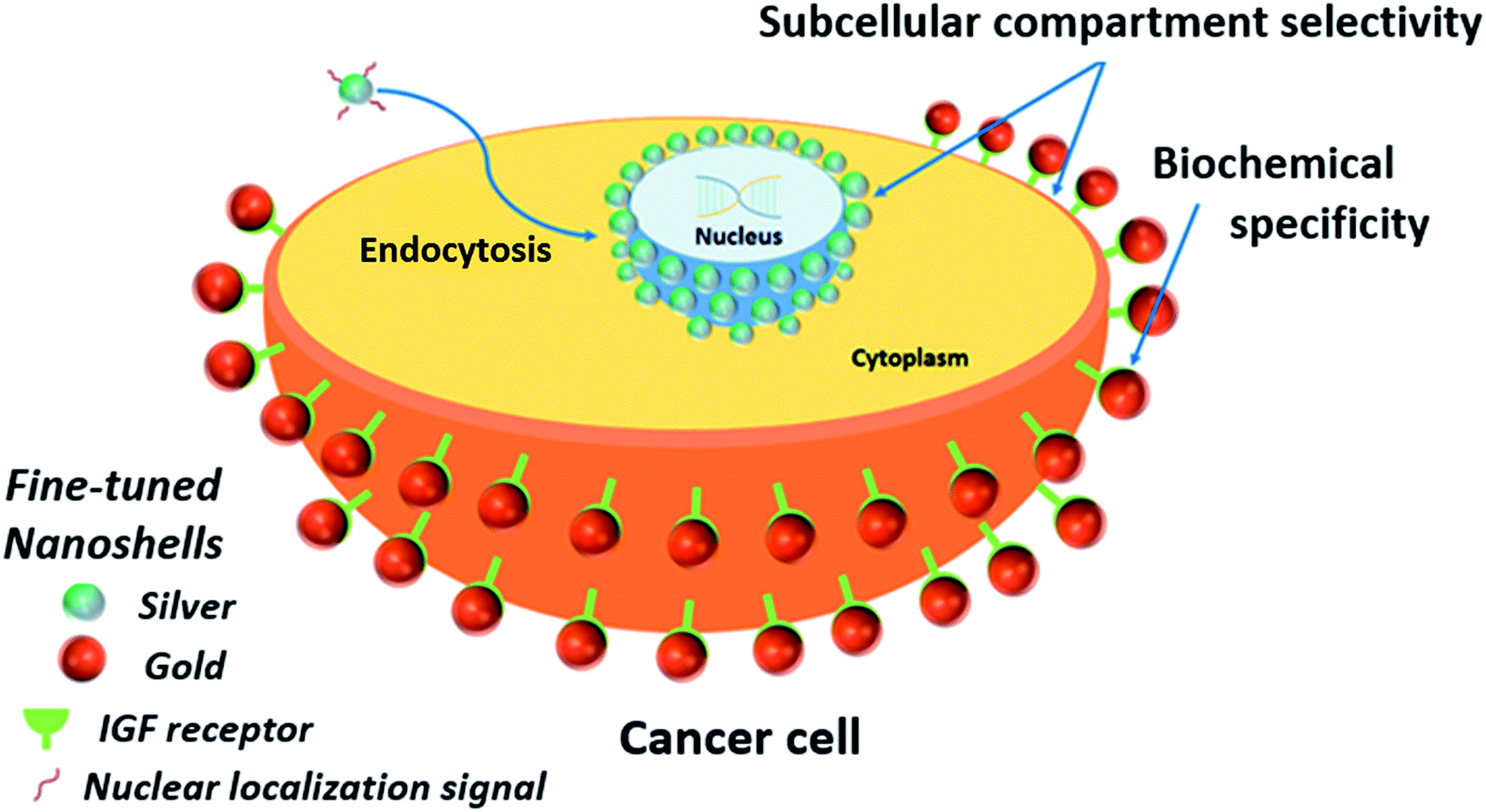 | ||
| Scheme 2 Labeling of subcellular compartments by ultramonodisperse nanoshells. Silver nanoshells bioconjugated to a nuclear localization signal (NLS) are internalized by the cells. The NLS peptide leads the particles to escape nonspecific endosomal and exocytic pathways and accumulate on the nuclear membrane. Gold nanoshells bioconjugated to an anti-IGFR antibody, target the insulin receptors (IGFR) localized on the plasma membrane of MCF-7 cells. Selectivity of the nanoshells for each subcellular compartment is evidenced, as well as the ability to target specific biochemical elements within such compartments, as showed for the insulin receptors and the antibody-coated gold nanoshells. | ||
Ag (18 nm)@SiO2 (50 nm) and Au (18 nm)@SiO2 (72 nm) were selected for the proof-of-principle cell labeling experiment, because of the good separation in their LSPR characteristics (as shown in Fig. 2). The Ag nanoshells were coated with the custom-made NLS (Ag-NLS) peptide while Au nanoshells were coated with the anti-IGFR antibody (Au-IGFR). Ag-NLS was resuspended in reduced-serum medium (opti-MEM®) and Au-IGFR was resuspended in PBS prior to incubation. Extinction spectra before and after bioconjugation are shown in Fig. SI-8,† where a shift in the LSPR peaks is evidenced due to the attachment of the biomolecules. Particle concentration for both samples was approximately 5 × 109 particles per milliliter. MCF-7 cells were plated onto sterile coverslips to a confluency of 30% and maintained in Dulbecco's modified eagles medium (DMEM) supplemented with 10% fetal bovine serum.
Nanoshells localization – combined microscopy of individual compartments
Individual incubations were performed with 1 mL of Au-IGFR for 30 minutes and 1 mL of Ag-NLS for 3 hours at 37 °C. Cells were washed with PBS after incubation. Staining and fixation protocols are described in the ESI file.† Coverslips were washed in PBS before being mounted onto microscopy slides for analysis. In order to unequivocally confirm the cellular localization of the nanoshells, an experiment combining fluorescence, conventional dark field and hyperspectral dark field microscopy was performed. The same field of view was analyzed by all three techniques. Counterstaining with fluorescent dyes was performed for each compartment (procedures in the ESI file†). Propidium iodide (λex = 535 nm, λem = 617 nm) was used for staining the nucleus and Palloidin CruzFluor® 488 (λex = 493 nm, λem = 517 nm) was used for the F-actin filaments on the plasma membrane. All images showed in this work were acquired in a Cytoviva Dual-Mode Fluorescence/Hyperspectral Dark Field microscope, allowing for easy interchangeable imaging modes for same-field analysis. Fluorescence filters were removed prior to dark field image acquisition. Fig. 4 displays the obtained results.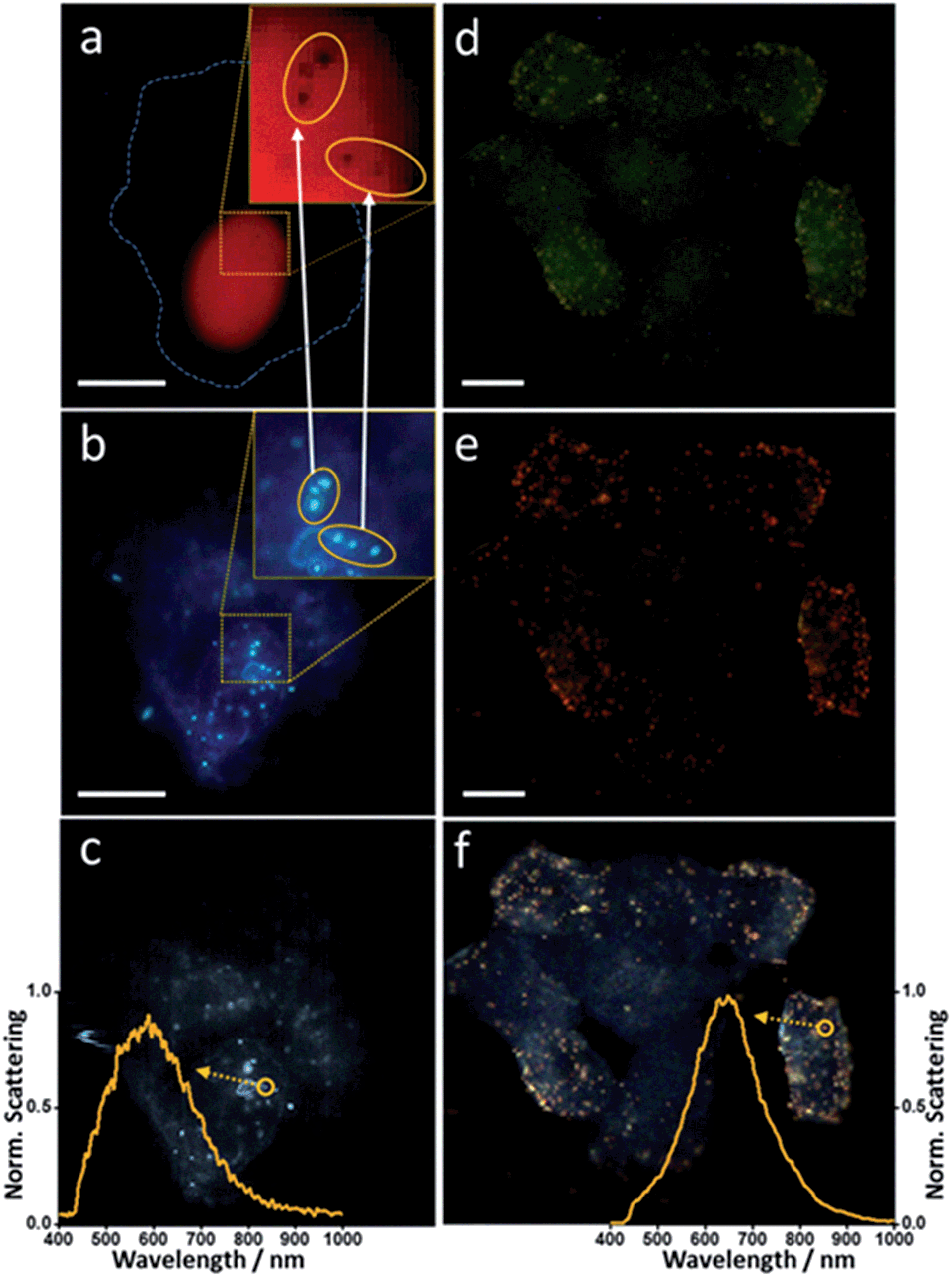 | ||
| Fig. 4 Combined microscopical (fluorescence, dark field and hyperspectral) analysis of nanoshell-labeling. (a) and (d) – fluorescence microscopy, (b) and (e) – conventional white light dark field microscopy, (c) and (f) – hyperspectral dark field microscopy. Inserts in (a) and (b) show magnified regions of interest on the nuclear membrane. Inserts in (c) and (f) evidence the scattering spectra of single silver (c) and gold (f) nanoshells. Cell membrane is outlined by dashed line in (a). Scale bar 10 μm. Measurement details in the ESI file.† | ||
The two inserts in Fig. 4a and b are magnifications of the same region of interest. Fig. 4a shows fluorescence imaging of the nuclear staining by propidium iodide. Fig. 4b is a dark field image of the same field. It is possible to notice the presence of dark spots on the magnified insert in Fig. 4a. The position of these spots overlaps with the position of the silver nanoshells shown on the insert in the dark field image (Fig. 4b). Fluorescence in these particular regions has been quenched due to the presence of the silver nanoshells, corroborating the localization of the nanoshells on the nuclear membrane. Two groups of silver nanoshells are evidenced in the inserts. Arrows indicate the precise match between the relative position of the nanoshells and the dark spots. Fig. 4c shows the hyperspectral image of the same field, where the scattering spectrum of a single nanoshell from the same group of particles is highlighted.
In Fig. 4d–f the counterstaining marks the inner portion of the plasma membrane. Focal depth was kept constant throughout all three measurements as to evidence the cell boundaries, while still showing the top portions of the plasma membranes. Notice that the nuclei cannot be visualized from this focal plane. The gold nanoshells can be easily seen in all three images. If compared to Fig. 4e and f, they are less noticeable in Fig. 4d, due to their much weaker ability to scatter light in this region of the spectrum, however, their visualization in this fluorescence channel is still possible due to their bigger size (diameter ∼ 108 nm). They are much more evident in Fig. 4e and f, however, and the perfect match in their relative positions becomes evident if compared among all three images. Similar to what is showed in Fig. 4c, the scattering spectrum for a single gold nanoshell is also highlighted in Fig. 4f. This shows that with these monodispersed nanoshells, not only we are capable of selectively labeling specific subcellular compartments at the single cell level, but, with the aid of hyperspectral microscopy, also have the ability to track and select single particles within those compartments.
Multiple compartments at the single cell-single particle level
Fig. 5 shows the selectivity of each sample for the desired subcellular compartment under conventional dark field illumination.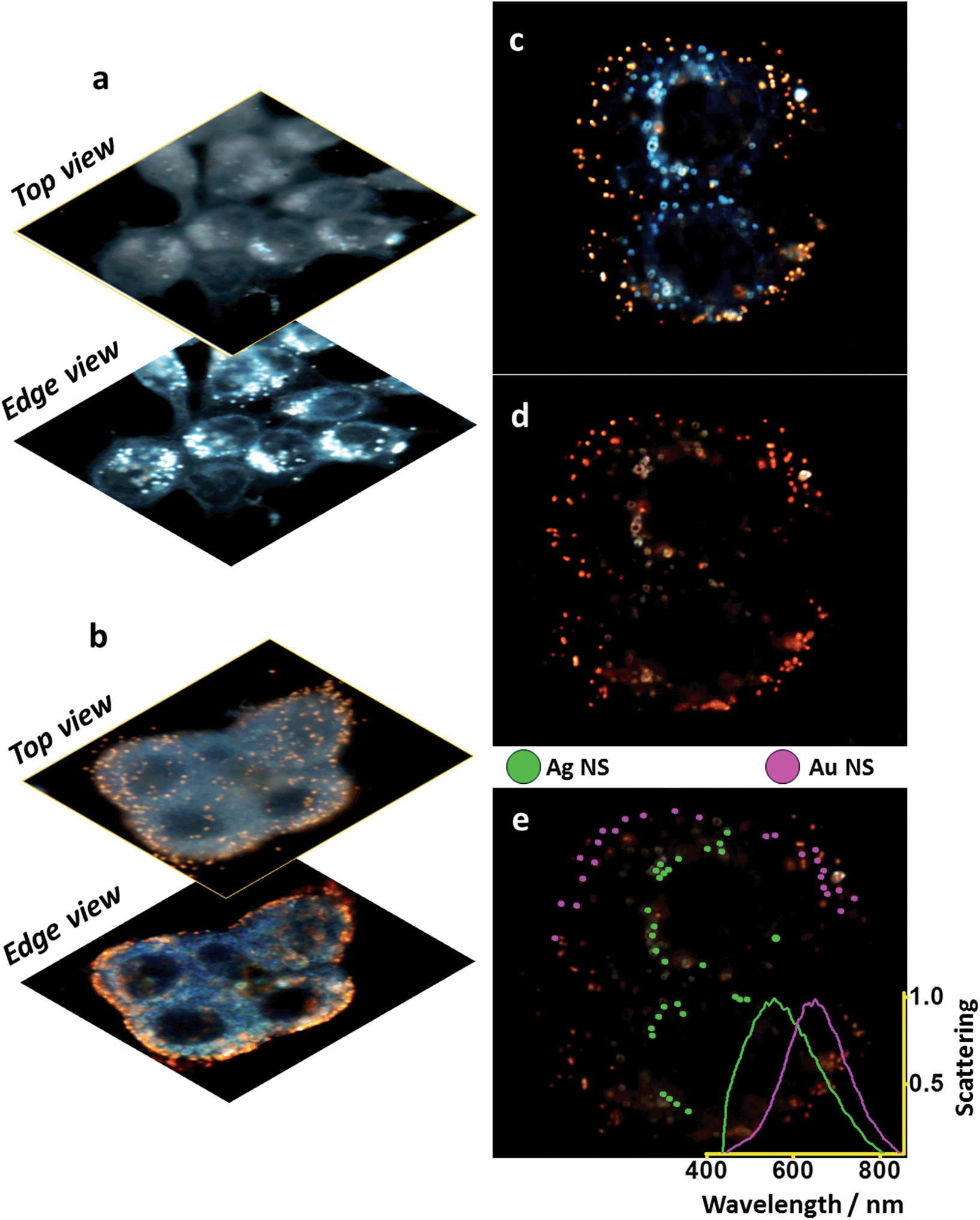 | ||
| Fig. 5 Multiplex-imaging different MCF-7 cell compartments with Ag and Au nanoshells. Different focal depths for: (a) Ag-NLS – targeting the nuclear membrane (b) Au-IGFR – targeting the plasma membrane; same field: (c) regular dark field image, (d) hyperspectral dark field image, (e) hyperspectral sorting between Ag and Au nanoshells – Ag (18 nm)@SiO2 (50 nm) labeling the nuclear membrane and Au (18 nm)@SiO2 (72 nm) labeling IGF receptors on the plasma membrane, insert shows averaged scattering spectra for the selected nanoshells. | ||
Different focal depths (top and edge) show that (Fig. 5a) Ag-NLSs accumulate in the nuclear membrane (edge view) and no particles are seen on the plasma membrane (top view). In the case of Au-IGFR (Fig. 5b) the nanoshells specifically target the plasma membrane and can be visualized in both top – homogeneously distributed over the cells, and edge view. Negative controls without the presence of a NLS peptide (for Ag nanoshells), and on IGFR-negative SKBR-3 cells (for Au nanoshells) are displayed in Fig. SI-9.† Ag50 without NLS do not accumulate on the nuclear membrane. SKBR-3 cells present low expression levels of IGFR and the sparse number of gold nanoshells seen in Fig. SI-9† confirms the specificity of Au-IGFR for the insulin receptors on the MCF-7 cells. That also shows that nonspecific binding does not pose a problem.
It is clear from Fig. 5a, the efficient internalization of Ag-NLS by the cells. Their overall absence from the cell membranes indicate that the Ag-NLS particles that came in contact with the cell membrane were internalized. At higher magnification (Fig. SI-9†) the nanoshells are seen inside the cell surrounding the nucleus, suggesting that they have not accumulated in other subcellular compartments such as the endoplasmic reticulum and the Golgi complex. In Fig. 5b, Au-IGFR nanoshells accumulate only on the plasma membrane and, as expected, no internalization is seen. These results attest to the efficacy of the bioconjugation process.
It is important to emphasize that sequential incubations were performed with 1 mL of Au-IGFR for 30 minutes, PBS washings followed by 1 mL of Ag-NLS for 3 hours. Cells were then washed with PBS and fixed with 4% paraformaldehyde before being mounted onto microscope slides. Conventional (Fig. 5c) and hyperspectral (Fig. 5d) dark field images of MCF-7 cells labelled with gold and silver nanoshells are displayed. Both images were acquired from the same field of view. The image in Fig. 5c was obtained under white light illumination with a regular CCD camera (no spectral resolution). Fig. 5d was acquired with a spectrograph-coupled CCD camera for the hyperspectral image (microscope setup in ESI file†). From Fig. 5c, the distinction between the gold and silver nanoshells is clear even to the naked eye, demonstrating that a regular dark field microscope (without hyperspectral capabilities) can be incorporated into the workflow of this platform. In Fig. 5e, gold (purple) and silver (green) nanoshells were sorted from the hyperspectral image based on their scattering profiles and the averaged scattering curves for each of the samples is shown on the inset. For the purpose of visualization, only 25 particles for each type of nanoshells were selected. These spectra confirm the localization and selectivity of the nanoshells, and the separation between them corroborates the multiplex capabilities of this platform.
Despite the long incubation time (3 h 30 min total), the Au-IGFR nanoshells stay on the plasma membrane and are not internalized by the cells. In all fields evaluated both samples were selective to their designated subcellular compartment. Cell viability was determined using the Trypan Blue exclusion test.38 Cells were analyzed 48 h post-incubation, and no significant variations were seen compared to cells that did not receive nanoshells (Table SI-2†). The intracellular toxicity of pristine Ag nanoparticles has been shown to be lessened by the presence of capping agents.39 Here, we credit the absence of significant cytotoxic effects to the use of an amine-rich NLS peptide that can act as an effective capping agent due to the strong interaction between amine groups and the metallic surface. It is important to point out that, although the results in Fig. 5 were obtained using the nanoshells with the best spectral separation obtained in this work, other combinations from our nanoshell library could also be used. This is possible due to the relative narrow LSPR of our samples (Fig. 2 and 3). For instance, Fig. SI-10† shows a similar experiment as in Fig. 5, but using two nanoshells of the same metal, Au50-NLS and Au80-IGF. In that case, the colours of the two types of nanoparticles viewed from white light scattering in a dark field microscope were similar. However, their LSPR characteristics were separated enough to allow differentiation in a hyperspectral experiment.
Cell samples labelled with nanoshells can be frozen and kept, similarly to what is adopted for other techniques, such as fluorescence microscopy. One of the main advantages of using nanoshells as labels is that some of the problems that are inherent to the use of fluorophores (photobleaching, quenching, etc.) will not be observed, eliminating the need for re-labeling cell samples over time.
Long-term photostability
In order to assess how the nanoshells stand against the method of reference for cell labelling (fluorescence imaging), an experiment was performed comparing the photostability of the nanoshells with that of fluorescence staining. The plasma membrane of MCF-7 cells was double-labelled with Au-IGFR and Palloidin CruzFluor® 488. The same field of view was then kept under illumination and images were acquired at different time intervals. Fig. 6 shows the obtained results. After 3 hours under illumination, the fluorescence image almost completely fades as the dye photodegrades (Fig. 6a–d). This shows the superiority of the nanoshells as a platform for long-term live imaging. Several cellular processes happen on the scale of minutes to hours. A typical cell cycle for human cell lines is on the 8 hour range. A photostable platform that can exceed the 24 hour mark, coupled to the capacity to track and identify at the single particle level will bring important progress to the field of cell analysis.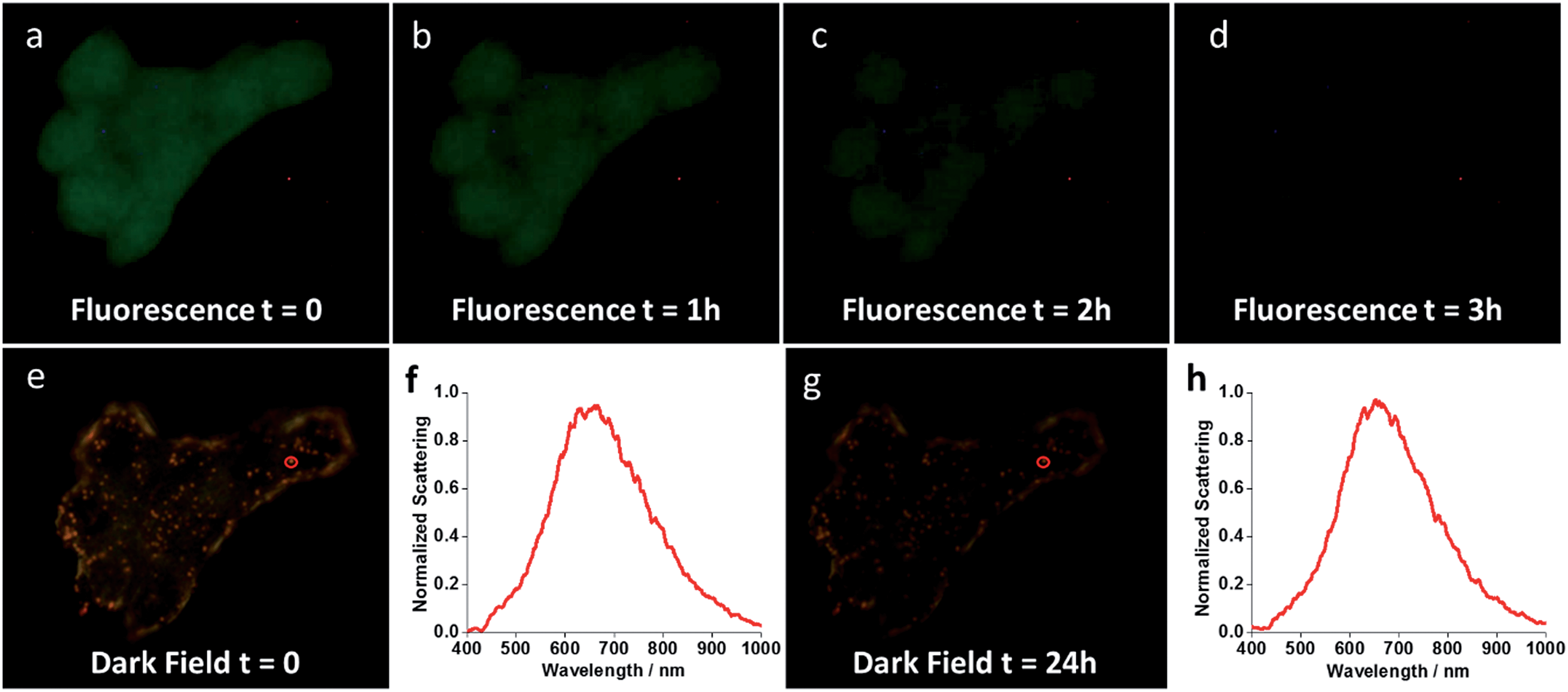 | ||
| Fig. 6 Long-term photostability of nanoshells. Time-lapse images show fluorescent phalloidin photodegrading over the course of a few hours under illumination (a–d), whereas Au-IGFR nanoshells remain active even after 24 h under illumination (e and g). (f and h) show the single scattering spectrum of the same gold nanoshell (red circular inserts in e and g) at t = 0 and t = 24 h. | ||
Conclusions
Hyperspectral analysis using nanoshells is potentially a powerful tool for advanced cellular investigations, such as real-time studies, nanoparticle tracking, LSPR-sensing and biomarker quantification. Due to the chemical and biological stability of noble metals, no alterations in cell activity should be expected when using nanoshells as labelling agents. However, stable and monodispersed nanoparticles must be developed for the full implementation of dark field methods. In this work, we have reformulated the entire synthesis of gold and silver nanoshells into a simpler method that requires only basic lab equipment. The process generates ultramonodisperse particles, resulting in samples of unseen quality whose narrow spectral bands are in close agreement with that for a single particle. Several nanoshell samples were prepared having their LSPRs tuned across the visible range. Their distinctive spectral features were explored in a multiplex experiment, labelling different subcellular compartments in MCF-7 breast cancer cells at the single cell-single particle level. Ag nanoshells targeted the nuclear membrane and Au nanoshells targeted IGF receptors on the plasma membrane of MCF-7 cells. A fluorescence counterstaining experiment unequivocally showed the localization of the nanoshells in different compartments. Hyperspectral images corroborated the selectivity and specificity of the nanoshells for bioanalytical applications. Photostability of these particles was investigated against the standard fluorescent dye phalloidin and surpassed the latter by several hours, hitting the 24 h mark without measurable degradation. Further investigations will include the use of these nanoshells to assess variations in protein expression and metabolic processes in cancer cells under physical and chemical stress.Acknowledgements
We thank Dr Patrick Nahirney and Brent Gowen for their kind assistance on the TEM.Notes and references
- L. Y. Chou, K. Zagorovsky and W. C. Chan, Nat. Nanotechnol., 2014, 9, 148–155 CrossRef CAS PubMed.
- A. M. Brito-Silva, C. B. de Araujo, F. A. Brayner, S. S. Santos, A. Galembeck and E. R. Milet, Polym. Eng. Sci., 2010, 50, 2350–2355 CAS.
- A. M. Brito-Silva, R. G. Sobral-Filho, H. A. Mejía, Y.-H. Wang, P. Wang, G. Machado, E. L. Falcão-Filho, C. B. de Araújo and A. G. Brolo, J. Phys. Chem. C, 2014, 118, 18372–18376 CAS.
- R. Bardhan, S. Lal, A. Joshi and N. J. Halas, Acc. Chem. Res., 2011, 44, 936–946 CrossRef CAS PubMed.
- Y. Choi, Y. Park, T. Kang and L. P. Lee, Nat. Nanotechnol., 2009, 4, 742–746 CrossRef CAS PubMed.
- L. Zhang, Y. Li, D. W. Li, C. Jing, X. Chen, M. Lv, Q. Huang, Y. T. Long and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 6789–6792 CrossRef CAS PubMed.
- A. G. Brolo, Nat. Photonics, 2012, 6, 709–713 CrossRef CAS.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- S. Zhang, H. Gao and G. Bao, ACS Nano, 2015, 9, 8655–8671 CrossRef CAS PubMed.
- C. M. MacLaughlin, N. Mullaithilaga, G. Yang, S. Y. Ip, C. Wang and G. C. Walker, Langmuir, 2013, 29, 1908–1919 CrossRef CAS PubMed.
- W. Pan, T. Zhang, H. Yang, W. Diao, N. Li and B. Tang, Anal. Chem., 2013, 85, 10581–10588 CrossRef CAS PubMed.
- X. Huang, M. Swierczewska, K. Y. Choi, L. Zhu, A. Bhirde, J. Park, K. Kim, J. Xie, G. Niu and K. C. Lee, Angew. Chem., Int. Ed., 2012, 51, 1625–1630 CrossRef CAS PubMed.
- W. Zhou, D. Li, C. Xiong, R. Yuan and Y. Xiang, ACS Appl. Mater. Interfaces, 2016, 21, 13303–13308 Search PubMed.
- B. van den Broek, B. Ashcroft, T. H. Oosterkamp and J. van Noort, Nano Lett., 2013, 13, 980–986 CrossRef CAS PubMed.
- T. Büchner, D. Drescher, V. Merk, H. Traub, P. Guttmann, S. Werner, N. Jakubowski, G. Schneider and J. Kneipp, Analyst, 2016, 141, 5096–5106 RSC.
- S. Westcott, J. Jackson, C. Radloff and N. Halas, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155431 CrossRef.
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419–422 CrossRef CAS PubMed.
- K. Fu, J. Sun, L. R. Bickford, A. W. Lin, N. J. Halas, T.-K. Yu and R. A. Drezek, Nanotechnology, 2008, 19(4), 045103–045108 CrossRef PubMed.
- A. M. Brito-Silva, R. G. Sobral-Filho, R. Barbosa-Silva, C. B. de Araújo, A. Galembeck and A. G. Brolo, Langmuir, 2013, 29, 4366–4372 CrossRef CAS PubMed.
- M. Kallumadil, M. Tada, T. Nakagawa, M. Abe, P. Southern and Q. A. Pankhurst, J. Magn. Magn. Mater., 2009, 321, 1509–1513 CrossRef CAS.
- J. Eastoe, M. J. Hollamby and L. Hudson, Adv. Colloid Interface Sci., 2006, 128, 5–15 CrossRef PubMed.
- R. P. Bagwe, L. R. Hilliard and W. Tan, Langmuir, 2006, 22, 4357–4362 CrossRef CAS PubMed.
- E. Falcão-Filho, R. Barbosa-Silva, R. Sobral-Filho, A. Brito-Silva, A. Galembeck and C. B. de Araújo, Opt. Express, 2010, 18, 21636–21644 CrossRef PubMed.
- W. Shi, Y. Sahoo, M. T. Swihart and P. Prasad, Langmuir, 2005, 21, 1610–1617 CrossRef CAS PubMed.
- S. J. Oldenburg, S. L. Westcott, R. D. Averitt and N. J. Halas, J. Chem. Phys., 1999, 111, 4729–4735 CrossRef CAS.
- J. C. Kah, N. Phonthammachai, R. C. Wan, J. Song, T. White, S. Mhaisalkar, I. Ahmadb, C. Shepparda and M. Olivoc, Gold Bull., 2008, 41, 23–36 CrossRef CAS.
- M. Swami, Nat. Rev. Cancer, 2010, 10, 597 CrossRef CAS PubMed.
- L. A. Walsh and S. Damjanovski, Cell Commun. Signaling, 2011, 9(1), 10–20 CrossRef CAS PubMed.
- Y. Zhang, M. Moerkens, S. Ramaiahgari, H. de Bont, L. Price, J. Meerman and B. van de Water, Breast Cancer Res., 2011, 13(3), R52 CrossRef CAS PubMed.
- A. Albanese, C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, ACS Nano, 2014, 8, 5515–5526 CrossRef CAS PubMed.
- R. V. Benjaminsen, H. Sun, J. R. Henriksen, N. M. Christensen, K. Almdal and T. L. Andresen, ACS Nano, 2011, 5, 5864–5873 CrossRef CAS PubMed.
- B. Yameen, W. I. Choi, C. Vilos, A. Swami, J. Shi and O. C. Farokhzad, J. Controlled Release, 2014, 190, 485–499 CrossRef CAS PubMed.
- M. Mahmoudi, J. Meng, X. Xue, X. J. Liang, M. Rahman, C. Pfeiffer, R. Hartmann, P. R. Gil, B. Pelaz and W. J. Parak, Biotechnol. Adv., 2014, 32, 679–692 CrossRef CAS PubMed.
- C. Yang, J. Uertz, D. Yohan and B. Chithrani, Nanoscale, 2014, 6, 12026–12033 RSC.
- C.-W. Chang, R. M. Couñago, S. J. Williams, M. Boden and B. Kobe, Plant Signaling Behav., 2013, 8, e25976 CrossRef PubMed.
- Y. Cesbron, U. Shaheen, P. Free and R. Lévy, PLoS One, 2015, 10, e0121683 Search PubMed.
- H.-Y. Wang, C. Li, W.-J. Yi, Y.-X. Sun, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, Bioconjugate Chem., 2011, 22, 1567–1575 CrossRef CAS PubMed.
- W. Strober, Current Protocols in Immunology, 2001, pp. A3.B.1–A3.B.3 Search PubMed.
- I.-L. Hsiao, Y.-K. Hsieh, C.-F. Wang, I.-C. Chen and Y.-J. Huang, Environ. Sci. Technol., 2015, 49, 3813–3821 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, methods and instruments, table for different SiO2 cores, DLS measurements, TEM and optical images. See DOI: 10.1039/c6sc04127b |
| ‡ Current address - Biophotonics Research Unit, Gloucestershire Hospitals NHS Foundation Trust, Gloucester, UK. |
| This journal is © The Royal Society of Chemistry 2017 |