
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereodivergent, Diels–Alder-initiated organocascades employing α,β-unsaturated acylammonium salts: scope, mechanism, and application†
Mikail E.
Abbasov
a,
Brandi M.
Hudson
 b,
Dean J.
Tantillo
*b and
Daniel
Romo
*a
b,
Dean J.
Tantillo
*b and
Daniel
Romo
*a
aDepartment of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, Texas 76798, USA
bDepartment of Chemistry, University of California-Davis, One Shields Avenue, Davis, California 95616, USA. E-mail: djtantillo@ucdavis.edu
First published on 25th October 2016
Abstract
Chiral α,β-unsaturated acylammonium salts are novel dienophiles enabling enantioselective Diels–Alder-lactonization (DAL) organocascades leading to cis- and trans-fused, bicyclic γ- and δ-lactones from readily prepared dienes, commodity acid chlorides, and a chiral isothiourea organocatalyst under mild conditions. We describe extensions of stereodivergent DAL organocascades to other racemic dienes bearing pendant secondary and tertiary alcohols, and application to a formal synthesis of (+)-dihydrocompactin is described. A combined experimental and computational investigation of unsaturated acylammonium salt formation and the entire DAL organocascade pathway provide a rationalization of the effect of Brønsted base additives and enabled a controllable, diastereodivergent DAL process leading to a full complement of possible stereoisomeric products. Evaluation of free energy and enthalpy barriers in conjunction with experimentally observed temperature effects revealed that the DAL is a rare case of an entropy-controlled diastereoselective process. NMR analysis of diene alcohol–Brønsted base interactions and computational studies provide a plausible explanation of observed stabilization of exo transition-state structures through hydrogen-bonding effects.
Introduction
The Diels–Alder (DA) cycloaddition is arguably one of the most useful transformations in organic synthesis for the rapid introduction of complexity including stereochemical information.1–3 The rich history,2 utility, simplicity of operation, and continued evolution of strategies that broaden the scope and improve the stereoselectivity of the venerable DA reaction makes this cycloaddition arguably the most versatile and powerful transform in chemical synthesis.3 In particular, catalytic asymmetric DA reactions are unparalleled in their ability to rapidly and efficiently generate optically active, architecturally complex, and densely functionalized heterocycles and carbocycles from simple achiral substrates. Furthermore, enantioselective organocatalytic DA variants have recently been established using iminium,4,5 enamine,6,7 bifunctional acid–base,8 and hydrogen-bonding catalysis.9,10 MacMillan and co-workers employed both α,β-unsaturated aldehydes4 and ketones5 in DA cycloadditions through iminium-activated chiral dienophiles 2, whereas unsaturated aldehydes9 and indolinones10 were activated through hydrogen-bonding catalysis (3) by Rawal and Barbas, respectively (Fig. 1a). Two extensions of the DA reaction that have received little attention are strategies for accessing the full complement of stereoisomers of a given cycloadduct and stereodivergent DA cycloadditions.11–19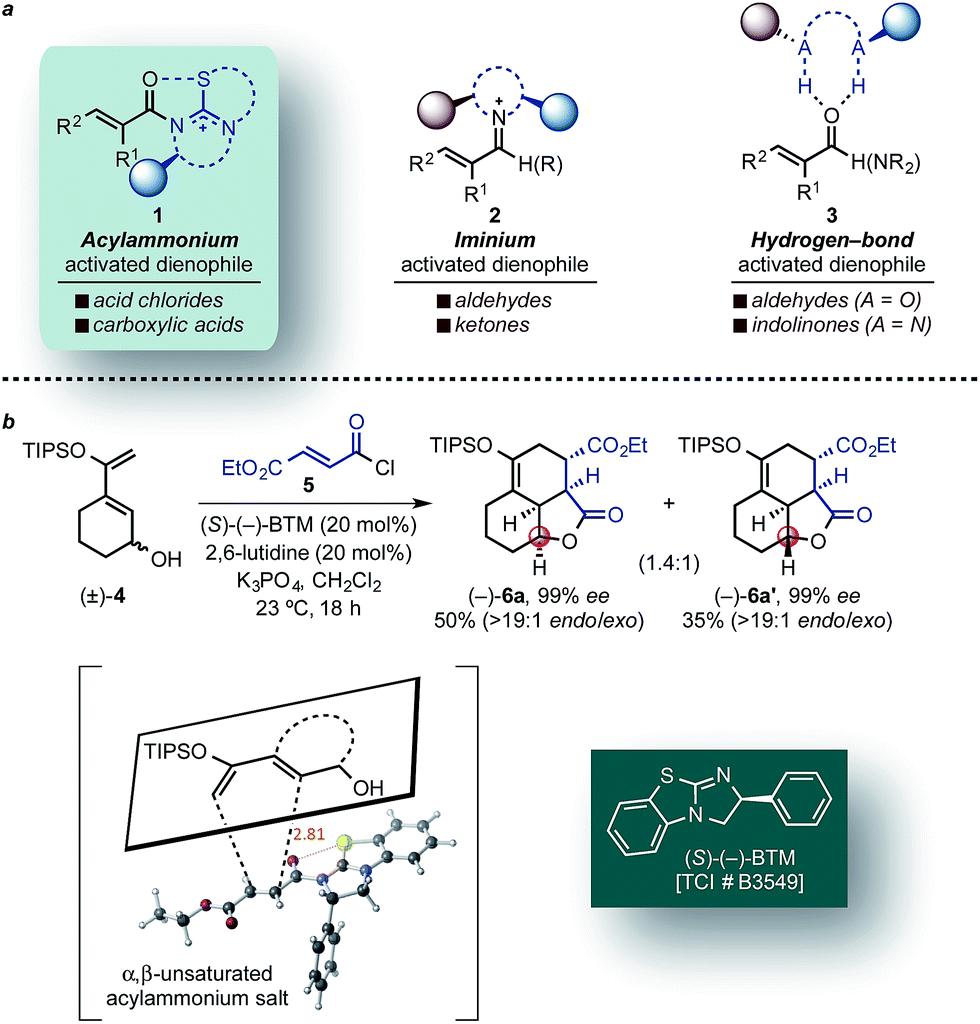 | ||
| Fig. 1 (a) Representative activation modes of α,β-unsaturated carbonyl compounds for organocatalytic asymmetric DA reactions: acylammonium salt- (this work), iminium-, and hydrogen bond-activated dienophiles. (b) Representative example of a DA-mediated, stereodivergent resolution of a racemic diene (e.g. (±)-4) employing in situ generated α,β-unsaturated acylammonium salt from acid chlorides (e.g. 5) and isothiourea catalysts (e.g. (S)-(−)-BTM) (inset, previously reported TS arrangement). | ||
The development of organic transformations that provide access to a full stereochemical array of a particular adduct bearing multiple stereocenters, i.e. diastereodivergent processes, remains a notable challenge in chemical synthesis.20 Such processes have impact beyond the realm of synthetic chemistry given that biological properties of organic molecules correlate to their three-dimensional architecture and thus the relative and absolute stereochemical configuration of each stereocenter in a molecule.21 In particular, the ability to access all stereoisomers of a target molecule allows evaluation of stereochemical structure–activity relationships. To date, this has been realized through a limited set of reactions including conjugate addition,12 Mannich reaction,13,14 intramolecular allylic substitution,15 deracemization,16 thio-Michael addition,17 hydrohydroxyalkylation,18 and α-allylation of aldehydes.19 The complex stereoselectivity issues inherent to the venerable DA cycloaddition leading to cycloadducts with up to four stereocenters makes this a challenging reaction to develop into a highly selective diastereodivergent process that would enable access to all possible diastereomers. While enantiomeric cycloadducts are readily obtained using the optical antipode of catalysts, altering diastereoselectivity through relative stereochemical control in DA cycloadditions is a much more challenging endeavour.
Synthetic methods that efficiently transform racemic mixtures into complex, enantioenriched products are important components of modern organic chemistry but remain scarce.1–3 These include underutilized stereodivergent processes, which convert racemic starting materials to non-enantiomeric products (e.g. diastereomers).11 Catalytic asymmetric variants of these reactions with racemic substrates represent a relatively unexploited strategy toward accessing a full complement of stereoisomers. Sarpong described an elegant example of a stereodivergent process applied to natural product synthesis;22 however, the majority of these reactions suffer from the crucial, practical issue of inseparable, diastereomeric products.11
We recently reported a novel class of dienophiles that utilize covalent,23 organocatalytic activation of α,β-unsaturated acid chlorides and carboxylic acid tosyl anhydrides enabling an asymmetric Diels–Alder-lactonization (DAL) organocascade.24 Importantly, this was the first example of a DA-initiated, stereodivergent organocascade (Fig. 1b) delivering complex and stereochemically diverse scaffolds found in bioactive compounds with excellent relative and absolute stereocontrol. Herein, we describe additional examples of stereodivergent DAL processes leading to complex, polycyclic adducts, new applications toward formal syntheses of natural products or core structures, and the potential of this reaction for diversity-oriented synthesis through diastereodivergent DAL processes. The latter is made possible by the discovery that diastereoselectivity of the DAL can be altered by judicious choice of Brønsted-base additive. Finally, we provide evidence that these organocascades may be rare examples of reactions in which diastereoselectivity is entropy-driven.
The potential of α,β-unsaturated acylammonium salts as catalytic intermediates (Fig. 2)25,26 was first demonstrated by Fu employing α,β-unsaturated acyl fluorides in a net [3 + 2] annulation promoted by a chiral 4-pyrrolidinopyridine catalyst.27 Building on this early work, the Lupton group reported an additional [3 + 2] cycloaddition,28 and the Smith group29 recently utilized α,β-unsaturated mixed anhydrides in an enantioselective tandem Michael-enol-lactonization. Furthermore, we demonstrated the full potential of chiral, triply reactive, α,β-unsaturated acylammonium salts derived from commodity acid chlorides for the rapid assembly of complex cyclopentanes through a nucleophile-catalyzed Michael-aldol-β-lactonization organocascade (NCMAL).30 Optically active γ-lactams and piperidones could also be prepared through a Michael-proton transfer-lactamization (NCMPL)31 process utilizing these intermediates. Most recently, Matsubara described the first example of a highly enantioselective net [4 + 3] cycloaddition to afford 1,5-benzothiazepines by utilizing α,β-unsaturated acylammonium intermediates generated by a chiral isothiourea catalyst.32
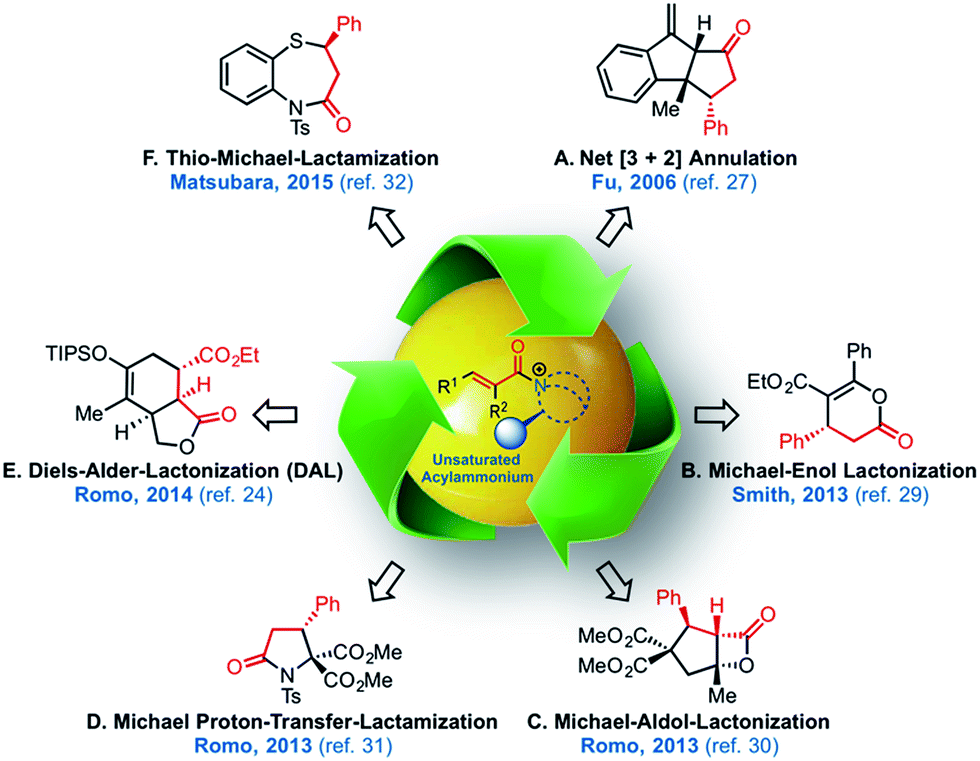 | ||
| Fig. 2 The expanding utility of α,β-unsaturated acylammonium salt-mediated, organocascade catalysis. | ||
The continued interest in diversity-oriented synthesis (DOS) to access structurally complex and diverse small-molecule libraries33 is premised on its value for drug discovery,34–36 chemical genetics37 and identification of small-molecule modulators of challenging biological targets.38,39 In particular, synthetic methods that rapidly generate stereochemical complexity40,41 are important for drug lead discovery and a recent success for drug development is exemplified by the antimalarial agent, NITD609, currently in phase IIa clinical trials.42,43 Furthermore, natural product-inspired libraries are beginning to provide good success rates in identifying more potent and drug-like molecules.44,45 As described herein, the use of a chiral isothiourea catalyst capable of exercising high relative and absolute stereocontrol in DAL organocascade processes in combination with a Brønsted base, which was found to profoundly impact endo/exo selectivity, points to the potential of accessing all possible diastereomers through a DA cycloaddition which may find utility in DOS.24
Our interest in understanding the origins of Lewis base catalyst-Brønsted base synergy in DAL organocascades led us to undertake both experimental and computational studies. Most known asymmetric reactions possess temperature-dependent diastereodifferentiation and thus are performed at low temperatures to maximize the impact of ΔΔH‡ induced by steric repulsion, structural strain, or electronic interactions in the transition state. From a synthetic perspective, entropy-controlled asymmetric transformations with sufficient ΔΔS‡ are preferable due to their temperature independence, however entropy-driven, diastereoselective reactions are rare.46–52
Results and discussion
Stereodivergent Diels–Alder-lactonization organocascades
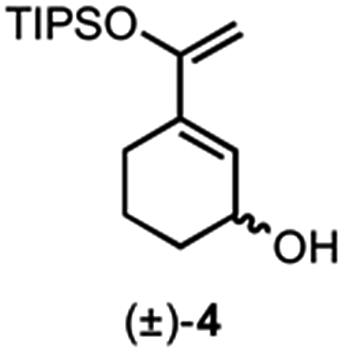
We previously reported a single example of a stereodivergent DAL organocascade with a cyclohexyl hydroxydiene (±)-4 (see Fig. 1b). To further explore the scope of stereodivergent DAL processes, we targeted additional complex, bicyclic γ-lactones under conditions previously described.24 These are ubiquitous structural motifs found in bioactive natural products that could be accessed in a single operation through the DAL cascade (Fig. 3a). Previous strategies toward this class of optically active complex, bicyclic γ-lactones include exo-selective intramolecular DA cycloadditions with optically active dienyl esters 10,53 for example those obtained by esterification with optically active dienyl alcohols 7 or through enzymatic resolution of racemic esters 10 by transesterification (Fig. 3b).54 We envisioned the use of racemic dienes bearing a pendant, stereogenic carbinol (e.g (±)-7, R6 ≠ R7, blue circles) for a terminating lactonization step for stereodivergent DAL cascades introducing up to four additional stereocenters through catalyst control independent of the existing carbinol center. The lactonization step would deliver diastereomeric cycloadducts 11 with distinct topologies that could facilitate separation (Fig. 3). | ||
| Fig. 3 (a) Selected structures of naturally occurring and biologically active terpenoids containing γ-substituted, cis-fused bicyclic-γ-lactones. (b) Comparison of conventional strategies employing optically active dienyl alcohols 7 or enzymatic resolution and the described DAL organocascade to access bicyclic γ- and δ-lactones 11 (n = 0, 1). Use of racemic dienes (±)-7 bearing a pendant carbinol stereocenter (blue circle) enables a stereodivergent organocascade that introduces up to four additional stereocenters (blue asterisk) through catalyst control independent of the resident carbinol stereocenter. | ||
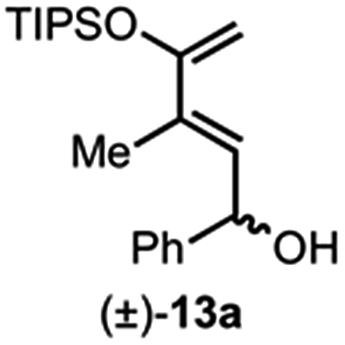
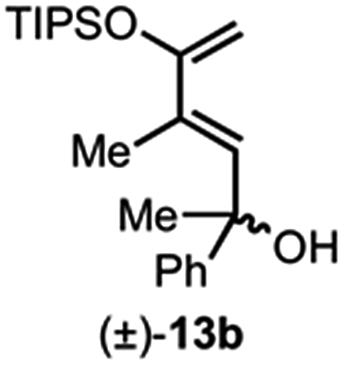
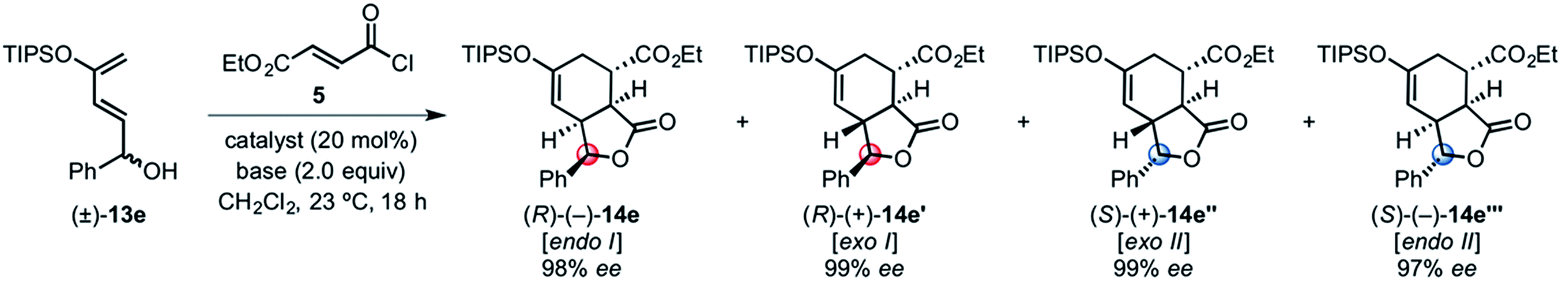
Building on our previous studies of stereodivergent DAL organocascades,24 we investigated additional racemic silyloxydienes (±)-13a–c and the previously described diene (±)-4 (ref. 24) bearing pendant, secondary alcohols (Table 1). Ethyl fumaroyl chloride (5) was employed as dienophile given its higher reactivity and (S)-(−)-BTM was used as the chiral Lewis base. Use of silyloxydiene (±)-13a delivered a readily separable 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture of bicyclic γ-lactones (−)-14a (99% ee) and (+)-14a′ (98% ee) in 48% and 31% yield, respectively (Table 1, entry 1). Similarly, diene (±)-13b bearing a pendant, tertiary benzylic alcohol afforded a separable 1.8:1 diastereomeric mixture of cycloadducts (+)-14b (41% yield, 99% ee) and (+)-14b′ (23% yield, 99% ee) bearing four contiguous stereocenters, including a quaternary carbon on gram-scale (entry 2).
:1 diastereomeric mixture of bicyclic γ-lactones (−)-14a (99% ee) and (+)-14a′ (98% ee) in 48% and 31% yield, respectively (Table 1, entry 1). Similarly, diene (±)-13b bearing a pendant, tertiary benzylic alcohol afforded a separable 1.8:1 diastereomeric mixture of cycloadducts (+)-14b (41% yield, 99% ee) and (+)-14b′ (23% yield, 99% ee) bearing four contiguous stereocenters, including a quaternary carbon on gram-scale (entry 2).
|
|
|||
|---|---|---|---|
| Entry | Diene | Acid chloride | Cycloadducts % yield (endo:exo, % ee) |
| a Unless otherwise specified, all reactions were performed with diene (1.0 equiv.), acid chloride (1.5 equiv.), K3PO4 (3.0 equiv.), 2,6-lutidine (20 mol%), and (S)-(−)-BTM (20 mol%) at 23 °C for 18 h. Yields are based on isolated, purified cycloadducts. Enantiomeric excess was determined by chiral phase HPLC. | |||
| 1 |
|
12a |

|
| 2 |
|
12a |

|
| 3 |

|
12a |
|
| 4 |
|
12b |
|
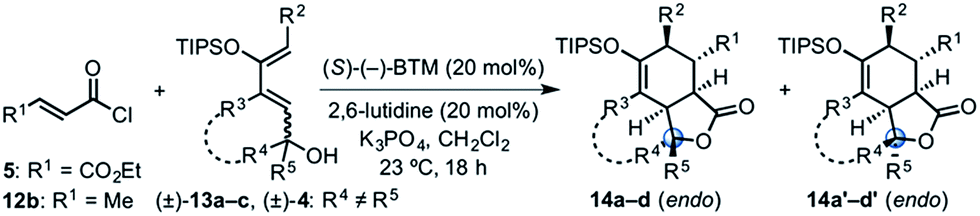
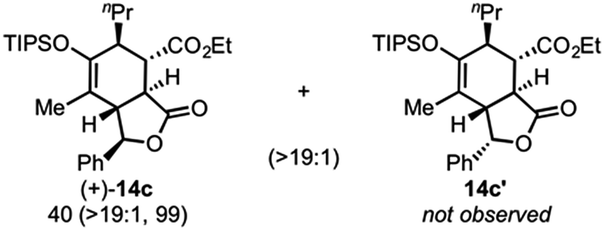
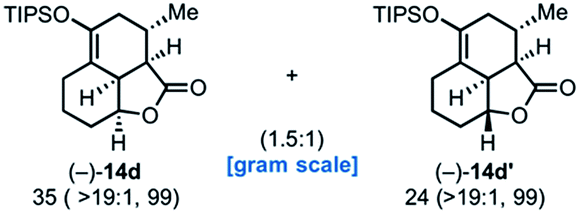
Use of racemic silyloxydiene (±)-13c possessing a (Z,Z)-configured diene provided trans-fused bicyclic γ-lactone (+)-14c as a single diastereomer with five contiguous stereocenters in 40% yield (99% ee) despite the cis-substituent that typically impedes effective cycloaddition (Table 1, entry 3).55 Presumably this diene leads to a kinetic resolution due to the presence of the endo-disposed phenyl substituent which may preclude terminating lactonization delivering the exo adduct 14c′. To the best of our knowledge, this is the first report of a catalytic DA cycloaddition with a cis-substituted diene that occurs at ambient temperature (23 °C) with high enantioselectivity.56 We again studied the racemic cyclohexanol diene (±)-4, but with the less reactive commodity dienophile, crotonoyl chloride (12b). This DAL process gave fused, tricyclic 6,6,5-adducts on gram-scale as separable diastereomers (−)-14d and (−)-14d′ in 35% (99% ee) and 24% yield (99% ee), respectively, from trans-β-methyl acryloyl chloride and the previously described diene (±)-4 (ref. 24) (entry 4). The relative and absolute configuration of crystalline cycloadduct (−)-14a was previously determined by X-ray analysis24 while cycloadduct (−)-14a′ required ring opening of γ-lactone with 4-bromobenzylamine (ESI, Fig. S2†). The relative and absolute configurations of cycloadducts 14a–d and 14a′–d′ was assigned premised on these analogous systems in conjunction with detailed NMR analysis. In general, low yields in these stereodivergent DAL reactions were due to desilylation of dienes at ambient temperature (23 °C) upon prolonged reaction times and competitive, irreversible esterification of the tethered alcohol moiety. Importantly, subsequent intramolecular DA reactions of the resulting esters do not proceed at ambient temperature, therefore potential non-selective background processes are precluded. Sterically demanding α,β- and β,β-disubstituted acid chlorides and non-silyloxy-substituted dienes do not undergo DAL organocascades under comparable conditions (see ESI, Table S1†).
Synthetic applications of the DAL organocascade process
Toward demonstrating the utility of the DAL process, we targeted the core structure of (+)-dihydrocompactin (Fig. 4), a potent hypocholesterolemic agent first isolated by a group at Merck.57 This natural product, along with mevinolin, is historically significant in providing the original information for eventual development of the well-known statin drugs, lovastatin (Mevacor®) and simvastatin (Zocor®). In one approach to dihydrocompactin by Hagiwara,58 the bicyclic ketodiol 15 was employed in racemic form. We recognized that the tricyclic lactone (−)-14d could be fashioned into this precursor to dihydrocompactin with a few additional synthetic manipulations. The sequence entailed reduction of lactone (−)-14d with LiAlH4 to the corresponding diol followed by desilylation of the silyl enol ether with tetrabutylammonium fluoride (TBAF). Under the basic conditions of the deprotection, thermodynamic equilibration occurred to provide the desired trans-decalin of the targeted ketodiol (+)-15 which previously served as a precursor to dihydrocompactin.58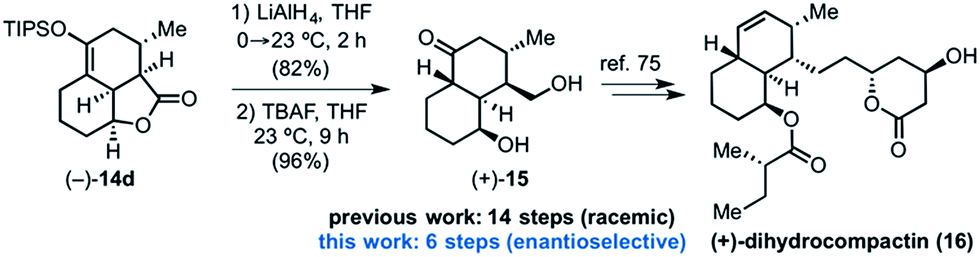 | ||
| Fig. 4 Conversion of the tricyclic γ-lactone (−)-14d to ketodiol (+)-15 representing a formal synthesis of (+)-dihydrocompactin (16). | ||
Effects of Brønsted base on acylammonium salt formation and initial Diels–Alder step
During our previous screening of Brønsted bases, we determined that certain tertiary amines exert a profound effect on the endo/exo selectivities of the DAL process. We reasoned that the Brønsted base may be playing a dual role of deprotonation of the pendant alcohol during lactonization through initial hydrogen-bonding and also ensuring the free-base form of the catalyst. However, certain tertiary amine Brønsted bases can also act as Lewis base catalysts leading to racemic product.59 Thus, computational studies were pursued to determine the extent to which various Brønsted bases could compete effectively with the optically active Lewis base leading to achiral acylammonium dienophiles and nonselective DAL processes. Results from quantum chemical calculations (see ESI† for details) of acylammonium salt formation between ethyl fumaroyl chloride (5) and various tertiary amine Brønsted bases indicate that only pyridine and Et3N, with predicted free energy barriers of 12.4 and 13.1 kcal mol−1, respectively, would plausibly compete with BTM (barrier of 13.0 kcal mol−1) (Fig. 5a). However, both reactions are endergonic, with energy barriers for reversion to the corresponding acid chlorides of only 7.1–9.5 kcal mol−1, and thus are readily reversible.24,60 | ||
| Fig. 5 (a) Comparison of free energies for acylammonium salt formation (I, DTBP; II, 2,6-lutidine; III, Hünig's base; IV, Et3N; V, (S)-(−)-BTM; VI, pyridine) between ethyl fumaroyl chloride (5) and various achiral amines and (S)-(−)-BTM. Free energies of transition state structures (TSSs) and products (shown in kcal mol−1 relative to free energies of separated reactants) computed using SMD(DCM)-M06-2X/6-31G(d). (b) Calculated TSSs (I–VI) for the formation of acylammonium salts with various Brønsted bases optimized at the M06-2X/6-31G(d) level with an implicit solvent model [SMD (dichloromethane)]. Selected bond distances are shown (Å). (c) Section of the 1H–13C gHMQC NMR spectrum of the acylammonium salt 17 in CD2Cl2 formed from a 1:1 mixture of (S)-(−)-BTM and ethyl fumaroyl chloride (5). | ||
With these computational results in hand, we next sought to provide experimental support through a brief screen of selected Brønsted bases employing diene 13d23 and acid chloride 5 with (S)-(−)-BTM as catalyst (Table 2). Triethylamine, pyridine and even Hünig's base (iPr2NEt) led to greatly reduced enantioselectivity (60–85% ee) compared to 2,6-lutidine (99% ee) and 2,6-di-tert-butylpyridine (DTBP, 99% ee). In that we predict that a nonselective background pathway involving achiral acylammonium salts would not compete effectively with the enantioselective DAL process, we suspect that the reduced enantioselectivity is due to the hydrogen-bonding effects discussed below. Interestingly, acylammonium salt formation through chloride ion exchange reactions with both (S)-(−)-BTM and tertiary-amine Brønsted bases were predicted to proceed by attack at the carbonyl carbon and displacement of chloride without formation of a tetrahedral intermediate as a minimum (Fig. 5b, I–VI). Instead, these reactions appear to proceed by a concerted SN2-type mechanism, consistent with previous computational studies indicating that some reaction coordinates for substitution of acyl derivatives lack minima corresponding to the tetrahedral intermediates expected for an addition–elimination process.61–63
|
|
||||
|---|---|---|---|---|
| Entry | Base | dea (%) | eeb,c (%) | Conversiond (%) |
| a Determined by 1H NMR (500 MHz) analysis of crude reaction mixtures. b Determined by chiral HPLC analysis (see ESI for details). c Enantiomeric excess of the major (−)-14f (endo) diastereomer (ee values for the exo diastereomer were similar). d Yields in parentheses refer to isolated, purified yields of cycloadducts. | ||||
| 1 | Et3N | 71 | 60 | >95(60) |
| 2 | Pyridine | 76 | 85 | >95(46) |
| 3 | Hünig's base [EtN(iPr)2] | 71 | 65 | >95(55) |
| 4 | 2,6-Lutidine | 60 | 99 | >95(68) |
| 5 | 2,6-Di-tert-butylpyridine [DTBP] | 95 | 99 | >95(43) |

We postulated previously that activation of the DA reaction upon acylammonium salt formation would originate from inductive effects propagated through the σ-framework, which could ultimately be revealed through reduced electron density at the β-carbon.64 We therefore performed a 1H–13C gHMQC experiment and measured the 13C NMR chemical shifts in CD2Cl2 at 23 °C for the acylammonium salt 17 formed through chloride ion exchange reaction of the acid chloride 5 with the Lewis base, (S)-(−)-BTM (Fig. 5c). However, no significant change in the chemical shift of the β-carbon of acylammonium 17 (δ 136.7 ppm) was observed compared to the acid chloride 5 (δ 136.8 ppm). Comparison of the chemical shift of the carbonyl carbons revealed only a slight upfield shift in acylammonium salt 17 (δ 163.6 ppm) compared to the acid chloride 5 (δ 164.1 ppm) possibly due to the close proximity of the phenyl ring of (S)-(−)-BTM to the carbonyl carbon.
Thus, isothiourea-catalyzed acylammonium formation may not lead to dramatic LUMO-lowering activation, but rather a significant decrease in the rate of intermolecular, nucleophilic substitution at the carbonyl carbon by the pendant alcohol of the diene, enabling the DA-initiated organocascade. In addition, a comparison of energy barriers for the initial DA step between the BTM-derived dienophile and various achiral Lewis base-derived dienophiles (e.g. pyridine, Et3N, 2,6-lutidine) indicates a lower barrier for the enantioselective DA cycloaddition compared to the background nonselective DA (ΔΔG‡ ∼ 13–43 kcal mol−1) and is consistent with the generally observed excellent enantioselectivities in the DAL process (for computed energies see ESI (Comp studies), Table S3†).
Effects of Brønsted base on the origins of the diastereoselectivity in the Diels–Alder-initiated cascades
Based on the aforementioned experimental and computational results suggesting that bulky Brønsted bases could not effectively compete with BTM as Lewis base promoter during the initial DA cycloaddition, we considered other roles that the Brønsted base could be serving during the cycloaddition. On the basis of previous studies of alcohol–amine complexes,65 we envisaged a hydrogen-bonded complex between these amino Brønsted bases and the alcohol moiety of the dienes employed. We postulated that evidence for such an interaction might be detected by 13C NMR through increased electron density at the carbinol-carbon of diene due to inductive effects propagated through the σ-framework. We performed standard 13C NMR (500 MHz) experiments in CD2Cl2 (0.1 M) at 23 °C and measured the changes in chemical shifts of the carbinol carbon of diene 13d (Fig. 6a) upon addition of an equimolar amount of Brønsted bases. Addition of 1.0 equiv. of DTBP to diene 13d leads to negligible change in the 13C NMR spectrum (Fig. 6b) indicating that under these conditions a hydrogen-bonded complex does not form or is highly reversible favouring a non-hydrogen bonded complex.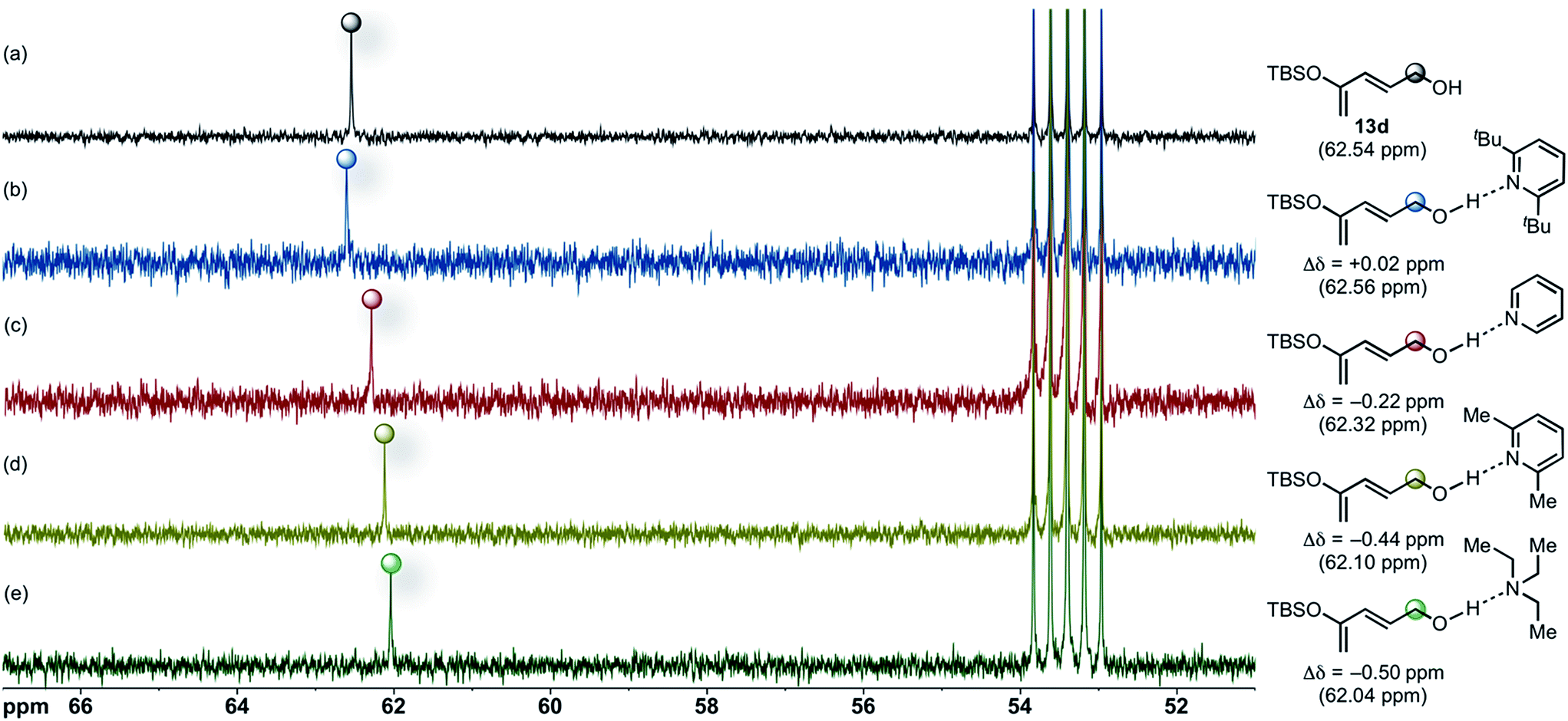 | ||
| Fig. 6 Partial 13C NMR (500 MHz, δ 51–67 ppm) spectra in CD2Cl2 at 23 °C of an equimolar mixture of diene 13d (a) and DTBP (b), pyridine (c), 2,6-lutidine (d), and Et3N (e). | ||
Furthermore, the chemical shift (62.56 ppm) of the carbinol-carbon (Δδ = +0.02) of diene 13d (62.54 ppm) is relatively unchanged upon addition of DTBP (Fig. 6a vs. b). The inability of the nitrogen atom in DTBP to participate in hydrogen-bonding is anticipated due to steric hindrance66 imposed by the adjacent tert-butyl substituents, and this is proposed to be largely responsible for its low relative basicity.67
In contrast, the 13C NMR spectrum of an equimolar mixture of diene 13d and pyridine shows a pronounced upfield chemical shift (Δδ = −0.22 ppm) for the carbinol carbon, supporting formation of a hydrogen-bonded complex (Fig. 6c). The extent of complexation was particularly evident in the 13C NMR spectrum of diene 13d with added 2,6-lutidine resulting in a significant upfield chemical shift (Δδ = −0.44 ppm) of the carbinol carbon (Fig. 6d) and likewise with Et3N (Δδ = −0.50 ppm). These upfield shifts qualitatively correlate with hydrogen-bond strength and Brønsted basicity. In particular, pKa values (in DMSO) and Δδ differences follow the order: DTBP (0.9,68–70 +0.02) < pyridine (3.4,70 −0.22) < 2,6-lutidine (4.46,69 −0.44) < Et3N (9.0,68 −0.50).
Based on the potential that hydrogen-bonded complexes may participate in this organocascade process, TSSs for the DA cycloaddition were recalculated with 2,6-lutidine complexed to silyloxydiene 13d (Fig. 7). A manual conformational search sampling numerous possible orientations of 2,6-lutidine resulted in the lowest energy endo and exo TSS conformations as shown in Fig. 7b. As indicated, 2,6-lutidine can indeed participate in hydrogen bonding with the terminal alcohol of the diene and simultaneously engage in CH–π and π–π stacking interactions with the benzotetramisole moiety of the BTM-bound acylammonium salt. These interactions lower the free energy barrier for the exo cycloaddition (12.0–10.3 kcal mol−1)71,72 to a greater extent than the endo cycloaddition (10.7–10.0 kcal mol−1), thereby leading to the prediction that such complexation would reduce diastereoselectivity as observed (95 vs. 60% de, DTBP vs. 2,6-lutidine, Table 2). Differences in relative stabilization of the DA TSSs by 2,6-lutidine are associated with the respective orientations of the BTM-bound acylammonium salt, 2,6-lutidine, and the terminal alcohol of the diene. In the exo TSS, 2,6-lutidine appears to be unable to maximize hydrogen-bonding, CH–π, and π–π stacking interactions simultaneously. The BTM-bound acylammonium salt and 2,6-lutidine are involved in a displaced π–π stacking interaction, whereas, in the exo TSS, π–π stacking of the BTM-bound acylammonium salt and 2,6-lutidine occurs over a larger area while allowing the base to maintain a hydrogen-bonding interaction with the terminal alcohol of the diene. While these complexation geometries bear on future design, the extremely similar predicted free energy barriers for endo and exo cycloadducts is not consistent with the magnitude of the experimentally observed diastereoselectivity (60% de, cf.Table 2), which prompted further investigations of the origins of this apparent discrepancy.
 | ||
| Fig. 7 (a) Free energies (normal text) and enthalpies (bold, italic) calculated at SMD(DCM)-M06-2X/6-31G(d) are in kcal mol−1 relative to separated reactant species shown. Values inside parentheses represent free energies without explicit base. An explicit base (2,6-lutidine) was modelled to study stereoelectronic effects on TSSs involved in the initial DA cycloaddition. (b) Optimized TSSs leading to endo and exo cycloadducts showing π–π stacking and CH–π interactions between the BTM-bound acylammonium salt and the hydrogen-bonded Brønsted base–diene complex. Selected bond distances are shown (Å). | ||
Entropy-controlled diastereodifferentiation in Diels–Alder-initiated cascades
While the predicted diastereoselectivity for the Brønsted base-free DA cycloaddition was found not to depend greatly on ΔS corrections, the BTM-promoted cycloaddition was found, based on calculations, to change significantly upon consideration of ΔH versus ΔG (Fig. 8a). Unfortunately, the direction and magnitude of this change varied with the level of theory used, likely a result of known difficulties in accurately computing entropy corrections73 and dispersion interactions.74 Nevertheless, these results led us to consider the possibility that the diastereoselectivity was not controlled by enthalpy (i.e. predicted ΔΔH‡s are insignificant), but rather by entropy.46–52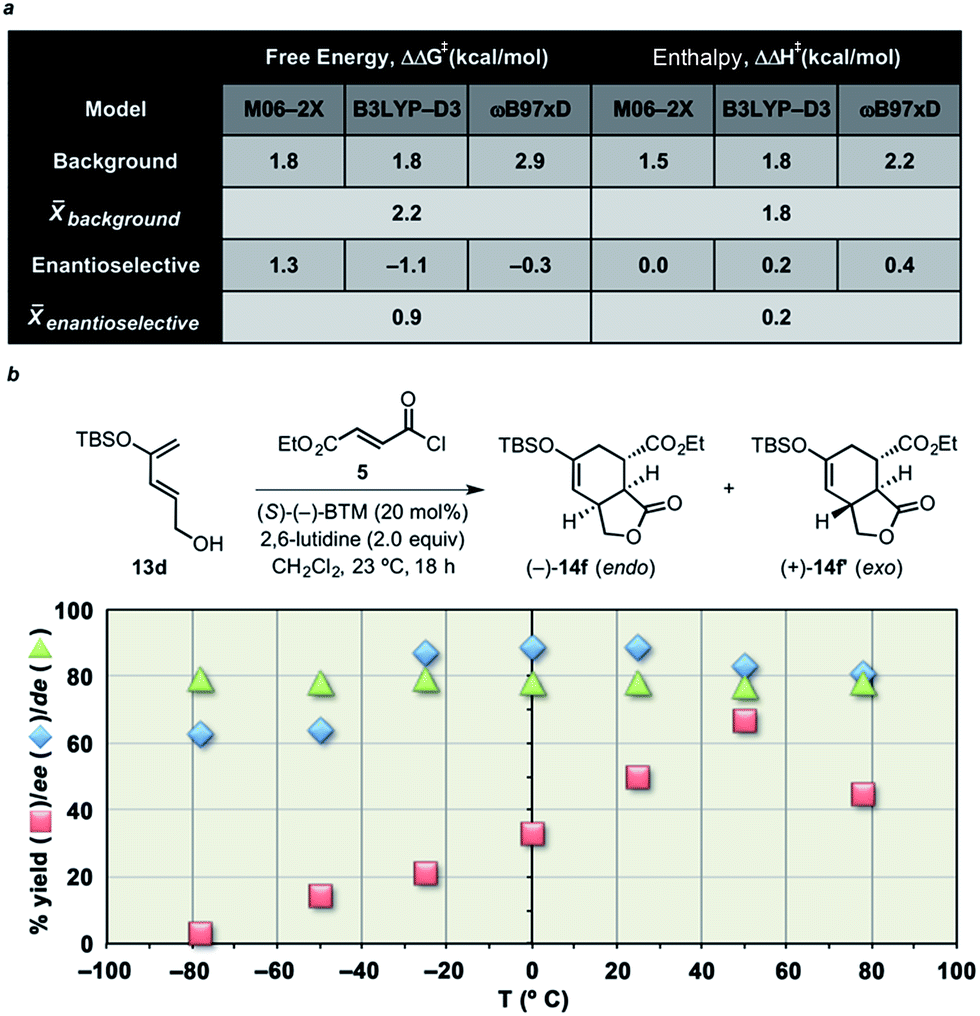 | ||
| Fig. 8 (a) Differences in free energies and enthalpies of TSSs from the non-selective background and enantioselective DA cycloadditions computed using multiple model chemistries and the 6-31G(d) basis set (see ESI† (Comp studies) for details). Energies shown in kcal mol−1 relative to separated reactants. (b) Plots of yield and enantiomeric and diastereomeric excess as a function of temperature for DAL reactions employing diene 13d and acid chloride 5. Enantiomeric excess was determined by chiral-phase HPLC and is only shown for the major (endo) diastereomer (ee values for the exo diastereomer were similar). | ||
We therefore investigated the enantio- and diastereoselectivity of the DAL organocascade of silyloxydiene 13d with ethyl fumaroyl chloride (5) over a wide range of temperatures (Fig. 8b). Plots of yield and enantio- and diastereomeric excess as a function of temperature are depicted in Fig. 8b. The data indicates the dominance of the background racemic reaction at the extremities of temperature, likely due to inefficient acylammonium formation at temperatures below −20 °C and adequately competent background reaction above +50 °C. In the case of diastereoselectivity in the temperature range −78 to +80 °C, the data clearly shows an invariance in diastereoselectivity. Reactions were analyzed after 18 h, and the relative endo/exo ratios and enantioselectivity were concurrently determined by chiral-phase HPLC of the crude reaction mixtures. The chemical yields were determined after flash chromatography on silica gel. The “flat” temperature dependency observed in this temperature range (ΔT ≈ 160 °C) provides evidence that the diastereoselectivity of the enantioselective DA cycloaddition (with or without modelled Brønsted base) is predominantly controlled by the differential activation entropy ΔΔS‡ rather than ΔΔH‡.
Computed potential energy surface for the lactonization step of the DAL
Plausible lactonization pathways are depicted in Fig. 9a. The results of our computations show that initial deprotonation of the alcohol leads, not surprisingly to attack on the acylammonium carbonyl with a low barrier (no barrier for the endo case; Fig. 9b). Again, well-defined tetrahedral intermediates are not found (a minimum for the endo case was found but it has a negligible barrier for fragmentation; Fig. 9b), consistent with a concerted but highly asynchronous lactonization reaction.75–77 The lactonization pathway involving attack of the neutral alcohol on the acylammonium salt intermediate showed this path is unlikely given the computed free energy barrier of ∼26 kcal mol−1; attempts to model this stepwise nucleophilic addition of the alcohol and subsequent deprotonation by 2,6-lutidine proved unsuccessful. Due to the difficulty of accurately modelling deprotonation/protonation steps,73 especially considering the presence of base, shuttle base, and solvent in solution, full pathways proposed in Fig. 9a were not computed; instead, ketene and enolate intermediates resulting from proposed pathways were optimized and their relative energies compared. The ketene intermediate (+ BTM) was found to be ∼17 kcal mol−1 higher in energy than the initial DA adduct (pathway a), making this pathway energetically feasible, with deprotonation and ketene formation with concomitant catalyst regeneration likely occurring in a concerted process (see ESI, Scheme S3†). The enolate intermediate was found to be ∼7 kcal mol−1 lower in energy than the deprotonated alcohol of the DA adduct (INT1, Fig. 9b, pathway b), making enolate formation also energetically feasible. However, both the ketene pathway and intramolecular proton transfer pathway would result in scrambling of the stereochemistry, which was not experimentally observed; therefore, it is likely that deprotonation and lactone formation occur in a coordinated process (pathway c).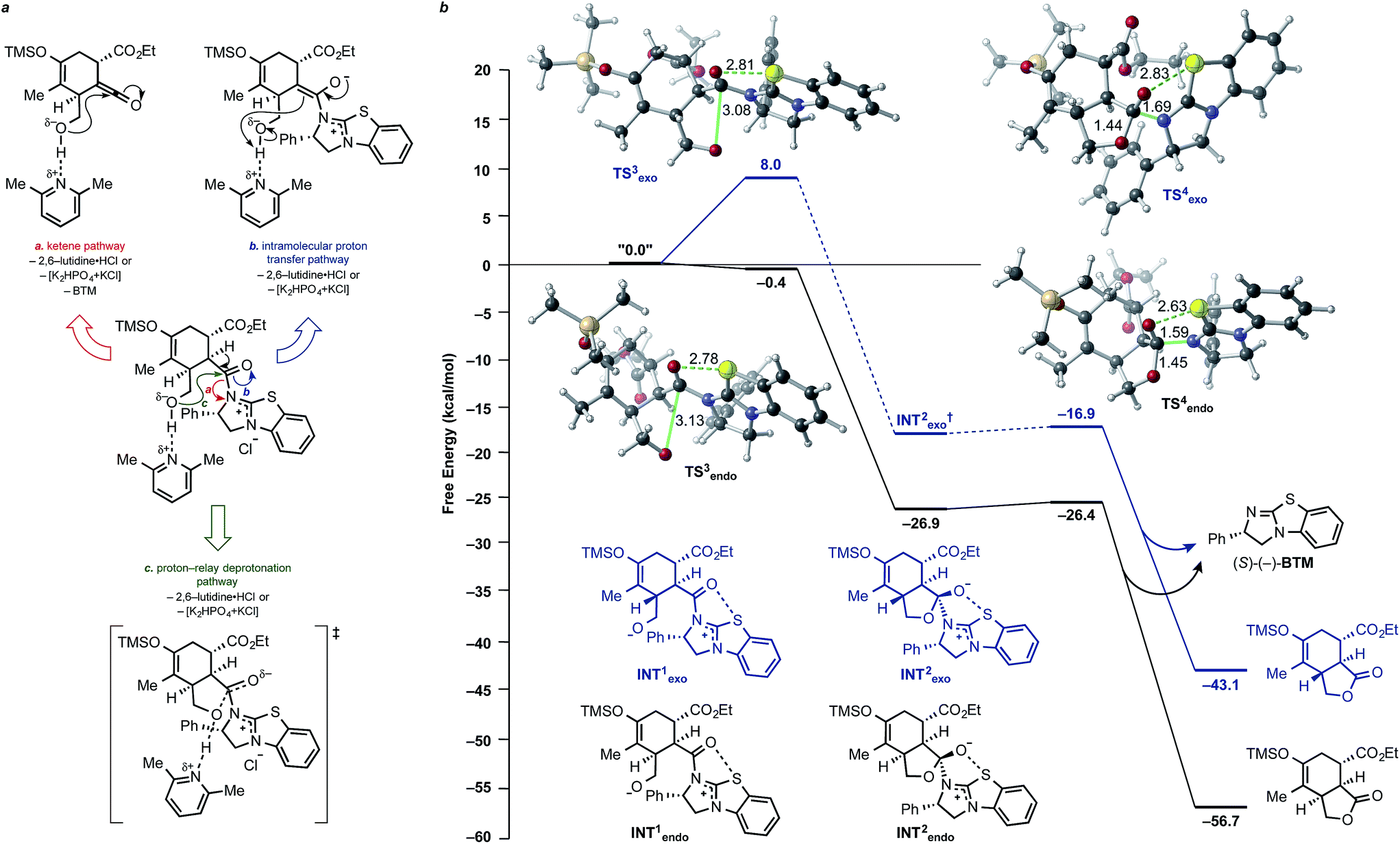 | ||
| Fig. 9 (a) Possible lactonization pathways for the DAL organocascade process. (b) Computed potential energy surface for the lactonization step of the DAL. Free energies (kcal mol−1) were calculated employing SMD(DCM)-M06-2X/6-31G(d). Intermediates and optimized TSSs for both endo (black) and exo (blue) pathways are shown. Selected bond distances are shown (Å). | ||
Switching diastereoselection: toward achieving the full complement of possible stereoisomeric products in a Diels–Alder cycloaddition
Based upon aforementioned computations suggesting selective stabilization of the exo TSS by CH–π and π–π stacking interactions between the Brønsted and Lewis bases employed, we reasoned that installation of an electron-withdrawing substituent at the C7 position of the benzothiazole moiety of BTM might enhance these interactions.78,79 Conversely, an electron-donating substituent also would be expected to impact the interaction, thus altering the endo/exo selectivity. In addition, a closer look at the optimized TSSs revealed the potential for an n → π* interaction80–83 between the hydroxyl group and imidazolium cation in both exo (3.04 Å) and endo (2.79 Å) TSSs (Fig. 10a).84–86 | ||
| Fig. 10 (a) Catalysts designed to potentially alter diastereoselection in DAL organocascades, and TSSs depicting potential stabilization by n → π* interaction optimized with SMD(DCM)-M06-2X/6-31G(d), modelled with an explicit Brønsted base (2,6-lutidine). Selected bond distances are shown (Å). (b) Preparative synthesis of electronically tuned BTM-based catalysts. | ||
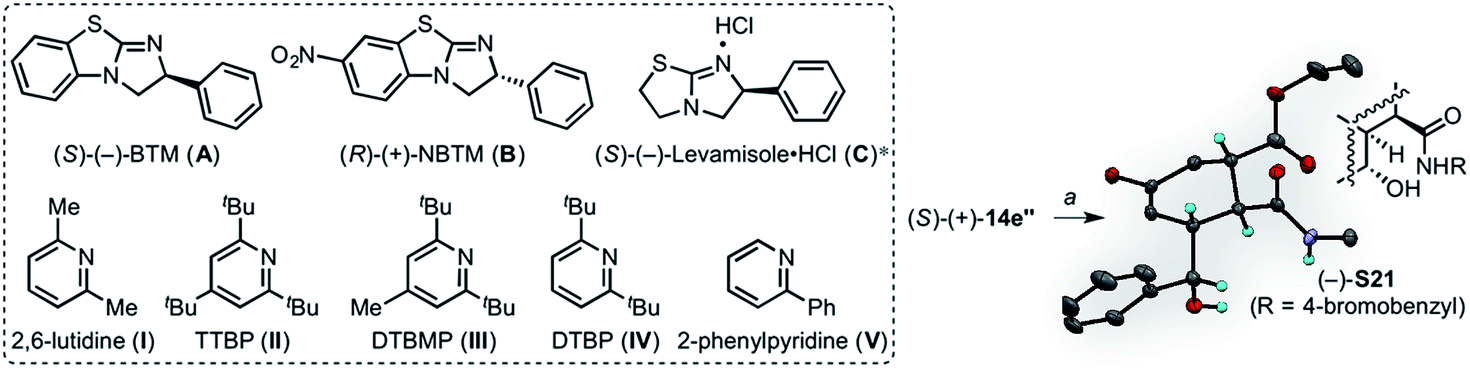
We chose to examine a highly electron-withdrawing nitro group and an electron-donating pyrrolidinyl group, reminiscent of 4-pyrrolidinopyridine (4-PPY) (Fig. 10). The synthesis of these catalysts commenced with nitration of 2-chlorobenzothiazole (18) with a mixture of concentrated sulfuric acid and fuming nitric acid to provide the nitrothiazole 19,87 which was used directly in the next step without purification. Employing Smith's recently improved, scalable two-step protocol,88 nitrothiazole 20 was subjected to condensation with (R)-phenylglycinol in neat diisopropylethylamine to furnish alcohol 20, which was directly treated with methanesulfonyl chloride. Refluxing a dichloromethane solution of the resultant mesylate in the presence of triethylamine and methanol overnight provided 7-nitrobenzotetramisole, (R)-(+)-NBTM in 36% yield (over 3 steps). The nitro group was reduced with iron powder in ethanol to afford the corresponding amino benzene derivative, which upon treatment with 2,5-dimethoxytetrahydrofuran (DMTHF) and sodium borohydride underwent reductive amination to provide the desired pyrrolidinylbenzotetramisole, (R)-(+)-PBTM in 75% yield (over 2 steps).
Given the ability to access both endo and exo transition states using particular Brønsted bases, we studied the possibility of a fully stereodivergent version of the DAL with a racemic diene to access all possible stereoisomers of a particular family of cycloadducts. Employing racemic silyloxydiene (±)-13e bearing a pendant secondary benzylic alcohol, ethyl fumaroyl chloride (5), and (S)-(−)-BTM (20 mol%) and 2,6-lutidine (2.0 equiv.), four separable diastereomers (−)-14 (27% yield, 98% ee), (+)-14′ (22% yield, 99% ee), (+)-14′′ (25% yield, 99% ee) and (−)-14′′′ (18% yield, 97% ee) in 92% combined yield were produced (entry 1, Table 3). This reaction could be performed on a preparative scale with only 10 mol% (S)-(−)-BTM providing 76% yield of these diastereomers.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | (R)-(−)-14ea (%) | (R)-(+)-14e′a (%) | (S)-(+)-14e′′a (%) | (S)-(−)-14e′′′a (%) |
| a Yields and diastereomeric ratios are based on isolated, purified cycloadducts. Enantiomeric excess was determined by chiral phase-HPLC. *Employed in free-base form. Inset is a single crystal X-ray structure in ORTEP format of adduct (R)-(+)-14′′ following ring opening with 4-bromobenzylamine (50% probability; TIPS and 4-bromobenzyl groups are removed for clarity, see ESI Fig. S1). a4-BrC6H4CH2NH2, THF, 23 °C, 36 h (46%). | ||||||
| 1 | A | I | 27 | 22 | 25 | 18 |
| 2 | A | II | 24 | — | 23 | 15 |
| 3 | A | III | 27 | — | — | 21 |
| 4 | A | IV | 29 | — | 20 | — |
| 5 | B | V | — | — | 26 | 19 |
| 6 | B | IV | — | — | 18 | — |
| 7 | C | IV | 22 | — | — | — |
|
||||||
The use of commercially available pyridines as Brønsted bases bearing electron-withdrawing substituents, such as 2- and 3-bromopyridine, and 2,6-dibromopyridine, were ineffective in delivering cycloadducts, presumably due to their reduced basicity. In contrast, pyridines with electron-donating groups, such as 3- and 4-methoxypyridine delivered cycloadducts, but led to negligible changes in diastereoselectivity. On the other hand, 2,4,6-tri-tert-butylpyridine (TTBP) selectively suppressed formation of the exo I diastereomer, (+)-14′ (entry 2, Table 3), whereas 2,6-di-tert-butyl-4-methylpyridine (DTBMP) deterred formation of both exo I and II diastereomers, (+)-14′ and (+)-14′′ (entry 3, Table 3). In addition, 2,6-di-tert-butylpyridine (DTBP) induced preferential formation of both endo I and exo II diastereomers, (−)-14 and (+)-14′′, in 29% and 20% yields, respectively (entry 4, Table 3).
We next studied the BTM-based catalysts in conjunction with various substituted pyridine bases. Accordingly, the highly nucleophilic, electron-rich (R)-(+)-PBTM catalyst accelerated the formation of the corresponding cycloadducts, likely due to faster formation of the resultant acylammonium salt, however without noticeable deviations in diastereoselection. In contrast, use of the (R)-(+)-NBTM catalyst and 2-phenylpyridine selectively impeded the reactivity of the (R)-enantiomer of (±)-14, consequently resulting in formation of both exo II and endo II, diastereomers, (+)-14′′ and (−)-14′′′, in 26% and 19% yields, respectively (entry 5, Table 3).
A single exo II diastereomer, (+)-14′′, was obtained in 18% yield (99% ee) through use of (R)-(+)-NBTM and 2,6-di-tert-butylpyridine (entry 6, Table 3). In addition, a single endo I diastereomer, (−)-14, was obtained in 22% yield (99% ee) by a combination of 2,6-di-tert-butylpyridine with the free-base form of levamisole hydrochloride (entry 7, Table 3). The relative and absolute configuration of a ring-opened derivative of cycloadduct (+)-14′′ was verified by X-ray analysis (see ESI, Fig. S1†) enabling assignment of all cycloadducts (−)-14, (+)-14′ and (−)-14′′′ through comparative 2-D NMR analysis. It should be noted that use of (Z)-(±)-13e and the optical antipode of the catalyst would theoretically enable access to the remaining diastereomeric and enantiomeric members (16 total) of this family of cycloadducts. While varying the chiral Lewis base catalyst and Brønsted base enabled control of DA diastereomers produced, the yields are not yet serviceable when single diastereomers are obtained (entries 6 and 7; Table 3). Indeed, variations in diastereoselectivity could be a reflection of the rate of the subsequent lactonization rather than inherent diastereoselectivity of the DA cycloaddition. However, these preliminary results point to the possibility of developing a fully stereodivergent DAL organocascade that can access all possible stereoisomers of a given family through variations of Lewis and Brønsted bases employed.
Conclusion
Further examples, applications, and mechanistic studies of the Diels–Alder-lactonization organocascade employing α,β-unsaturated acylammonium salts are presented. Factors affecting the selectivity of stereodivergent, Diels–Alder-initiated organocascades were investigated systematically with a view to understanding, predicting, and tuning the stereochemical outcome. An evaluation of various experimental parameters, guided by the results of computational studies of the DAL process, was undertaken in order to derive a more detailed understanding of the origins of selectivity. In addition, the substrate scope of the stereodivergent organocascade was extended to tethered secondary and tertiary racemic alcohols leading to the corresponding optically active γ-substituted cis- and trans-fused bicyclic γ-lactones in good yields and with excellent enantiocontrol. The combined experimental and theoretical results described herein allow a detailed picture of the full catalytic cycle of the DAL process to emerge. While the described organocascade demonstrates reasonable scope, it currently has limitations. e.g. the type of dienes that participate in cycloaddition, typically requiring more electron rich dienes.The utility of this methodology was showcased through the formal synthesis of a member of the fungus-derived and widely marketed statin drugs, (+)-dihydrocompactin.
Computational studies indicate that benzotetramisole-derived acylammonium formation proceeds by an exergonic, concerted SN2-type mechanism and provided insights into the role of Brønsted base in these cycloadditions. These studies revealed the potential of n → π*, CH–π and π–π stacking inter-actions in controlling selectivity, and point to the possibility of a rare entropy-controlled stereoselectivity. The experimentally observed temperature independence in the studied range of −78 to +80 °C supports the idea that the diastereoselectivity of these cycloadditions is indeed predominantly controlled by the differential activation entropy, ΔΔS‡.
Lastly, we documented the potential of developing a fully stereodivergent DA organocascade that could enable access to all stereoisomeric members of a given family of cycloadducts through judicious choice of Lewis and Brønsted bases. To date, this goal has only been reached for a small set of reactions and not for the DA process. Continued studies directed toward understanding the subtleties of the DAL organocascade process and applications of this process toward bioactive natural product synthesis are ongoing in our laboratories.
Acknowledgements
Support from NSF (CHE-1112397 to D. R.; CHE-030089 with XSEDE to D. J. T.), the Robert A. Welch Foundation (AA-1280 to D. R.), ACS PRF (52801-ND4 to D. J. T.), and partial support from NIH (GM 052964 to D. R.) is gratefully acknowledged. This work was initiated at Texas A & M University, Department of Chemistry. Drs Nattamai Bhuvanesh and Joe Reibenspies (Center for X-ray Analysis, TAMU) secured X-ray data, and Dr Bill Russell (Laboratory for Biological Mass Spectrometry, TAMU) provided mass data.Notes and references
- P. Merino, E. Marques-Lopez, T. Tejero and R. P. Herrera, Enantioselective Organocatalytic Diels–Alder Reactions, Synthesis, 2010, 1 CrossRef CAS.
- O. Diels and K. Alder, Synthesis in the hydro-aromatic tier, Liebigs Ann. Chem., 1928, 460, 98 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, The Diels–Alder reaction in total synthesis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels–Alder reaction, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- A. B. Northrup and D. W. C. MacMillan, The First General Enantioselective Catalytic Diels–Alder Reaction with Simple α,β-Unsaturated Ketones, J. Am. Chem. Soc., 2002, 124, 2458 CrossRef CAS PubMed.
- R. Thayumanavan, B. Dhevalapally, K. Sakthivel, F. Tanaka and C. F. Barbas, Amine-catalyzed direct Diels–Alder reactions of alpha,beta-unsaturated ketones with nitro olefins, Tetrahedron Lett., 2002, 43, 3817 CrossRef CAS.
- Z. J. Jia, H. Jiang, J. L. Li, B. Gschwend, Q. Z. Li, X. A. Yin, J. Grouleff, Y. C. Chen and K. A. Jorgensen, Trienamines in Asymmetric Organocatalysis: Diels–Alder and Tandem Reactions, J. Am. Chem. Soc., 2011, 133, 5053 CrossRef CAS PubMed.
- Y. Wang, H. M. Li, Y. Q. Wang, Y. Liu, B. M. Foxman and L. Deng, Asymmetric Diels–Alder reactions of 2-pyrones with a bifunctional organic catalyst, J. Am. Chem. Soc., 2007, 129, 6364 CrossRef CAS PubMed.
- Y. Huang, A. K. Unni, A. N. Thadani and V. H. Rawal, Hydrogen bonding: Single enantiomers from a chiral-alcohol catalyst, Nature, 2003, 424, 146 CrossRef CAS PubMed.
- B. Tan, G. Hernández-Torres and C. F. Barbas, Highly Efficient Hydrogen-Bonding Catalysis of the Diels–Alder Reaction of 3-Vinylindoles and Methyleneindolinones Provides Carbazolespirooxindole Skeletons, J. Am. Chem. Soc., 2011, 133, 12354 CrossRef CAS PubMed.
- L. C. Miller and R. Sarpong, Divergent reactions on racemic mixtures, Chem. Soc. Rev., 2011, 40, 4550 RSC.
- B. Wang, F. Wu, Y. Wang, X. Liu and L. Deng, Control of diastereoselectivity in tandem asymmetric reactions generating nonadjacent stereocenters with bifunctional catalysis by cinchona alkaloids, J. Am. Chem. Soc., 2007, 129, 768 CrossRef CAS PubMed.
- X. X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X. L. Hou and Y. D. Wu, Highly Diastereoselective Switchable Enantioselective Mannich Reaction of Glycine Derivatives with Imines, J. Am. Chem. Soc., 2008, 130, 14362 CrossRef CAS PubMed.
- A. Nojiri, N. Kumagai and M. Shibasaki, Linking Structural Dynamics and Functional Diversity in Asymmetric Catalysis, J. Am. Chem. Soc., 2009, 131, 3779 CrossRef CAS PubMed.
- M. Morgen, S. Bretzke, P. F. Li and D. Menche, Stereodivergent Synthesis of 1,3-syn- and -anti-Tetrahydropyrimidinones, Org. Lett., 2010, 12, 4494 CrossRef CAS PubMed.
- M. Luparia, M. T. Oliveira, D. Audisio, F. Frebault, R. Goddard and N. Maulide, Catalytic Asymmetric Diastereodivergent Deracemization, Angew. Chem., Int. Ed., 2011, 50, 12631 CrossRef CAS PubMed.
- X. Tian, C. Cassani, Y. K. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, Diastereodivergent Asymmetric Sulfa-Michael Additions of alpha-Branched Enones using a Single Chiral Organic Catalyst, J. Am. Chem. Soc., 2011, 133, 17934 CrossRef CAS PubMed.
- E. L. McInturff, E. Yamaguchi and M. J. Krische, Chiral-Anion-Dependent Inversion of Diastereo- and Enantioselectivity in Carbonyl Crotylation via Ruthenium-Catalyzed Butadiene Hydrohydroxyalkylation, J. Am. Chem. Soc., 2012, 134, 20628 CrossRef CAS PubMed.
- S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Enantio- and Di Aereodivergent Dual Catalysis: alpha-Allylation of Branched Aldehydes, Science, 2013, 340, 1065 CrossRef CAS PubMed.
- C. S. Schindler and E. N. Jacobsen, A New Twist on Cooperative Catalysis, Science, 2013, 340, 1052 CrossRef CAS PubMed.
- S. K. Branch M. Eichelbaum, M., B. Testa and A. Somogyi, Stereochemical aspects of drug action and disposition, Springer, Berlin, New York, 2003 Search PubMed.
- L. C. Miller, J. M. Ndungu and R. Sarpong, Parallel Kinetic Resolution Approach to the Cyathane and Cyanthiwigin Diterpenes Using a Cyclopropanation/Cope Rearrangement, Angew. Chem., Int. Ed., 2009, 48, 2398 CrossRef CAS PubMed.
- M. E. Abbasov and D. Romo, The ever-expanding role of asymmetric covalent organocatalysis in scalable, natural product synthesis, Nat. Prod. Rep., 2014, 31, 1318 RSC.
- M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, Acylammonium salts as dienophiles in Diels–Alder/lactonization organocascades, J. Am. Chem. Soc., 2014, 136, 4492 CrossRef CAS PubMed.
- K. N. Van, L. C. Morrill, A. D. Smith and D. Romo, Lewis Base Catalysis in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2016 Search PubMed.
- S. Vellalath and D. Romo, The Emerging Utility of Unsaturated Acylammonium Salts for Organocatalysis, Angew. Chem., Int. Ed., 2016, 55, 13934 CrossRef CAS PubMed.
- E. Bappert, P. Muller and G. C. Fu, Asymmetric [3 + 2] annulations catalyzed by a planar-chiral derivative of DMAP, Chem. Commun., 2006, 2604 RSC.
- S. Pandiancherri, S. J. Ryan and D. W. Lupton, 1,3-Dipolar cycloaddition of unstabilised azomethine ylides by Lewis base catalysis, Org. Biomol. Chem., 2012, 10, 7903 CAS.
- E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Anhydrides as α,β-unsaturated acyl ammonium precursors: isothiourea-promoted catalytic asymmetric annulation processes, Chem. Sci., 2013, 4, 2193 RSC.
- G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Rapid assembly of complex cyclopentanes employing chiral, α,β-unsaturated acylammonium intermediates, Nat. Chem., 2013, 5, 1049 CrossRef CAS PubMed.
- S. Vellalath, K. N. Van and D. Romo, Direct Catalytic Asymmetric Synthesis of N-Heterocycles from Commodity Acid Chlorides by Employing α,β-Unsaturated Acylammonium Salts, Angew. Chem., Int. Ed., 2013, 52, 13688 CrossRef CAS PubMed.
- Y. Fukata, K. Asano and S. Matsubara, Facile Net Cycloaddition Approach to Optically Active 1,5-Benzothiazepines, J. Am. Chem. Soc., 2015, 137, 5320 CrossRef CAS PubMed.
- S. L. Schreiber, Target-oriented and diversity-oriented organic synthesis in drug discovery, Science, 2000, 287, 1964 CrossRef CAS PubMed.
- J. A. Frearson and I. T. Collie, HTS and hit finding in academia – from chemical genomics to drug discovery, Drug Discovery Today, 2009, 14, 1150 CrossRef PubMed.
- S. Dandapani and L. A. Marcaurelle, Grand challenge commentary: Accessing new chemical space for ‘undruggable’ targets, Nat. Chem. Biol., 2010, 6, 861 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Impact of high-throughput screening in biomedical research, Nat. Rev. Drug Discovery, 2011, 10, 188 CrossRef CAS PubMed.
- C. J. O'Connor, L. Laraia and D. R. Spring, Chemical genetics, Chem. Soc. Rev., 2011, 40, 4332 RSC.
- D. S. Tan, Diversity-oriented synthesis: exploring the intersections between chemistry and biology, Nat. Chem. Biol., 2005, 1, 74 CrossRef CAS PubMed.
- C. J. O’ Connor, H. S. Beckmann and D. R. Spring, Diversity-oriented synthesis: producing chemical tools for dissecting biology, Chem. Soc. Rev., 2012, 41, 4444 RSC.
- D. A. Annis, O. Helluin and E. N. Jacobsen, Stereochemistry as a diversity element: Solid-phase synthesis of cyclic RGD peptide derivatives by asymmetric catalysis, Angew. Chem., Int. Ed., 1998, 37, 1907 CrossRef CAS.
- B. A. Harrison, T. M. Gierasch, C. Neilan, G. W. Pasternak and G. L. Verdine, High-affinity muopioid receptor ligands discovered by the screening of an exhaustively stereodiversified library of 1,5-enediols, J. Am. Chem. Soc., 2002, 124, 13352 CrossRef CAS PubMed.
- M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. González-Páez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Spiroindolones, a Potent Compound Class for the Treatment of Malaria, Science, 2010, 329, 1175 CrossRef CAS PubMed.
- S. Dandapani, E. Comer, J. R. Duvall and B. Munoz, Hits, leads and drugs against malaria through diversity-oriented synthesis, Future Med. Chem., 2012, 4, 2279 CrossRef CAS PubMed.
- J. Clardy and C. Walsh, Lessons from natural molecules, Nature, 2004, 432, 829 CrossRef CAS PubMed.
- L.-M. Xu, Y.-F. Liang, Q.-D. Ye, Z. Yang, M. Foley, S. A. Snyder and D.-W. Ma, in Organic Chemistry – Breakthroughs and Perspectives, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, p. 1 Search PubMed.
- T. Sugimura, T. Tei, A. Mori, T. Okuyama and A. Tai, Temperature-independent stereocontrolled [2 + 2] cycloaddition. Potential of the 2,4-pentanediol tether in asymmetric reactions as a differential activation entropy promoter, J. Am. Chem. Soc., 2000, 122, 2128 CrossRef CAS.
- M. Lombardo, S. Fabbroni and C. Trombini, Entropy-controlled selectivity in the vinylation of a cyclic chiral nitrone. An efficient route to enantiopure polyhydroxylated pyrrolidines, J. Org. Chem., 2001, 66, 1264 CrossRef CAS PubMed.
- B. Giese, Basis and Limitations of the Reactivity-Selectivity Principle, Angew. Chem., Int. Ed., 1977, 16, 125 CrossRef.
- H. Buschmann, H.-D. Scharf, N. Hoffmann and P. Esser, The Isoinversion Principle—a General Model of Chemical Selectivity, Angew. Chem., Int. Ed., 1991, 30, 477 CrossRef.
- G. Cainelli, D. Giacomini and P. Galletti, Temperature and solvent effects in facial diastereoselectivity of nucleophilic addition: entropic and enthalpic contribution, Chem. Commun., 1999, 567 RSC.
- Y. Sohtome, B. Shin, N. Horitsugi, R. Takagi, K. Noguchi and K. Nagasawa, Entropy-Controlled Catalytic Asymmetric 1,4-Type Friedel–Crafts Reaction of Phenols Using Conformationally Flexible Guanidine/Bisthiourea Organocatalyst, Angew. Chem., Int. Ed., 2010, 49, 7299 CrossRef CAS PubMed.
- A. Garzan, A. Jaganathan, N. Salehi Marzijarani, R. Yousefi, D. C. Whitehead, J. E. Jackson and B. Borhan, Solvent-Dependent Enantiodivergence in the Chlorocyclization of Unsaturated Carbamates, Chem.–Eur. J., 2013, 19, 9015 CrossRef CAS PubMed.
- L. S. M. Wong and M. S. Sherburn, IMDA-radical cyclization approach to (+)-himbacine, Org. Lett., 2003, 5, 3603 CrossRef CAS PubMed.
- S. Akai, K. Tanimoto and Y. Kita, Lipase-catalyzed domino dynamic kinetic resolution of racemic 3-vinylcyclohex-2-en-1-ols/intramolecular Diels–Alder reaction: One-pot synthesis of optically active polysubstituted decalins, Angew. Chem., Int. Ed., 2004, 43, 1407 CrossRef CAS PubMed.
- J. Sauer, Diels–Alder-Reactions. I. New Preparative Aspects, Angew. Chem., Int. Ed., 1966, 5, 211 CrossRef CAS.
- W. R. Roush and D. A. Barda, Highly Selective Lewis Acid Catalyzed Diels–Alder Reactions of Acyclic (Z)-1,3-Dienes, J. Am. Chem. Soc., 1997, 119, 7402 CrossRef CAS.
- Y. K. Tonylam, V. P. Gullo, R. T. Goegelman, D. Jorn, L. Huang, C. Deriso, R. L. Monaghan and I. Putter, Dihydrocompactin, a New Potent Inhibitor of 3-Hydroxy-3-Methylglutaryl Coenzyme-a Reductase from Penicillium-Citrinum, J. Antibiot., 1981, 34, 614 CrossRef.
- H. Hagiwara, M. Konno, T. Nakano and H. Uda, Total Synthesis of (+)-Dihydrocompactin, J. Chem. Soc., Perkin Trans. 1, 1994, 2417 RSC.
- J. Ammer, M. Baidya, S. Kobayashi and H. Mayr, Nucleophilic reactivities of tertiary alkylamines, J. Phys. Org. Chem., 2010, 23, 1029 CrossRef CAS.
- A. J. Wagner and S. D. Rychnovsky, Kinetic analysis of the HBTM-catalyzed esterification of an enantiopure secondary alcohol, Org. Lett., 2013, 15, 5504 CrossRef CAS PubMed.
- D. J. Tantillo and K. N. Houk, Fidelity in hapten design: How analogous are phosphonate haptens to the transition states for alkaline hydrolyses of aryl esters?, J. Org. Chem., 1999, 64, 3066 CrossRef CAS PubMed.
- J. M. Fox, O. Dmitrenko, L. A. Liao and R. D. Bach, Computational studies of nucleophilic substitution at carbonyl carbon: the S(N)2 mechanism versus the tetrahedral intermediate in organic synthesis, J. Org. Chem., 2004, 69, 7317 CrossRef CAS PubMed.
- M. Adler, S. Adler and G. Boche, Tetrahedral intermediates in reactions of carboxylic acid derivatives with nucleophiles, J. Phys. Org. Chem., 2005, 18, 193 CrossRef CAS.
- S. Vellalath, K. N. Van and D. Romo, Utility and NMR studies of alpha,beta-unsaturated acylammonium salts: synthesis of polycyclic dihydropyranones and a dihydropyridone, Tetrahedron Lett., 2015, 56, 3647 CrossRef CAS.
- B. Saikia, I. Suryanarayana, B. K. Saikia and I. Haque, Hydrogen-Bond Formation between Aliphatic-Alcohols and Tertiary-Amines, Spectrochim. Acta, Part A, 1991, 47, 791 CrossRef.
- D. Farcasiu, M. Lezcano and A. Vinslava, Solvent effects on N-15 NMR chemical shifts of 2,6-di-tert-butylpyridine. Absence of hydrogen bonding with the nitrogen atom, New J. Chem., 2000, 24, 199 RSC.
- E. M. Arnett and B. Chawla, Complete Thermodynamic Analysis of the Hydration of 13 Pyridines and Pyridinium Ions – Special Case of 2,6-Di-Tert-Butylpyridine, J. Am. Chem. Soc., 1979, 101, 7141 CrossRef CAS.
- I. M. Kolthoff, M. K. Chantoon and S. Bhowmik, Dissociation Constants of Uncharged and Monovalent Cation Acids in Dimethyl Sulfoxide, J. Am. Chem. Soc., 1968, 90, 23 CrossRef CAS.
- R. L. Benoit, M. Frechette and D. Lefebvre, 2,6-Di-Tert-Butylpyridine – an Unusually Weak Base in Dimethylsulfoxide, Can. J. Chem., 1988, 66, 1159 CrossRef CAS.
- F. G. Bordwell, Equilibrium acidities in dimethyl sulfoxide solution, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- J. H. Aasheim, H. Fliegl, E. Uggerud, T. Bonge-Hansen and O. Eisenstein, Stereoselectivity through a network of non-classical CH weak interactions: a prospective study of a bicyclic organocatalytic scaffold, New J. Chem., 2014, 38, 5975 RSC.
- S. E. Wheeler and J. W. G. Bloom, Toward a More Complete Understanding of Noncovalent Interactions Involving Aromatic Rings, J. Phys. Chem. A, 2014, 118, 6133 CrossRef CAS PubMed.
- R. E. Plata and D. A. Singleton, A Case Study of the Mechanism of Alcohol-Mediated Morita Baylis–Hillman Reactions. The Importance of Experimental Observations, J. Am. Chem. Soc., 2015, 137, 3811 CrossRef CAS PubMed.
- J. P. Wagner and P. R. Schreiner, London Dispersion in Molecular Chemistry—Reconsidering Steric Effects, Angew. Chem., Int. Ed., 2015, 54, 12274 CrossRef CAS PubMed.
- D. J. Tantillo, Recent excursions to the borderlands between the realms of concerted and stepwise: carbocation cascades in natural products biosynthesis, J. Phys. Org. Chem., 2008, 21, 561 CrossRef CAS.
- T. Haven, G. Kubik, S. Haubenreisser and M. Niggemann, Calcium-Catalyzed Cyclopropanation, Angew. Chem., Int. Ed., 2013, 52, 4016 CrossRef CAS PubMed.
- H. S. Rzepa and C. Wentrup, Mechanistic Diversity in Thermal Fragmentation Reactions: A Computational Exploration of CO and CO2 Extrusions from Five-Membered Rings, J. Org. Chem., 2013, 78, 7565 CrossRef CAS PubMed.
- S. E. Wheeler, Local Nature of Substituent Effects in Stacking Interactions, J. Am. Chem. Soc., 2011, 133, 10262 CrossRef CAS PubMed.
- S. E. Wheeler, Understanding substituent effects in noncovalent interactions involving aromatic rings, Acc. Chem. Res., 2013, 46, 1029 CrossRef CAS PubMed.
- K. Wozniak, H. He, J. Klinowski, W. Jones and E. Grech, Attractive Inter- and Intramolecular N⋯O Interactions in N,N-Dipicrylamine and Its Ionic Complexes, J. Phys. Chem., 1994, 98, 13755 CrossRef CAS.
- Z. Yin, L. Jiang, J. He and J.-P. Cheng, The first observation of nitrogen-carbonyl bonding: self-assembly of N-oxalyl 2,4-dinitroanilide assisted by a weak N⋯O
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interaction, Chem. Commun., 2003, 2326 RSC.
C interaction, Chem. Commun., 2003, 2326 RSC. - R. Paulini, K. Müller and F. Diederich, Orthogonal Multipolar Interactions in Structural Chemistry and Biology, Angew. Chem., Int. Ed., 2005, 44, 1788 CrossRef CAS PubMed.
- A. Bauza, T. J. Mooibroek and A. Frontera, Directionality of π-holes in nitro compounds, Chem. Commun., 2015, 51, 1491 RSC.
- A. Choudhary, C. G. Fry, K. J. Kamer and R. T. Raines, An n → π* interaction reduces the electrophilicity of the acceptor carbonyl group, Chem. Commun., 2013, 49, 8166 RSC.
- A. Choudhary, R. W. Newberry and R. T. Raines, n → π* Interactions Engender Chirality in Carbonyl Groups, Org. Lett., 2014, 16, 3421 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, A Key n → π* Interaction in N-Acyl Homoserine Lactones, ACS Chem. Biol., 2014, 9, 880 CrossRef CAS PubMed.
- J. Qi, M. S. Han, Y. C. Chang and C. H. Tung, Developing visible fluorogenic ‘click-on’ dyes for cellular imaging, Bioconjugate Chem., 2011, 22, 1758 CrossRef CAS PubMed.
- D. S. B. Daniels, S. R. Smith, T. Lebl, P. Shapland and A. D. Smith, A Scalable, Chromatography-Free Synthesis of Benzotetramisole, Synthesis, 2015, 47, 34 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization details for all new compounds including 1H and 13C NMR spectra, computational data, crystallographic data, chiral phase-HPLC traces. CCDC 972246. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04273b |
| This journal is © The Royal Society of Chemistry 2017 |