DOI:
10.1039/C6SC04129A
(Edge Article)
Chem. Sci., 2017,
8, 2942-2951
Side-chain modulation of dithienofluorene-based copolymers to achieve high field-effect mobilities†
Received
14th September 2016
, Accepted 20th January 2017
First published on 10th February 2017
Abstract
A ladder-type dithieno[3,2-b:6,7-b′]fluorene (DTF), where the central fluorene is fused with two outer thiophene rings at its 2,3- and 6,7-junctions, is developed. The pentacyclic DTF monomers were polymerized with dithienodiketopyrrolopyrrole (DPP) acceptors to afford three alternating donor–acceptor copolymers PDTFDPP16, PDTFDPP20, and PDTFDPP32 incorporating different aliphatic side chains (R1 group at DTF; R2 group at the DPP moieties). The side-chain variations in the polymers play a significant role in determining not only the intrinsic molecular properties but also the intermolecular packing. As evidenced by the 2-dimensional GIXS measurements, PDTFDPP16 with octyl (R1) and 2-ethylhexyl (R2) side chains tends to align in an edge-on π-stacking orientation, whereas PDTFDPP20 using 2-butyloctyl (R1) and 2-ethylhexyl (R2) adopts a predominately face-on orientation. PDTFDPP32 with the bulkiest 2-butyloctyl (R1) and 2-octyldodecyl (R2) side chains shows a less ordered amorphous character. The OFET device using PDTFDPP20 with a face-on orientation determined by GIXS measurements achieved a high hole-mobility of up to 5 cm2 V−1 s−1. The high rigidity and coplanarity of the DTF motifs play an important role in facilitating intramolecular 1-dimensional charge transport within the polymer backbones. The implementation of main-chain coplanarity and side-chain engineering strategies in this research provides in-depth insights into structure–property relationships for guiding development of high-mobility OFET polymers.
Introduction
Organic field-effect transistors (OFETs) have drawn much research attention on account of their promising advantages of good solution processibility, flexibility, and capability for large-area roll-to-roll fabrication.1 Solution-processable semiconducting conjugated polymers play a key role in promoting the advance of OFETs. It is generally believed that a high degree of crystallinity of the polymers with in-plane (edge-on) π-stacking is essential to achieve superior carrier transporting properties due to the existence of two charge-transporting pathways: intramolecular charge transport within the conjugated backbones, and intermolecular charge hopping through π-stacking on the condition that the polymer backbones or π-stacking directions are aligned parallel to the transistor channels.2 Poly(3-hexylthiophene),3 poly(3,3′-dialkyl-quaterthiophene),4 and poly(2,5-bis(thiophen-2-yl)thieno[3,2-b]thiophene) (pBTTT)5 are well-documented semiconducting polymers with high mobilities in this category. In constrast, a few emergent semiconducting donor–acceptor (D–A) copolymers lacking detectable long-range order or in-plane dominance yet exhibit mobilities close to or higher than 1 cm2 V−1 s−1, challenging the authority of the high crystallinity and in-plane packing model.6 The most representative polymer is the indacenodithiophene–benzothiadiazole (IDT–BT) copolymer exhibiting short-range and preferentially face-on crystallites, reaching a superior hole mobility of 3.6 cm2 V−1 s−1.7 McCulloch and DeLongchamp demonstrated that the backbone rigidity and coplanarity of the indacenodithiophene moiety in IDT–BT are important structural features to facilitate one-dimensional charge transport along the polymer backbones which is the primary mechanism that accounts for the exceptional OFET mobility.7 The high molecular weight of such polymers is also important to connect short-range crystallite domains.8 The understanding of structure–property relationships in the IDT–BT polymer prompts us to further implement a backbone coplanarity strategy for developing new high-mobility D–A copolymers. Ladder-type conjugated motifs with forced coplanarity therefore are suitable for this purpose.9 Tricyclic fluorene is a superior building block to construct a variety of organic semiconducting materials due to its good chemical stability and intrinsic hole-transporting property.10 Embedding a fluorene unit into a highly rigid and coplanar ladder-type conjugated framework would be a promising molecular design.11 To this end, we recently designed and synthesized a fluorene-based dithieno[3,2-b:6,7-b′]fluorene (DTF) in which the central fluorene is π-extended by fusing with two outer thiophene rings at its 2,3- and 6,7-junctions.12 Structurally analogous to IDT, the pentacyclic DTF exhibits high coplanarity and rigidity which reduces the energetic disorder of the backbone and thus enhances 1-dimensional intramolecular charge transport. Furthermore, in comparison with the IDT unit having two dialkyl sp3-carbon bridges in the cyclopentadiene (CP) moieties, the singly dialkyl substituted CP ring in the middle of DTF would minimize the inter- and intramolecular steric hindrance between the aliphatic side chains attached to the polymer backbones, which is beneficial to the strength of the intermolecular interactions and maintenance of main-chain coplanarity. To date, however, incorporation of DTF into D–A copolymers for organic optoelectronics has never been explored. It should also be emphasized that the choice of the aliphatic side chains attached to the polymer would dramatically influence the solubility, molecular packing and thin-film morphology which in turn would alter the mechanism of charge transport and thus the mobility.13 For instance, Fréchet and co-workers demonstrated that the edge-on and face-on orientations of conjugated polymers in thin films can be manipulated by the variation of their side chains.13d In this research, newly developed electron-rich DTF units were copolymerized with electron-deficient dithienodiketopyrrolopyrrole (DPP) units to afford a new class of alternating poly(dithienofluorene-alt-dithienoyldiketopyrrolopyrrole) copolymers (denoted as PDTFDPPs) given that the DPP moiety has a planar conjugated bicyclic structure and strongly electron-withdrawing polar amide groups to induce strong intermolecular interactions.14 With an aim to control the face-on/edge-on π-stacking orientations of the polymers through side-chain engineering, three PDFTDPPs (PDTFDPP16, PDTFDPP20 and PDTFDPP32) incorporating different aliphatic side chains were produced for systematic investigation. In spite of forming short-range crystallites with face-on π-stacking, PDTFDPP20 with the 2-butyloctyl group on DTF and the 2-ethylhexyl group on the DPP moieties achieved an unprecedentedly high OFET mobility of 5 cm2 V−1 s−1.
Results and discussion
Design and synthesis of stannylated DTFs and PDTFDPPs
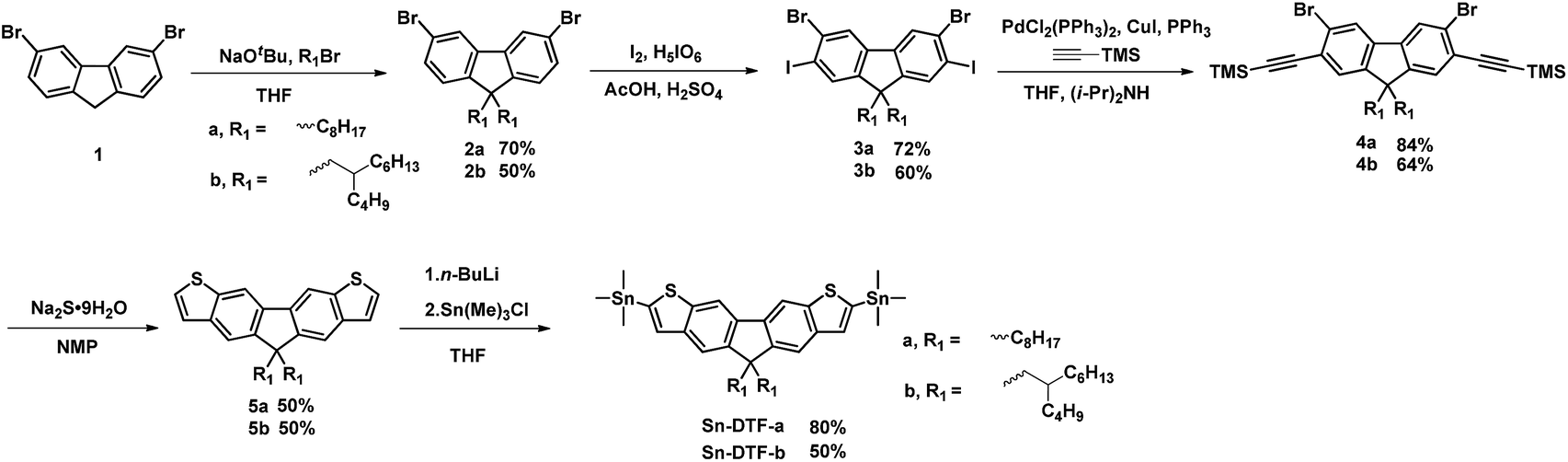
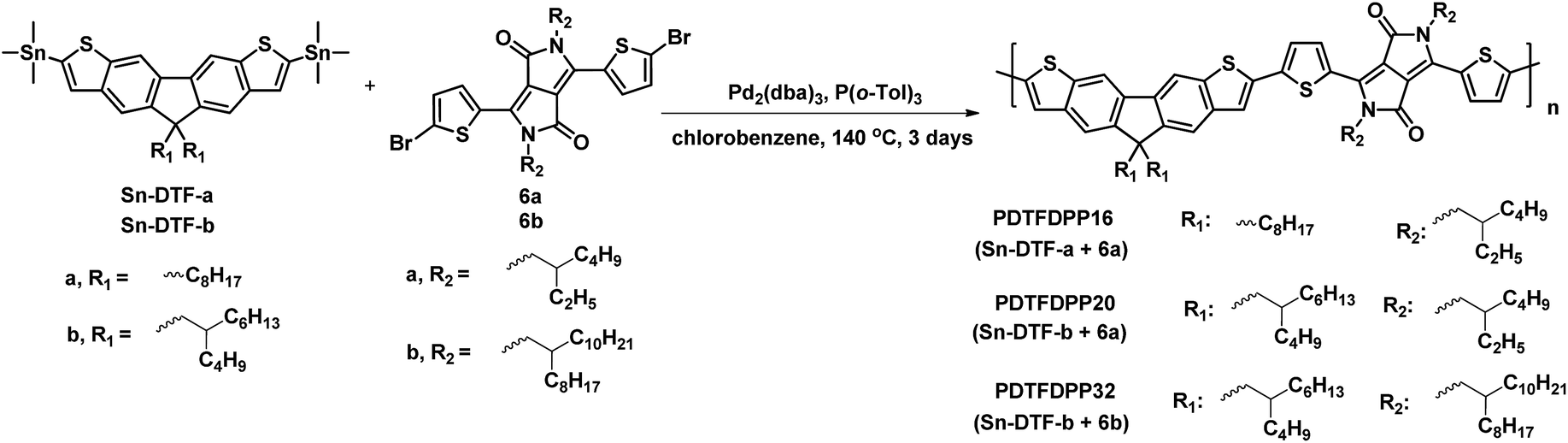
Linear octyl and branched 2-butyloctyl solubilizing groups are selected to be installed on the DTF unit in order to tailor the molecular packing of the resulting polymers. Synthesis of the stannylated DTF monomers is depicted in Scheme 1. 3,6-Dibromofluorene (1) was reacted with octyl bromide and 5-(bromomethyl)undecane under basic conditions to form 3,6-dibromo-9,9-dioctylfluorene (2a) and 3,6-dibromo-9,9-di(2-butyloctyl)fluorene (2b), respectively. Selective iodination of 2a and 2b at the 2,7-positions of the fluorenes yielded the key intermediates 3a and 3b, respectively. Sonogashira coupling of 3a and 3b regioselectively occurred at the 2,7-positions to afford 4a and 4b due to the higher reactivity of iodo over bromo. Reaction of 4a and 4b with Na2S generated the thiolate intermediates which underwent 5-endo-dig cyclization to form two fused thiophenes in 5a and 5b. Stannylation of 5a and 5b was carried out to afford the Sn-DTF-a and Sn-DTF-b monomers, respectively. Similarly, branched 2-ethylhexyl and 2-octyldodecyl groups with different levels of steric hindrance are introduced to the DPP monomer (6a and 6b, respectively). As shown in Scheme 2, Sn-DTF-a and Sn-DTF-b were copolymerized with 2,5-di(2-ethylhexyl)-3,6-di(5-bromothiophen-2-yl)diketopyrrolopyrrole (6a) to furnish two alternating D–A copolymers, PDTFDPP16 and PDTFDPP20, respectively (16 and 20 are the total carbon numbers of the aliphatic side chains in a repeating unit). Sn-DTF-b was polymerized with 2,5-di(2-octyldodecyl)-3,6-di(5-bromothiophen-2-yl)diketopyrrolopyrrole (6b) to yield PDTFDPP32. Different number average molecular weights (Mn) were determined by gel permeation chromatography (GPC) to be 4.5, 68, and 32 kg mol−1 for PDTFDPP16, PDTFDPP20, and PDTFDPP32, respectively (Table 1), indicating that the magnitude of Mn is essentially governed by the aliphatic side chains flanking the DTF and DPP units. PDTFDPP20 with branched side chains on both the DTF and DPP moieties has the highest Mn, whereas PDTFDPP16 with linear octyl groups on the DTF units shows the poorest solubility in chlorobenzene, which is responsible for its lowest Mn. PDTFDPP32 has a lower Mn than PDTFDPP20 due to the fact that 6b with the bulkiest 2-octyldodecyl group encounters more steric hindrance when reacting with Sn-DFT-b during the polymerization.
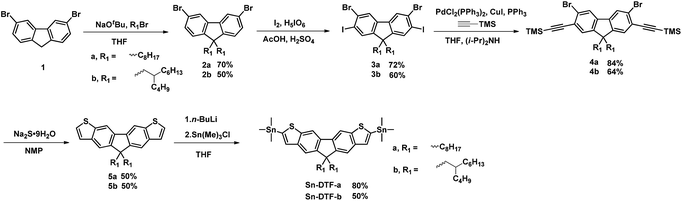 |
| | Scheme 1 Synthesis of Sn-DTF-a and Sn-DTF-b monomers. | |
 |
| | Scheme 2 Synthesis of the three D–A copolymers PDTFDPP16, PDTFDPP20, and PDTFDPP32. | |
Table 1 Summary of the intrinsic properties of PDTFDPP16, PDTFDPP20, and PDTFDPP32
|
|
M
n
(kDa) |
PDIb |
T
d
(°C) |
E
optg
(film) (eV) |
E
eleg
(eV) |
λ
max (nm) |
E
HOMO (eV) |
E
LUMO (eV) |
| CHCl3 |
Film |
|
M
n = number-average molar weight.
PDI = polydispersity index.
T
d = decomposition temperature at 5% weight loss.
E
optg was estimated from the onset of UV spectrum in thin film.
E
eleg was estimated from cyclic voltammetry.
|
|
PDTFDPP16
|
4.5 |
1.2 |
442 |
1.59 |
1.63 |
655 |
641 |
−5.23 |
−3.60 |
|
PDTFDPP20
|
68 |
2.1 |
456 |
1.65 |
1.76 |
658, 707 |
643, 704 |
−5.38 |
−3.62 |
|
PDTFDPP32
|
32 |
1.8 |
432 |
1.65 |
1.73 |
643, 707 |
643, 704 |
−5.39 |
−3.66 |
Thermal properties
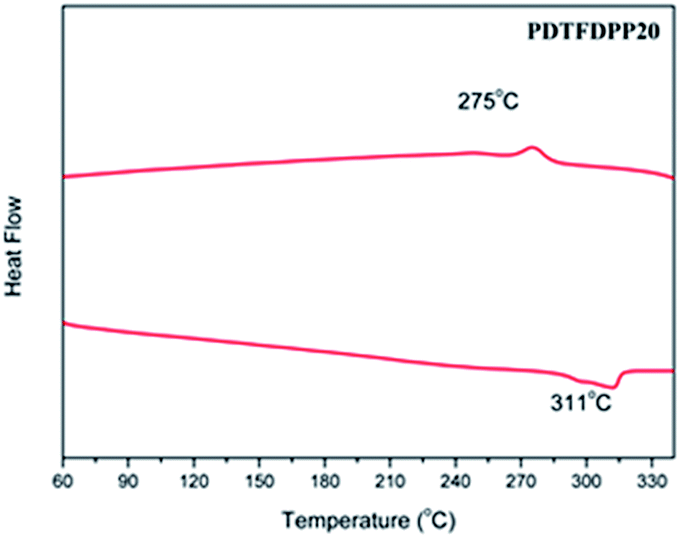
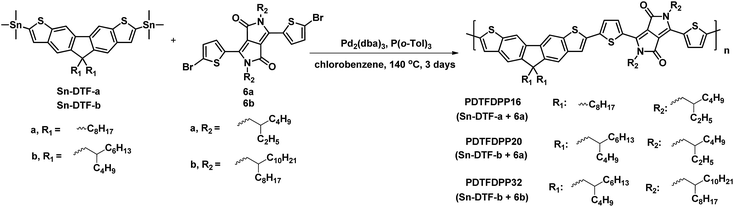
The thermal stability of the polymers was analyzed by thermogravimetric analysis (TGA). PDTFDPP16, PDTFDPP20, and PDTFDPP32 exhibited sufficiently high decomposition temperatures (Td) of 442, 456, and 432 °C, respectively (Fig. S1† and Table 1). No thermal transition in the differential scanning calorimetry measurements (DSC) was observed for PDTFDPP16 and PDTFDPP32, implying their less crystalline character within the range of scanning temperatures. Nevertheless, PDTFDPP20 exhibited a melting point at 311 °C upon heating and a crystallization point at 275 °C upon cooling, implying the semicrystalline nature of PDTFDPP20 (Fig. 1).
 |
| | Fig. 1 Differential scanning calorimetry of PDTFDPP20 with a ramping rate of 10 °C min−1. | |
Optical properties
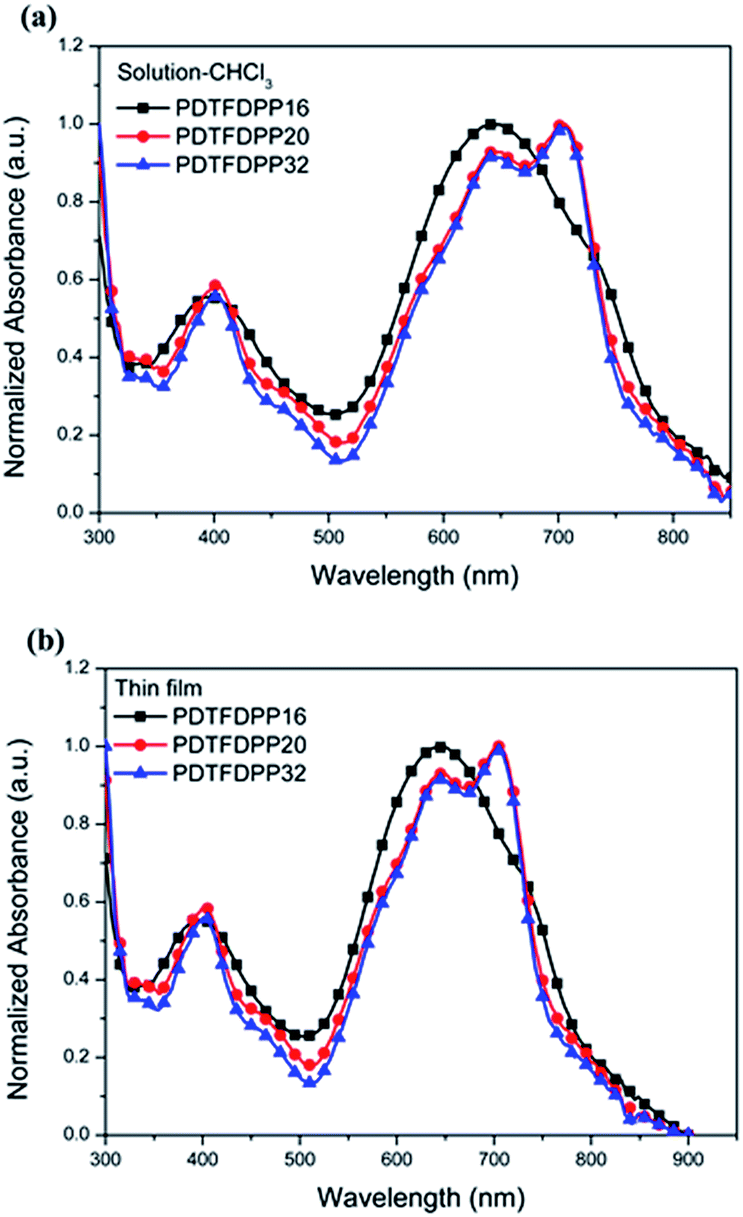
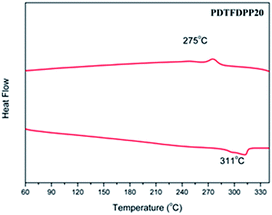
The UV-vis absorption spectra in CHCl3 and thin film are depicted in Fig. 2 and summarized in Table 1. It is expected that the three polymers should possess analogous absorption profiles due to their identical backbone composition. Nevertheless, PDTFDPP20 and PDTFDPP32 exhibited a pronounced vibronic structure, which is much less noticeable for PDTFDPP16. PDTFDPP20 and PDTFDPP32 showed lower-lying HOMO energy levels of −5.38 and −5.39 eV, respectively, while PDTFDPP16 exhibited a higher HOMO level of −5.23 eV. The higher HOMO level may be associated with the linear side chain in PDTFDPP16, which results in less distorted polymer chains. PDTFDPP32 might already reach a saturation of the effective conjugated length. Therefore, PDTFDPP20 with higher molecular weight shows an almost identical absorption profile to PDTFDPP32.
 |
| | Fig. 2 Normalized UV-vis absorption spectra of PDTFDPP16, PDTFDPP20, and PDTFDPP32 in (a) chloroform solutions; (b) thin films. | |
Electrochemical properties
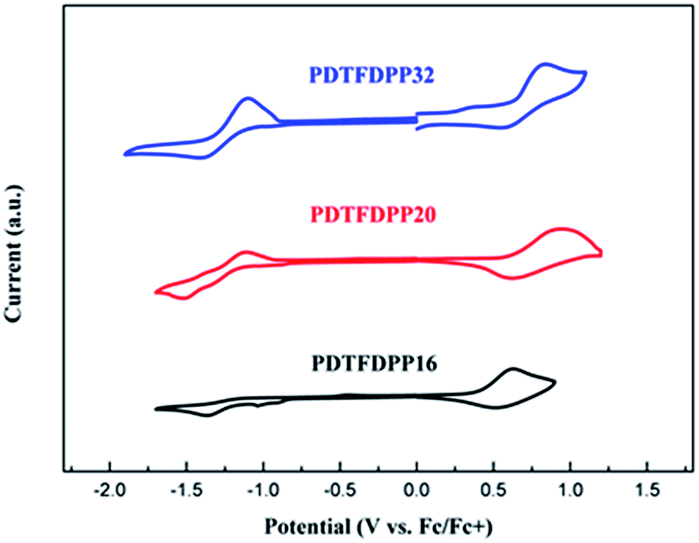
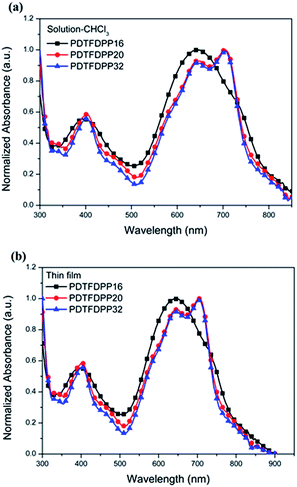
Cyclic voltammetry (CV) was employed to determine the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies of the polymers (Fig. 3, Table 1). PDTFDPP20 and PDTFDPP32 showed lower-lying HOMO energy levels of −5.38 and −5.39 eV, respectively, while PDTFDPP16 exhibited a higher HOMO level of −5.23 eV. This discrepancy might result from the variation in the HOMO electron-density distribution. As for the LUMO, the three polymers have comparable values, which approximate to −3.60 eV.
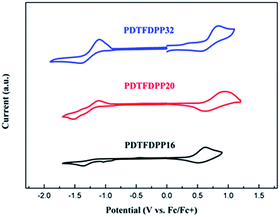 |
| | Fig. 3 Cyclic voltammograms of PDTFDPP16, PDTFDPP20, and PDTFDPP32 in thin films at a scan rate of 100 mV s−1. | |
GIXS measurements
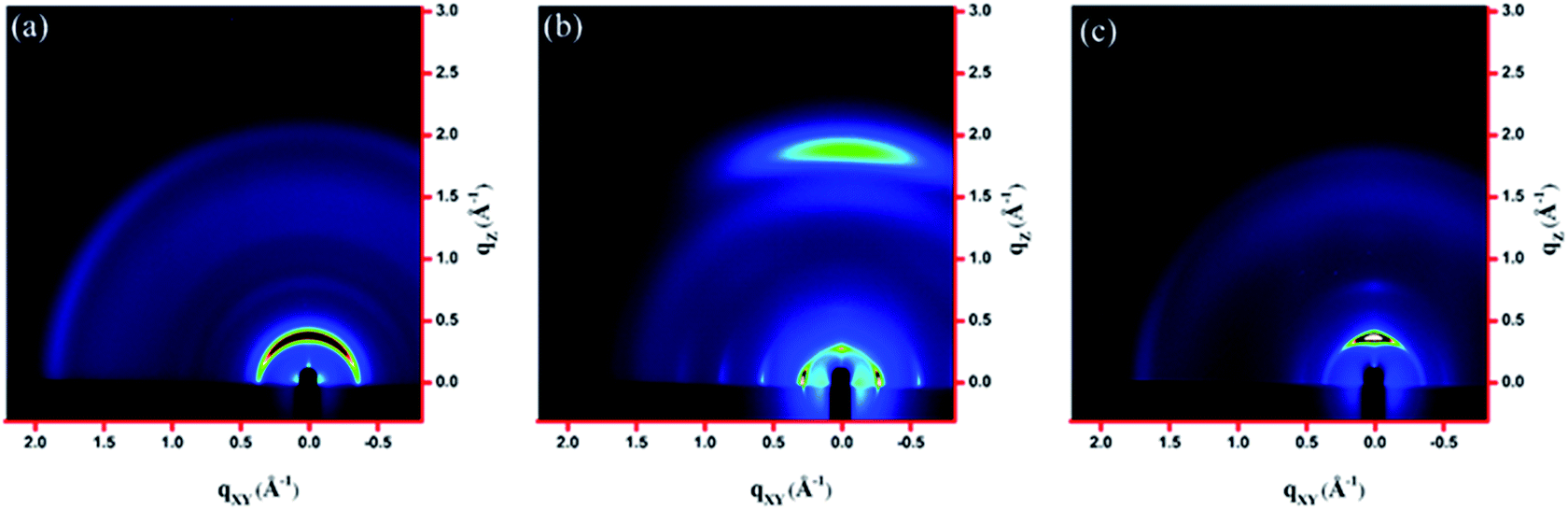
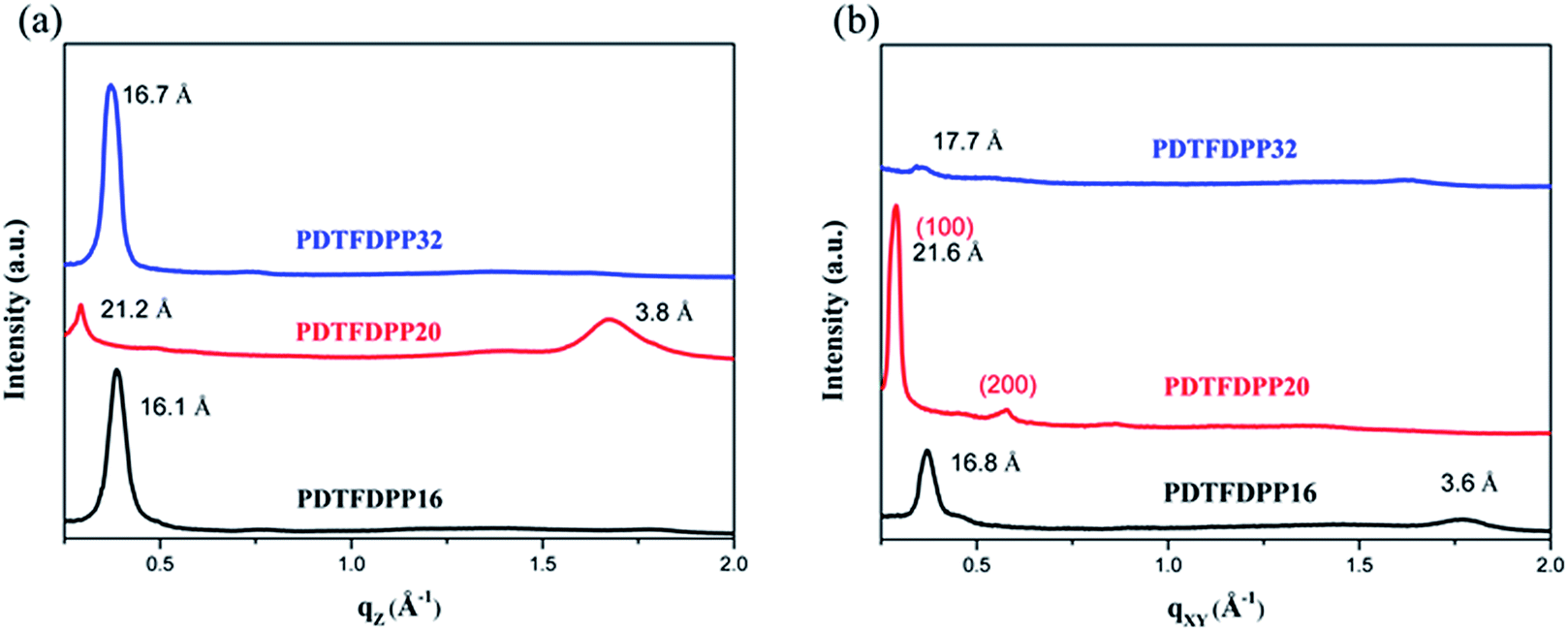
The molecular packing of the thin films on an Si/SiO2 substrate modified by a self-assembled monolayer (SAM) of octadecyltrichlorosilane (ODTS) was investigated using 2-dimensional grazing incidence X-ray scattering (GIXS). The 2-D GIXS images and the corresponding 1-dimensional out-of-plane (qz) and in-plane (qxy) diffraction patterns of the polymer thin films annealed at 200 °C for 10 minutes are illustrated in Fig. 4 and 5, respectively. The PDTFDPP20 film exhibited in-plane (h00) signals assigned to a lamellar spacing (dl) of ca. 21.6 Å corresponding to side-chain interdigitation (Fig. 5b). Meanwhile, PDTFDPP20 also showed an obvious (010) out-of-plane peak at qz = 1.7 Å−1 corresponding to periodic π–π stacking with a distance (dπ) of ca. 3.8 Å between the two facing conjugated backbones (Fig. 5a). The out-of-plane (010) peak also reveals that the PDTFDPP20 adopts a predominately face-on orientation with the backbone plane approximately parallel to the substrate. Conversely, judging from the out-of-plane lamellar stacking (qz = 0.4 Å−1 corresponding to dl = 16.1 Å, Fig. 5a) and the in-plane π-stacking (qxy = 1.7 Å−1 corresponding to dπ = 3.6 Å, Fig. 5b) peaks, PDTFDPP16 tends to align preferably in an edge-on π-stacking orientation with the polymer backbone roughly perpendicular to the substrate.
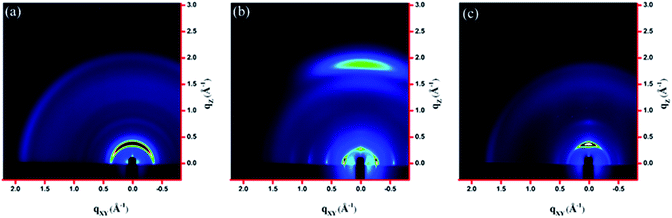 |
| | Fig. 4 2-Dimensional GIXS images of the thin films (a) PDTFDPP16, (b) PDTFDPP20, and (c) PDTFDPP32 on the ODTS-treated Si/SiO2 substrate. | |
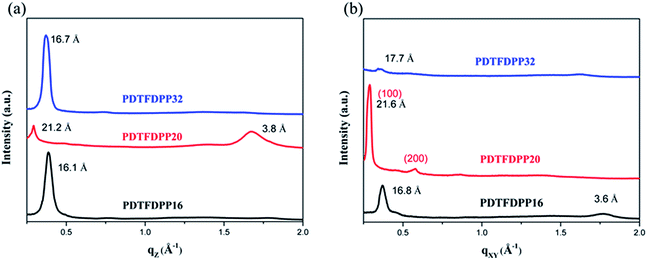 |
| | Fig. 5 1-Dimensional (a) out-of-plane and (b) in-plane GIXS patterns of PDTFDPP16, PDTFDPP20, and PDTFDPP32. The spacings are listed near the peaks. | |
As for PDTFDPP32, no evident π-stacking peak could be found in the direction of either qz or qxy, indicative of its amorphous character.
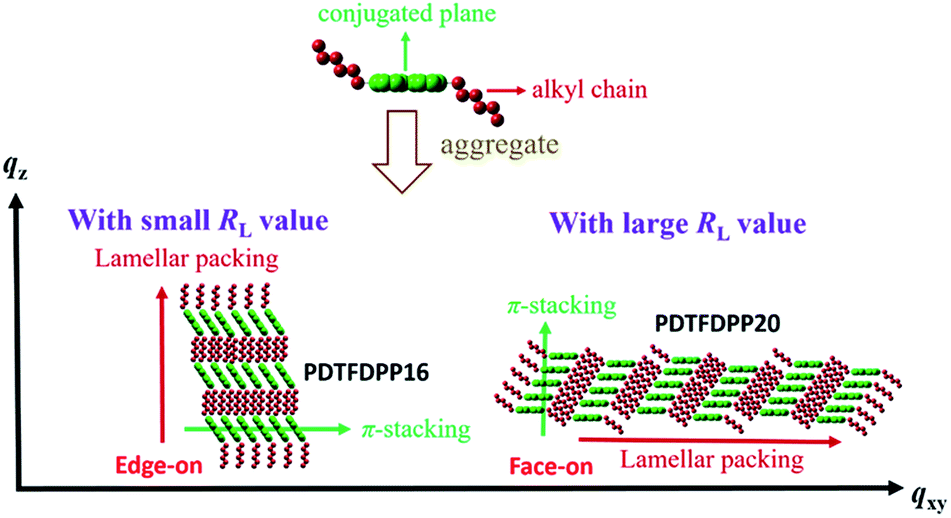
The correlation length (Lc) derived from the full width at half maximum (FWHM) of a X-ray diffraction peak in GIXS based on Scherrer's equation can be used to estimate the size of a crystallite.1f,7,13d By combining the lamellar-stacking diffraction peak in the qxy or qz direction and the π-stacking peak in the corresponding perpendicular direction (i.e. qz or qxy, respectively) in the 2D-GIXS images, we can approximately acquire the vertical and horizontal correlation lengths (Lc) of a 2-dimensional crystallite. We realized that the ratio (RL) of lamellar stacking Lc to π-stacking Lc is an important parameter in dictating the face-on/edge-on orientations of the PDTFDPPs. The PDTFDPP16 film with a π-stacking Lc of 4.0 nm (qxy) and a lamellar Lc of 10.1 nm (qz) leads to a RL of 2.5, while the PDTFDPP20 film with a π-stacking Lc of 3.4 nm (qz) and a lamellar Lc of 19.5 nm (qxy) yields a RL of 5.7. The relevant parameters are summarized in Table 2. It can be reasonably inferred that a crystallite with a smaller RL value prefers to align in an edge-on π-stacking orientation perpendicular to the substrate. Increasing the RL value by lengthening the lamellar-stacking dimension changes the π-stacking from an edge-on alignment to a face-on orientation. The 2-dimensional molecular packing of PDTFDPP16 and PDTFDPP20 is illustrated in Fig. 6. This argument is also valid for other well-investigated polymer systems. It should also be noted that other factors such as side chain density,15 solvent selection16 and the branching point position of the side chain17 might also play important roles in determining the crystalline orientation. For example, P3HT and PBTTT-C12 (ref. 5a) with smaller RL values of 2.03 and 1.90, respectively, show edge-on orientations, whereas PDPP3F-BO with a large RL value of 7.86 favors the face-on packing.13d This observation is also consistent with the fact that a nanorod with a high aspect ratio (length divided by width) fails to stand vertically on a substrate.18 Additional GIXS data evidencing this argument are provided in the ESI.†
Table 2 Stacking parameters of the PDTFDPP16 and PDTFDPP20 thin films
|
|
π-Stacking |
Lamellar stacking |
R
L
|
|
d
(Å) |
L
c
(nm) |
d
(Å) |
L
c
(nm) |
|
Spacing of diffraction peak.
Correlation length based on Scherrer's equation.
Ratio of Lc (lamellar spacing) to Lc (π-stacking).
|
|
PDTFDPP16
|
3.6 |
4.0 |
16.8 |
10.1 |
2.5 |
|
PDTFDPP20
|
3.8 |
3.4 |
21.6 |
19.5 |
5.7 |
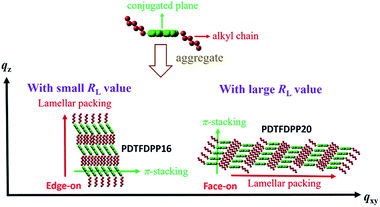 |
| | Fig. 6 Illustration of the 2-dimensional lamellar and π-stacking crystallites of PDTFDPP16 and PDTFDPP20 with RL of 2.5 and 5.7, respectively. | |
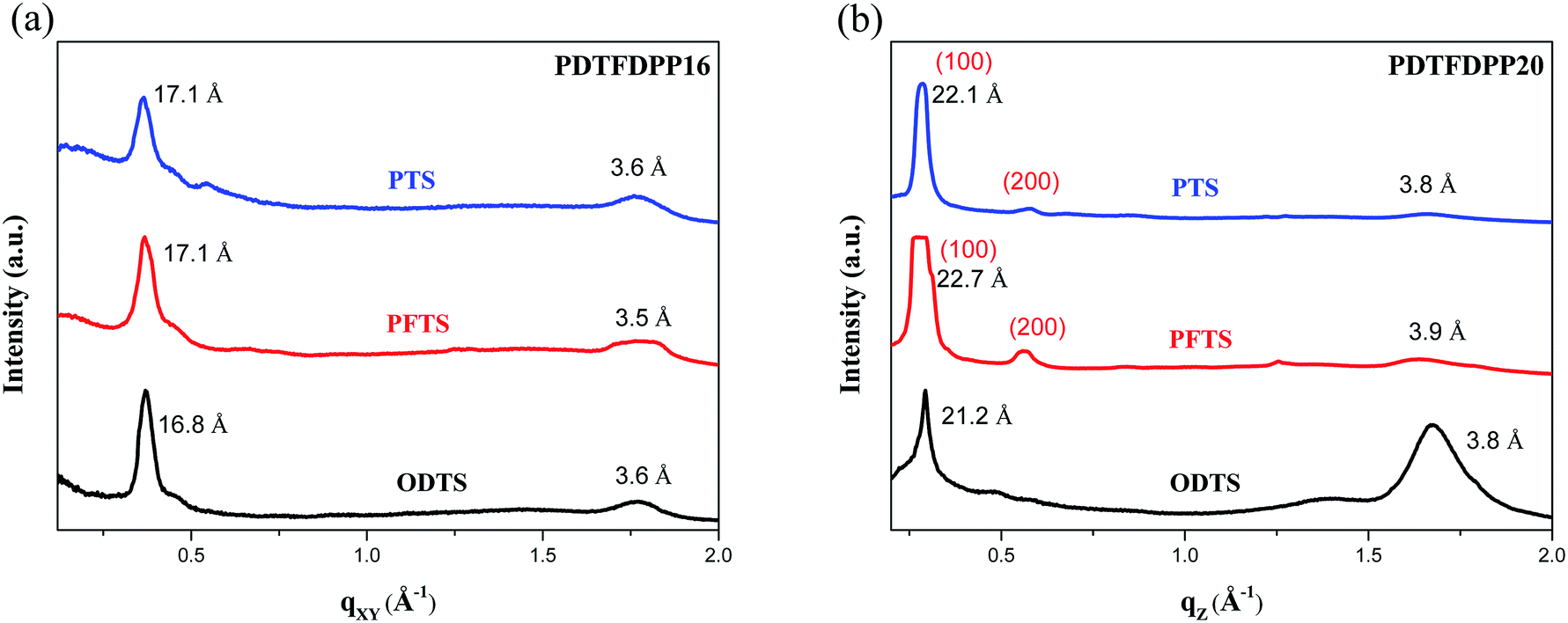
In addition to the crystallite dimensions, the interactions between the polymers and underlying self-assembled monolayer (SAM) on the Si/SiO2 substrate may also influence the polymer orientation. Thin films on trichloro(phenethyl)silane (PTS) and trichloro(1H,1H,2H,2H-perfluorooctyl)silane (PFTS) SAM layers were thus prepared and examined in comparison with ODTS. The GIXS measurements of PDTFDPP16 in the qxy direction (Fig. 7) reveal that the edge-on π-stacking remains essentially intact regardless of the SAM layer; however, the original face-on orientation of PDTFDPP20 on ODTS in the qz direction is almost diminished when the SAM layer is changed to PTS or PFTS.
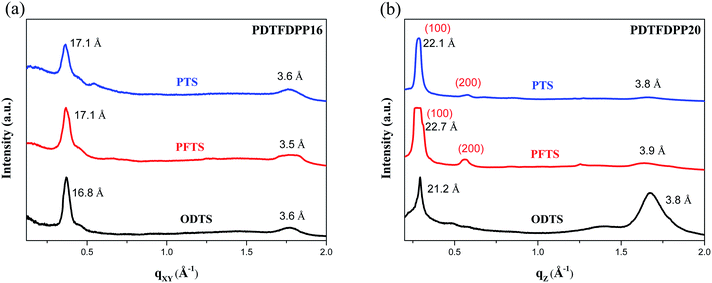 |
| | Fig. 7 1-Dimensional GIXS patterns of (a) PDTFDPP16 in the qxy and (b) PDTFDPP20 in the qz direction on the SAM layer of PTS, PFTS and ODTS. | |
The results indicate that the formation of face-on π-stacking is highly dependent on the underlying SAM layer, which may be attributed to the more intimate contact between the face-on π-conjugated plane and the underlying substrate. It should be also mentioned that the GIXS measurements only reflect the bulk orientations of the thin films. The polymer might adopt a different orientation at the semiconductor/dielectric interface which is the channel area where the charge transport occurs.19
Theoretical calculations
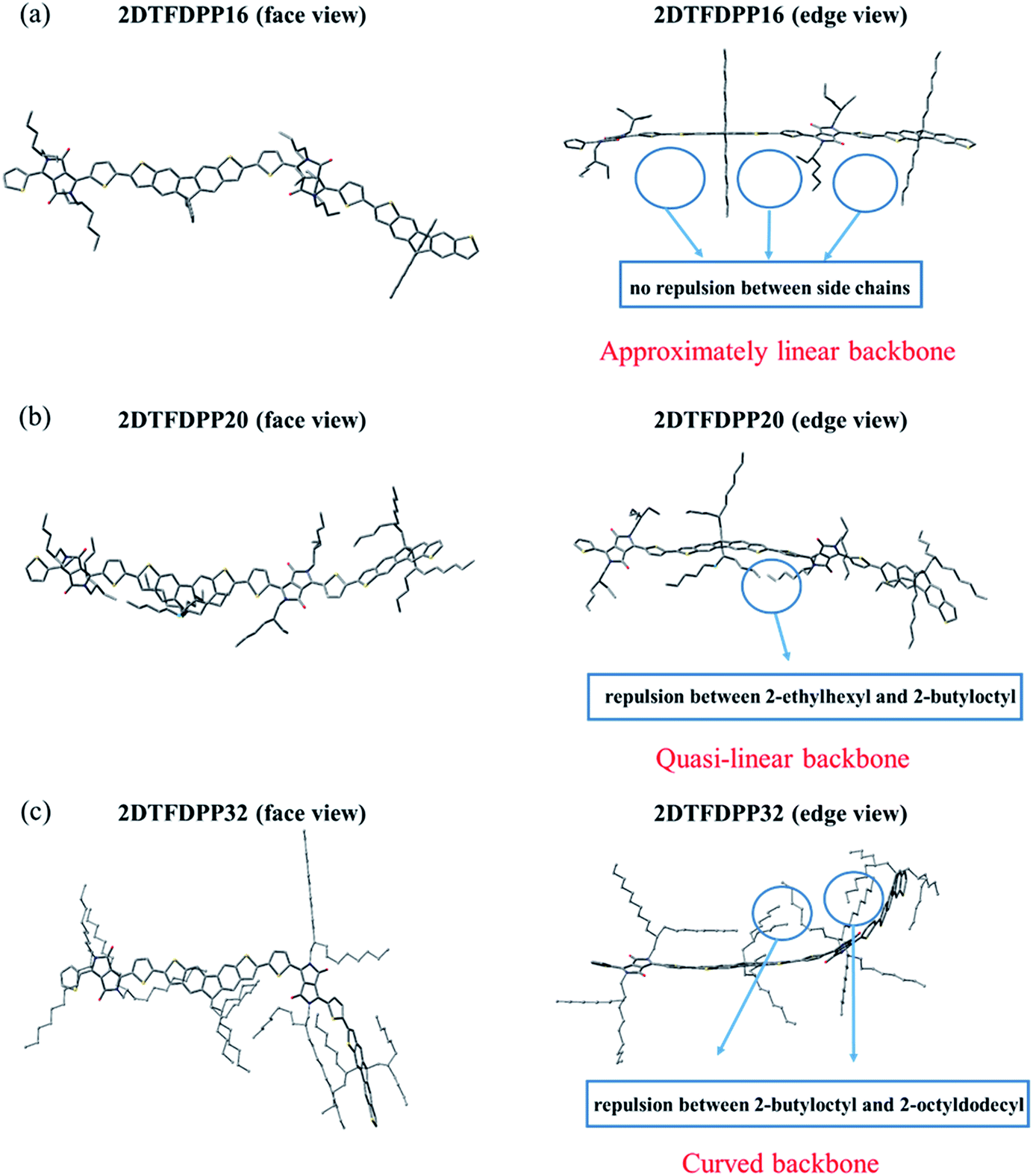
In order to gain insight into the steric effect derived from different side chains installed on PDTFDPP, our own n-layered integrated molecular orbital and molecular mechanics (ONIOM) calculations were performed. ONIOM approaches were developed to compute large molecules with high accuracy and reduced computational time.20 Within the target structure, two or three layers are defined and treated at different levels of theory. In this study, dimeric structures, 2DTFDPP16, 2DTFDPP20, and 2DTFDPP32 were used as the model compounds for simulating PDTFDPP16, PDTFDPP20, and PDTFDPP32, respectively. Geometry optimization was carried out with the B3LYP/6-31G(d) level of theory applied to the conjugated systems and the universal force field (UFF) applied to the alkyl side chains. The optimized structures are depicted in Fig. 8. It is evident from the edge-viewed structures that 2DTFDPP16 has the most planar and linear conjugated backbone among the three model compounds due to the low steric repulsion between the octyl and 2-ethylhexyl side chains in 2DTFDPP16. It is envisioned that PDTFDPP16 would have strong intermolecular π–π stacking, leading to poor solubility during polymerization and thus would have the lowest Mn. In contrast, steric repulsion between the 2-butyloctyl and 2-octyldodecyl groups in 2DFTDPP32 distorts its main-chain coplanarity and linearity, which is disadvantageous for intermolecular π–π stacking (Fig. 8c). The monomeric unit of PDTFDPP32 has the highest molecular weight among all; however, the Mn of PDTFDPP32 is significantly lower than that of PDTFDPP20. This side-chain repulsion obstructs the cross-coupling reaction between two monomeric units, accounting for the modest Mn of PDTFDPP32. 2DTFDPP20 shows a quasi-linear backbone. The 2-butyloctyl and 2-ethylhexyl moieties in PDTFDPP20 are sufficient to solubilize the resulting polymer without hampering the polymerization, leading to the highest Mn. Furthermore, the curved main-chain structure of 2DTFDPP32 is associated with the amorphous character of PDTFDPP32 suggested by the 2D-GIXS. As mentioned previously, the planar and linear conjugated plane might allow PDTFDPP16 to form a longer π-stacking Lc than PDTFDPP20. On the other hand, it is envisaged that PDTFDPP20 with the dense aliphatic chains would assemble into a longer lamellar packing Lc than PDTFDPP16. Accordingly, the variation in the crystallite dimensions results in the disparity in the thin-film orientation between PDTFDPP16 and PDTFDPP20.
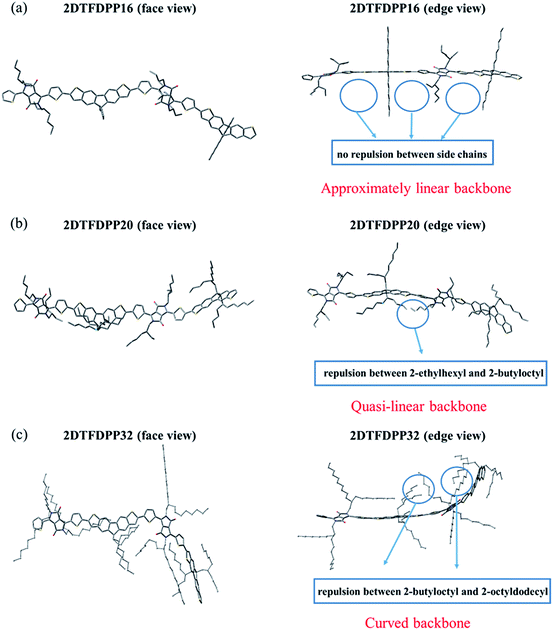 |
| | Fig. 8 Optimized geometry of dimeric structures (a) 2DTFDPP16; (b) 2DTFDPP20; (c) 2DTFDPP32 (face view and edge view). | |
Transistor characterization
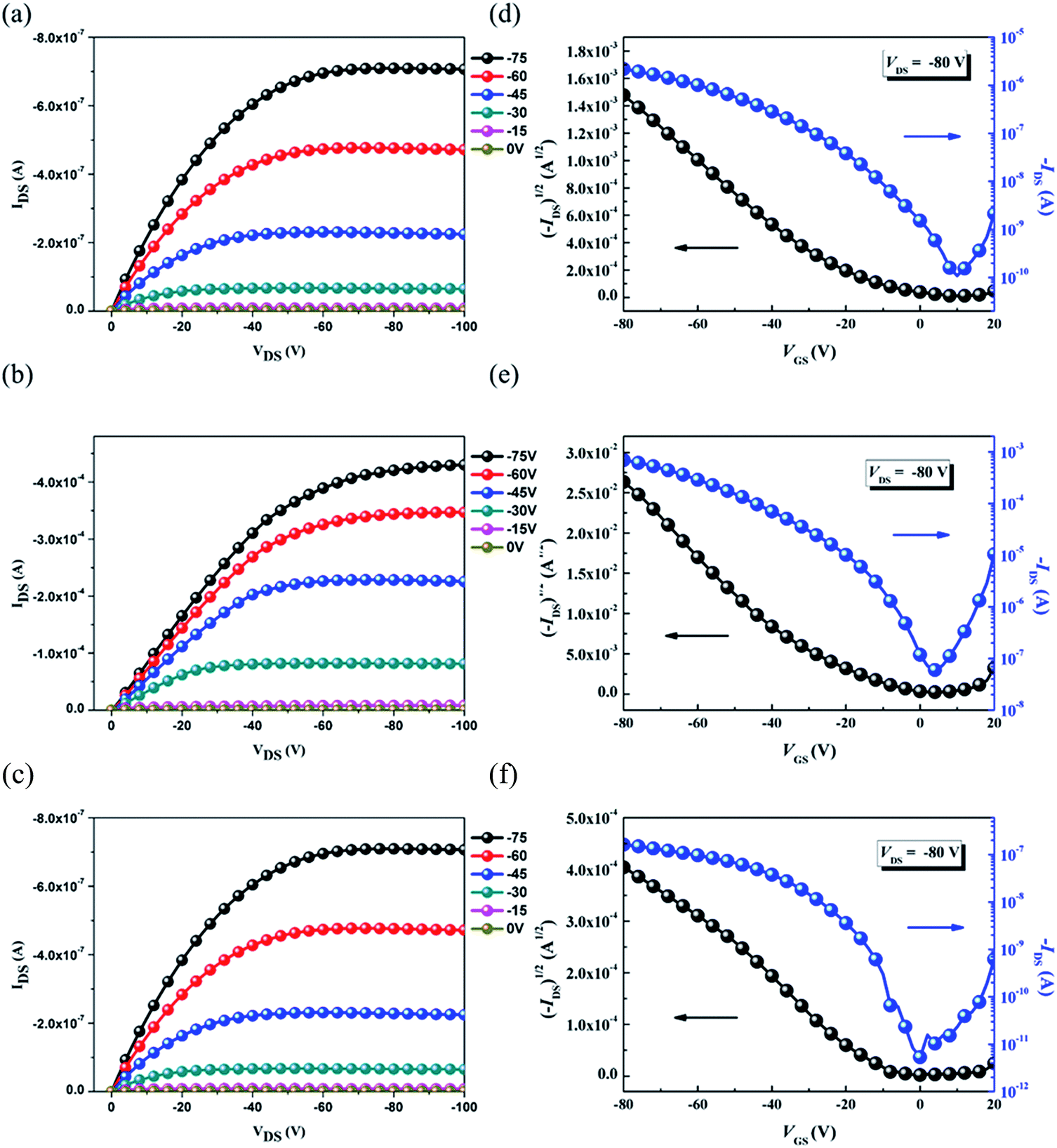
The OFET mobilities of the DTF-based polymers were measured by devices using a bottom-gate/top-contact configuration with evaporated gold source/drain electrodes (40 nm in thickness). The OFET devices used SiO2 as the gate dielectric whose surfaces were modified with a SAM layer of ODTS. All of the output and transfer plots of the devices exhibited typical p-type OFET characteristics (Fig. 9). The hole mobilities are calculated from the transfer characteristics of the devices in the saturation regime. As summarized in Table 3, the hole mobility of the PDTFDPP16 device was 1.20 × 10−2 cm2 V−1 s−1 with an on–off ratio of 4.98 × 105. PDTFDPP32 with more carbons in the aliphatic side chains exhibited the lowest mobility of 1.04 × 10−3 cm2 V−1 s−1. PDTFDPP20 achieved the highest hole mobility reaching 5.00 cm2 V−1 s−1 in the saturated regime. The mobility extracted from the linear regime showed an even higher value of 5.26 cm2 V−1 s−1. GIXS measurements reveal that PDTFDPP20 formed short-range and face-on crystallites in the thin film which is in opposition to the conventional point of view that long-range crystallinity and edge-on π-stacking are essential for high-performance OFET materials. According to the work carried out by Salleo and co-workers, efficient charge transport between the short-range crystallites in the polymer thin film can be realized on the condition that they are inter-connected.8 Compared to PDTFDPP16, the much higher Mn (68 kg mol−1) of PDTFDPP20 not only enhances intrachain charge transport but also enables sufficient connection between the different crystalline domains to realize a high OFET mobility. More importantly, the rigidity and coplanarity of the DTF units facilitate intramolecular 1-dimensional charge transport within the polymer backbones. Charge transport to adjacent chains via π-stack hopping is supposed to be the minor pathway.
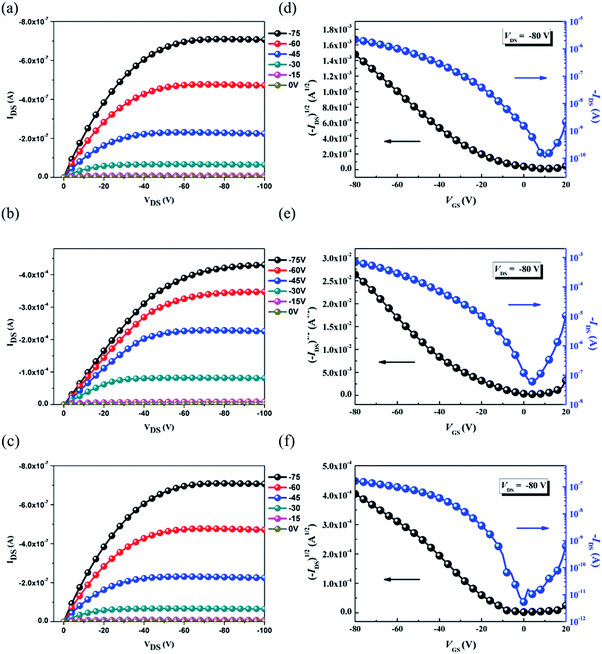 |
| | Fig. 9 Typical output curves (a, b and c) and transfer plots (d, e and f) of the bottom-gate/top-contact OFET devices for PDTFDPP16, PDTFDPP20, and PDTFDPP32, respectively. | |
Table 3 Bottom-gate/top-contact OFET characteristics of the polymer thin films
| Polymer |
SAM layer |
Annealing temp (°C) |
Mobilitya (cm2 V−1 s−1) |
On/off ratio |
V
t (V) |
|
Saturated mobility.
Linear mobility with Vds of −20 V.
|
|
PDTFDPP16
|
ODTS |
200 |
1.20 × 10−2 |
4.98 × 105 |
−19.0 |
|
PDTFDPP20
|
ODTS |
240 |
5.00 |
8.67 × 105 |
−26.0 |
|
PDTFDPP20
|
ODTS |
200 |
5.26b |
1.57 × 106 |
−1.6 |
|
PDTFDPP32
|
ODTS |
Pristine |
1.04 × 10−3 |
3.85 × 104 |
−13.1 |
The surface morphologies of the PDTFDPPs were investigated by atomic force microscopy (AFM). As shown in Fig. 10, PDTFDPP16 exhibited obvious aggregation with a high RMS roughness of 20.2 nm due to its poor solubility in chloroform. However, PDTFDPP32 with better solubility showed a more homogeneous thin-film structure with a dramatically reduced roughness of 2.64 nm. In particular, PDTFDPP20 with a roughness of 8.29 nm exhibited a fibrous texture in its phase image, which might be also associated with its highest charge mobility. These results again demonstrate that side-chain engineering of PDTFDPPs plays a pivotal role in determining the thin-film morphology.
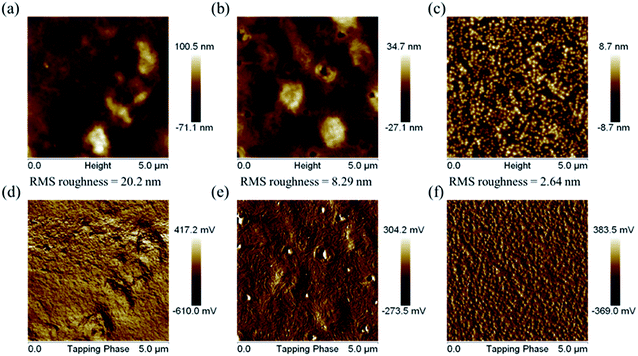 |
| | Fig. 10 Atomic force microscopy height (a, b and c) and phase (d, e and f) images for PDTFDPP16, PDTFDPP20, and PDTFDPP32, respectively. | |
Conclusions
We designed and synthesized a pentacyclic dithieno[3,2-b:6,7-b′]fluorene (DTF) where the central fluorene is π-extended by fusing with two outer thiophene rings at the 2,3- and 6,7-junctions. The newly developed DTF units were copolymerized with the DPP acceptors to yield three D–A copolymers PDTFDPP16, PDTFDPP20, PDTFDPP32 with different aliphatic side chains. The side-chain variations in the polymers play a key role in determining not only the intrinsic molecular properties but also the intermolecular packing. The face-on/edge-on π-stacking of the PDTFDPP-based polymers are associated with the ratio (RL) of the lamellar stacking correlation length to the π-stacking correlation length of the crystallites. Based on GIXS thin film measurements, PDTFDPP16 with a smaller RL tends to align in an edge-on π-stacking orientation, whereas PDTFDPP20 with a large RL adopts a predominately face-on orientation. Bottom-gate/top-contact OFET devices using PDTFDPP20 achieved high hole mobilities of up to 5 cm2 V−1 s−1. To the best of our knowledge, this value represents the highest p-type mobility for solution-processable conjugated polymers with a face-on orientation determined by GIXS. The rigidity and coplanarity of the DTF framework facilitate intramolecular 1-dimensional charge transport within the polymer backbones. A sufficiently high molecular weight to connect the short-range crystalline domains in PDTFDPP20 is also crucial for efficient charge transport.
This research verifies that edge-on dominance and long-range order to achieve high mobility might not be obligatory as long as the short-range crystallites can be interconnected and efficient intrachain 1D-charge transport along the backbone can be established. This work, implementing main-chain coplanarity and side-chain variations, will provide a new direction to guide the development of new high-mobility semiconducting materials.
Acknowledgements
We thank the Ministry of Science and Technology and the Ministry of Education in Taiwan for financial support. We thank the National Center for High-performance Computing (NCHC) in Taiwan for computer time and facilities. We also thank the National Synchrotron Radiation Research Center (NSRRC), and Dr U-Ser Jeng and Dr Chun-Jen Su at BL23A1 station for their help with the GIXS experiments. Y. J. C thanks the Golden-Jade fellowship of the Kenda Foundation, Taiwan for their support.
References
-
(a) A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed
 ;
(b) J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed ;
(c) G. Horowitz, Adv. Mater., 1998, 10, 365–377 CrossRef CAS ;
(d) Y. Zhou, W.-J. Liu, Y. Ma, H. Wang, L. Qi, Y. Cao, J. Wang and J. Pei, J. Am. Chem. Soc., 2007, 129, 12386–12387 CrossRef CAS PubMed ;
(e) J. A. Letizia, A. Facchetti, C. L. Stern, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 13476–13477 CrossRef CAS PubMed ;
(f) M.-H. Yoon, S. A. DiBenedetto, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 1348–1349 CrossRef CAS PubMed ;
(g) S. C. B. Mannsfeld, B. C. K. Tee, R. M. Stoltenberg, C. V. H. H. Chen, S. Barman, B. V. O. Muir, A. N. Sokolov, C. Reese and Z. Bao, Nat. Mater., 2010, 9, 859–864 CrossRef CAS PubMed ;
(h) S. Shinamura, I. Osaka, E. Miyazaki, A. Nakao, M. Yamagishi, J. Takeya and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 5024–5035 CrossRef CAS PubMed ;
(i) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed ;
(j) H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed ;
(k) T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS PubMed ;
(l) S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed ;
(m) A. Cravino, S. Roquet, O. Alévêque, P. Leriche, P. Frère and J. Roncali, Chem. Mater., 2006, 18, 2584–2590 CrossRef CAS ;
(n) C. Videlot, J. Ackermann, P. Blanchard, J. M. Raimundo, P. Frère, M. Allain, R. de Bettignies, E. Levillain and J. Roncali, Adv. Mater., 2003, 15, 306–310 CrossRef CAS .
;
(b) J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed ;
(c) G. Horowitz, Adv. Mater., 1998, 10, 365–377 CrossRef CAS ;
(d) Y. Zhou, W.-J. Liu, Y. Ma, H. Wang, L. Qi, Y. Cao, J. Wang and J. Pei, J. Am. Chem. Soc., 2007, 129, 12386–12387 CrossRef CAS PubMed ;
(e) J. A. Letizia, A. Facchetti, C. L. Stern, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 13476–13477 CrossRef CAS PubMed ;
(f) M.-H. Yoon, S. A. DiBenedetto, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 1348–1349 CrossRef CAS PubMed ;
(g) S. C. B. Mannsfeld, B. C. K. Tee, R. M. Stoltenberg, C. V. H. H. Chen, S. Barman, B. V. O. Muir, A. N. Sokolov, C. Reese and Z. Bao, Nat. Mater., 2010, 9, 859–864 CrossRef CAS PubMed ;
(h) S. Shinamura, I. Osaka, E. Miyazaki, A. Nakao, M. Yamagishi, J. Takeya and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 5024–5035 CrossRef CAS PubMed ;
(i) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed ;
(j) H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed ;
(k) T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS PubMed ;
(l) S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed ;
(m) A. Cravino, S. Roquet, O. Alévêque, P. Leriche, P. Frère and J. Roncali, Chem. Mater., 2006, 18, 2584–2590 CrossRef CAS ;
(n) C. Videlot, J. Ackermann, P. Blanchard, J. M. Raimundo, P. Frère, M. Allain, R. de Bettignies, E. Levillain and J. Roncali, Adv. Mater., 2003, 15, 306–310 CrossRef CAS .
-
(a) R. A. Street, J. E. Northrup and A. Salleo, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 165202 CrossRef ;
(b) R. J. Kline, D. M. DeLongchamp, D. A. Fischer, E. K. Lin, M. Heeney, I. McCulloch and M. F. Toney, Appl. Phys. Lett., 2007, 90, 062117 CrossRef ;
(c) A. Salleo, Mater. Today, 2007, 10, 38–45 CrossRef CAS .
-
(a) H. Sirringhaus, N. Tessler and R. H. Friend, Science, 1998, 280, 1741–1744 CrossRef CAS PubMed ;
(b) H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS ;
(c) S. Y. Son, Y. Kim, J. Lee, G.-Y. Lee, W.-T. Park, Y.-Y. Noh, C. E. Park and T. Park, J. Am. Chem. Soc., 2016, 138, 8096–8103 CrossRef CAS PubMed ;
(d) M. J. Panzer and C. D. Frisbie, Adv. Funct. Mater., 2006, 16, 1051–1056 CrossRef CAS ;
(e) Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108–4110 CrossRef CAS .
-
(a) B. S. Ong, Y. Wu, P. Liu and S. Gardner, J. Am. Chem. Soc., 2004, 126, 3378–3379 CrossRef CAS PubMed ;
(b) N. Zhao, G. A. Botton, S. Zhu, A. Duft, B. S. Ong, Y. Wu and P. Liu, Macromolecules, 2004, 37, 8307–8312 CrossRef CAS ;
(c) B. S. Ong, Y. Wu, P. Liu and S. Gardner, Adv. Mater., 2005, 17, 1141–1144 CrossRef CAS .
-
(a) M. L. Chabinyc, M. F. Toney, R. J. Kline, I. McCulloch and M. Heeney, J. Am. Chem. Soc., 2007, 129, 3226–3237 CrossRef CAS PubMed ;
(b) Z. Fei, P. Pattanasattayavong, Y. Han, B. C. Schroeder, F. Yan, R. J. Kline, T. D. Anthopoulos and M. Heeney, J. Am. Chem. Soc., 2014, 136, 15154–15157 CrossRef CAS PubMed ;
(c) I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed ;
(d) Y. Li, Y. Wu, P. Liu, M. Birau, H. Pan and B. S. Ong, Adv. Mater., 2006, 18, 3029–3032 CrossRef CAS ;
(e) M. Heeney, C. Bailey, K. Genevicius, M. Shkunov, D. Sparrowe, S. Tierney and I. McCulloch, J. Am. Chem. Soc., 2005, 127, 1078–1079 CrossRef CAS PubMed .
-
(a) H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed ;
(b) J. Rivnay, M. F. Toney, Y. Zheng, I. V. Kauvar, Z. Chen, V. Wagner, A. Facchetti and A. Salleo, Adv. Mater., 2010, 22, 4359–4363 CrossRef CAS PubMed ;
(c) H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. Shakya Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed ;
(d) J. Lee, A. R. Han, J. Kim, Y. Kim, J. H. Oh and C. Yang, J. Am. Chem. Soc., 2012, 134, 20713–20721 CrossRef CAS PubMed ;
(e) J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed ;
(f) S. G. Bucella, A. Luzio, E. Gann, L. Thomsen, C. R. McNeill, G. Pace, A. Perinot, Z. Chen, A. Facchetti and M. Caironi, Nat. Commun., 2015, 6, 8394 CrossRef CAS PubMed .
-
(a) X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 2238 Search PubMed ;
(b) W. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS PubMed .
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed .
-
(a) Q. Zheng, B. J. Jung, J. Sun and H. E. Katz, J. Am. Chem. Soc., 2010, 132, 5394–5404 CrossRef CAS PubMed ;
(b) L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697–9702 CrossRef CAS PubMed ;
(c) I. Osaka, T. Kakara, N. Takemura, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 8834–8837 CrossRef CAS PubMed ;
(d) J.-S. Wu, C.-T. Lin, C.-L. Wang, Y.-J. Cheng and C.-S. Hsu, Chem. Mater., 2012, 24, 2391–2399 CrossRef CAS ;
(e) S.-W. Cheng, C.-E. Tsai, W.-W. Liang, Y.-L. Chen, F.-Y. Cao, C.-S. Hsu and Y.-J. Cheng, Macromolecules, 2015, 48, 2030–2038 CrossRef CAS ;
(f) H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS PubMed ;
(g) H.-H. Chang, C.-E. Tsai, Y.-Y. Lai, W.-W. Liang, S.-L. Hsu, C.-S. Hsu and Y.-J. Cheng, Macromolecules, 2013, 46, 7715–7726 CrossRef CAS ;
(h) P. M. Burrezo, X. Zhu, S.-F. Zhu, Q. Yan, J. T. López Navarrete, H. Tsuji, E. Nakamura and J. Casado, J. Am. Chem. Soc., 2015, 137, 3834–3843 CrossRef CAS PubMed ;
(i) H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed .
-
(a) Y. Li, J. Ding, M. Day, Y. Tao, J. Lu and M. D'Iorio, Chem. Mater., 2004, 16, 2165–2173 CrossRef CAS ;
(b) M. Ranger, D. Rondeau and M. Leclerc, Macromolecules, 1997, 30, 7686–7691 CrossRef CAS ;
(c) D. Marsitzky, M. Klapper and K. Müllen, Macromolecules, 1999, 32, 8685–8688 CrossRef CAS ;
(d) J. Yang, C. Jiang, Y. Zhang, R. Yang, W. Yang, Q. Hou and Y. Cao, Macromolecules, 2004, 37, 1211–1218 CrossRef CAS ;
(e) S. H. Lee, T. Nakamura and T. Tsutsui, Org. Lett., 2001, 3, 2005–2007 CrossRef CAS PubMed ;
(f) K.-T. Wong, Y.-Y. Chien, R.-T. Chen, C.-F. Wang, Y.-T. Lin, H.-H. Chiang, P.-Y. Hsieh, C.-C. Wu, C. H. Chou, Y. O. Su, G.-H. Lee and S.-M. Peng, J. Am. Chem. Soc., 2002, 124, 11576–11577 CrossRef CAS PubMed ;
(g) J. Jacob, S. Sax, T. Piok, E. J. W. List, A. C. Grimsdale and K. Müllen, J. Am. Chem. Soc., 2004, 126, 6987–6995 CrossRef CAS PubMed ;
(h) U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS .
-
(a) J.-S. Wu, Y.-J. Cheng, M. Dubosc, C.-H. Hsieh, C.-Y. Chang and C.-S. Hsu, Chem. Commun., 2010, 46, 3259–3261 RSC ;
(b) C.-Y. Chang, Y.-J. Cheng, S.-H. Hung, J.-S. Wu, W.-S. Kao, C.-H. Lee and C.-S. Hsu, Adv. Mater., 2012, 24, 549–553 CrossRef CAS PubMed .
- C.-H. Lee, Y.-Y. Lai, S.-W. Cheng and Y.-J. Cheng, Org. Lett., 2014, 16, 936–939 CrossRef CAS PubMed .
-
(a) A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 2180–2185 CrossRef CAS PubMed ;
(b) I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed ;
(c) J. Shin, G. E. Park, D. H. Lee, H. A. Um, T. W. Lee, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2015, 7, 3280–3288 CrossRef CAS PubMed ;
(d) M. S. Chen, O. P. Lee, J. R. Niskala, A. T. Yiu, C. J. Tassone, K. Schmidt, P. M. Beaujuge, S. S. Onishi, M. F. Toney, A. Zettl and J. M. J. Fréchet, J. Am. Chem. Soc., 2013, 135, 19229–19236 CrossRef CAS PubMed ;
(e) O. Knopfmacher, M. L. Hammock, A. L. Appleton, G. Schwartz, J. Mei, T. Lei, J. Pei and Z. Bao, Nat. Commun., 2014, 5, 2954 Search PubMed .
-
(a) C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed ;
(b) Z. Liu, G. Zhang, Z. Cai, X. Chen, H. Luo, Y. Li, J. Wang and D. Zhang, Adv. Mater., 2014, 26, 6965–6977 CrossRef CAS PubMed ;
(c) J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed ;
(d) C. Cheng, C. Yu, Y. Guo, H. Chen, Y. Fang, G. Yu and Y. Liu, Chem. Commun., 2013, 49, 1998–2000 RSC ;
(e) J. Li, Y. Zhao, H. S. Tan, Y. Guo, C.-A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed ;
(f) Y. Qiao, Y. Guo, C. Yu, F. Zhang, W. Xu, Y. Liu and D. Zhu, J. Am. Chem. Soc., 2012, 134, 4084–4087 CrossRef CAS PubMed .
- X. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073–15084 CrossRef CAS PubMed .
- W.-Y. Lee, G. Giri, Y. Diao, C. J. Tassone, J. R. Matthews, M. L. Sorensen, S. C. B. Mannsfeld, W.-C. Chen, H. H. Fong, J. B. H. Tok, M. F. Toney, M. He and Z. Bao, Adv. Funct. Mater., 2014, 24, 3524–3534 CrossRef CAS .
- J. Mei, H.-C. Wu, Y. Diao, A. Appleton, H. Wang, Y. Zhou, W.-Y. Lee, T. Kurosawa, W.-C. Chen and Z. Bao, Adv. Funct. Mater., 2015, 25, 3455–3462 CrossRef CAS .
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425–2427 CrossRef CAS PubMed .
- S. Wang, A. Kiersnowski, W. Pisula and K. Müllen, J. Am. Chem. Soc., 2012, 134, 4015–4018 CrossRef CAS PubMed .
-
(a) S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 461–462, 1–21 CrossRef CAS ;
(b) T. Vreven, K. Morokuma, Ö. Farkas, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 2003, 24, 760–769 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04129a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Jhih-Yang
Hsu
a,
Po-Kai
Huang
a and
Yen-Ju
Cheng
b,
Jhih-Yang
Hsu
a,
Po-Kai
Huang
a and
Yen-Ju
Cheng