
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise observation and quantification and mixed matrix membrane separation of CO2 within a hydroxy-decorated porous host†
Christopher G.
Morris
ab,
Nicholas M.
Jacques
a,
Harry G. W.
Godfrey
a,
Tamoghna
Mitra
c,
Detlev
Fritsch
d,
Zhenzhong
Lu
a,
Claire A.
Murray
b,
Jonathan
Potter
b,
Tom M.
Cobb
b,
Fajin
Yuan
b,
Chiu C.
Tang
*b,
Sihai
Yang
*a and
Martin
Schröder
 *a
*a
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: Sihai.Yang@manchester.ac.uk; M.Schroder@manchester.ac.uk
bDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, UK. E-mail: chiu.tang@diamond.ac.uk
cDepartment of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK
dFraunhofer IAP, FB3, Geiselbergstrasse 69, Potsdam-Golm, 14476, Germany
First published on 27th February 2017
Abstract
The identification of preferred binding domains within a host structure provides important insights into the function of materials. State-of-the-art reports mostly focus on crystallographic studies of empty and single component guest-loaded host structures to determine the location of guests. However, measurements of material properties (e.g., adsorption and breakthrough of substrates) are usually performed for a wide range of pressure (guest coverage) and/or using multi-component gas mixtures. Here we report the development of a multifunctional gas dosing system for use in X-ray powder diffraction studies on Beamline I11 at Diamond Light Source. This facility is fully automated and enables in situ crystallographic studies of host structures under (i) unlimited target gas loadings and (ii) loading of multi-component gas mixtures. A proof-of-concept study was conducted on a hydroxyl-decorated porous material MFM-300(VIII) under (i) five different CO2 pressures covering the isotherm range and (ii) the loading of equimolar mixtures of CO2/N2. The study has successfully captured the structural dynamics underpinning CO2 uptake as a function of surface coverage. Moreover, MFM-300(VIII) was incorporated in a mixed matrix membrane (MMM) with PIM-1 in order to evaluate the CO2/N2 separation potential of this material. Gas permeation measurements on the MMM show a great improvement over the bare PIM-1 polymer for CO2/N2 separation based on the ideal selectivity.
1. Introduction
With increasing levels of anthropogenic CO2 emissions contributing to climate change, there is growing research interest in the field of carbon capture and sequestration (CCS).1 Capture of CO2 from flue gas via selective adsorption by porous adsorbents is particularly promising because of a number of advantages over the state-of-the-art carbon capture system based upon corrosive amine scrubbers.2 For example, most solid adsorbents have reduced toxicity in comparison to amines, and more importantly, their operation involves a much lower energy input for regeneration due to the comparatively weaker binding interactions between the adsorbents and CO2.3 Metal–organic frameworks (MOFs) are an emerging class of porous solids showing potential in a wide range of applications such as gas adsorption and storage,4–7 gas separation,8,9 catalysis,10,11 drug delivery,12 and sensing.13 MOFs, constructed by metal ions/clusters bridged by organic linkers, usually show crystalline extended structures with high internal surface areas and pore volumes. Within the field of gas adsorption, the identification of preferred binding sites at a molecular level within the MOF structure represents an important methodology for understanding the host–guest (MOF–gas) binding interactions and any observed selectivity. However, such studies are often very challenging because most porous adsorbents (e.g., activated carbons, porous silica, and large pore zeolites) lack structural order. In contrast, the highly crystalline nature of MOFs makes them particularly advantageous in this perspective since it enables the interrogation of preferred binding domains via advanced crystallographic investigations.3,8,9,14–22Although single crystal diffraction is the most powerful crystallographic technique to study the location of guests, full retention of single crystalline MOF samples upon desolvation at elevated temperatures, and with sequential guest loading under pressure is only achieved in exceptional cases.14–19 On the other hand, techniques such as powder X-ray diffraction (suitable for CO2, SO2) and neutron powder diffraction (suitable for H2 and hydrocarbons) are also being employed to probe in situ host–guest structures.3,8,20–25 However, most of the current experimental systems suffer from two major limitations. Firstly, precise control of gas loading can be highly problematic in manual operation, not least because it is time-consuming, and as a result, most of the studies rely on the characterisation of the bare and gas-loaded structures under one or two pressures (typically atmospheric pressure). Information on the gas uptake process over a wide pressure range is therefore lost. Secondly, precise dosing with mixtures of gases at desirable and variable ratios that reflect more closely real world conditions is difficult to achieve and thus molecular details on the competitive binding between two different gas species in the same pore environment are unclear. Previously, a manually operated system was used to perform such studies on the high resolution powder diffraction beamline I11 at Diamond Light Source.26 This system, however, suffers from the aforementioned issues regarding precise control, and is also time-consuming to operate because of the stringent “search activity” at synchrotron beamlines each time the hutch is opened for researcher access. Therefore, the development of programmable equipment to facilitate these studies with greater precision is essential. Automation of operations will afford more efficient use of beam time at such facilities by increasing sample capacity and turnover at gas different pressures. Herein we present the design and construction of a new remote control gas dosing system. When coupled with the diffraction beamline, it enables advanced crystallographic studies on the host materials with (i) unlimited single-component gas loadings with precise control and (ii) loadings of multicomponent gas mixtures at desirable ratios and pressures. More significantly, all of these operations and data collections can be carried out by automated computer programme without the need for researcher access to the beamline (except from setting up the sample at the beginning). A proof-of-concept study was conducted on a hydroxyl-decorated porous material MFM-300(VIII) (i) under five different CO2 pressures covering fully the isotherm range and (ii) on loading of an equimolar mixture of CO2 and N2 at different pressures. This study has captured successfully the details of the structural dynamics underpinning CO2 uptake over the entire surface coverage, and identified CO2 binding at crystallographic resolution. The results provide direct structural evidence for the observed high CO2/N2 adsorption selectivity for MFM-300(VIII).
Investigation of the separation selectivity of a given material under dynamic flow conditions can afford critical insights beyond the static isotherm selectivity. A potential method for utilising MOFs in CCS, whilst simultaneously reducing the financial impact of the current high cost of MOF production, is incorporating them in a mixed matrix membrane (MMM).27,28 MMMs are composite materials combining a polymeric matrix with an inorganic filler and has attracted significant attention in recent years although the concept has been known since the late 1980s, where components such as aluminosilicates and CaY zeolites were added to organic polymers to form films for separation of gases29 and saccharides.30
A membrane which exhibits the ideal properties for gas separation will have high adsorption selectivity but simultaneously having ultrahigh permeability to assist the gas diffusion. However, there is an inherent trade-off between selectivity and permeability as represented by the Robeson upper bound.31,32 Initially set in 1991, this upper bound demonstrated the trade-off between the two factors affecting the efficacy of these membranes.31 As membrane technology has evolved, this upper bound was revised in 2008.32 The majority of pure polymeric membranes are unable to exceed the upper bound limit.32 Inorganic membranes have been shown to have very good selectivity and permeability and therefore exceed the upper bound. However, they have been shown to be poorly reproducible, expensive and mechanically unstable.33 In contrast, MMMs show a good compromise between pure organic and inorganic polymer membranes and are considered to be promising candidates to fall above the Robeson upper bound whilst being more reproducible and mechanically stable. Although initial studies on MMMs focussed on zeolites,34 recent attention has turned to MOFs due to the increased affinity shown between the MOF particles and the polymer matrix.35 This increased affinity is due to the hybrid inorganic–organic nature of MOFs and has been shown to lead to fewer non-selective macro-voids at the interface between the MOF particles and polymer matrix, as commonly seen in zeolite-based MMMs.34 Such voids allow unrestricted gas permeation and dramatically decrease the separation capability of the MMM. Herein, we show that MFM-300(VIII) can be incorporated in a MMM with PIM-1 without generating macro-voids. Dynamic gas permeation measurements on MFM-300(VIII)/PIM-1 have shown great improvement over the bare PIM-1 polymer for CO2 separation based on the ideal selectivity.
2. Experimental
2.1. Design and construction of the automated gas dosing system at Diamond powder diffraction Beamline I11
A schematic view of the automated gas dosing system is shown in Fig. 1. The system consists of three Alicat MCS-1SLPM-D mass flow controllers (MFCs), an Alicat PCS-100PSIA-D pressure controller functioning as a programmable back-pressure regulator (BPR), and an Alicat PCDS-100PSIA-D dual valve pressure controller (DVPC) connected with 6 mm stainless steel Swagelok tubing and connectors. A pressure relief valve (5.5 bar absolute) protects the system components from over-pressurisation. Two Swagelok pneumatically-activated bellows-sealed valves are used to control the venting of the system to a COSHH level to a vacuum pump. The system is compatible with a wide range of gases, including CO2, N2, air, Ar, CH4, CO, C2H6, H2, He, N2O, Ne, O2, C3H8, n-C4H10, C2H2, C2H4, i-C4H10, Kr, Xe, SF6, NO, NF3, NH3, SO2 and H2S. | ||
| Fig. 1 (a) Schematic and (b) photographic views of the remote controlled gas dosing system at Diamond powder diffraction Beamline I11. | ||
To dose a sample, the target gas is first flowed through the MFCs into the main body and buffer cylinder to a pressure slightly higher than the desired final sample pressure. The sample cell is then dosed from this reservoir of gas at a programmable flow rate, with the system isolated from the gas cylinders by the MFCs. The gas dosing system has been fully integrated into the Generic Data Acquisition (GDA) software and the Experimental Physics and Industrial Control System (EPICS) at Diamond Light Source, and can be controlled either via EPICS GUI or GDA command line. Commands have been written for filling the system, flushing the system, and setting the sample pressure. It is important that any pressure changes that the sample is subjected to occur gradually to minimise sample movement inside the capillary. An example of the logic used in the GDA commands is shown in Fig. S1.†
2.2. Collection of in situ PXRD data for MFM-300(VIII) upon loading of CO2 and equimolar mixture of CO2/N2 at different pressures
A powder crystalline sample (particle size < 0.2 μm) of MFM-300(VIII) was loaded into a 0.7 mm borosilicate capillary which was secured into a static sample cell.26 The sample was activated at 393 K under dynamic vacuum (1 × 10−8 bar) for 2 h to remove the free solvents from the pore. The activated sample was then cooled to 273 K and PXRD data collected for 1 h with the high resolution MAC detector at Beamline I11.36 The bare sample was then dosed with 100, 300, 500, 750, and 1000 mbar of CO2 at 273 K, incrementally in 10 mbar steps to minimise the movement of the powder sample within the capillary. The gas-loaded sample was left for 30 min to achieve equilibration of the host–guest structure before PXRD data collection for 1 h at 273 K. After final data collection, the sample was re-activated at 393 K under dynamic vacuum (1 × 10−8 bar) for 30 min to fully remove adsorbed CO2 molecules. The re-activated sample was then dosed with N2 to 300, 500 and 1000 mbar with 15 minute data collections at 273 K with 30 min to allow equilibration for each dosing. Finally, the sample was re-activated and dosed with an equimolar mixture of CO2 and N2 to 600, 1000 and 2000 mbar with 1 h data collection at 273 K and 30 min to allow equilibration for each dosing. It is important to emphasise that all above operations and data collections (ca. 20 hours in total) were carried out by the programmable gas dosing and integrated data collection system without human interruption during the entire experiment.2.3. Details of the structural refinement of PXRD data
Structures and unit cells of MFM-300(VIII) over the range of loadings were refined using the Rietveld and Pawley methods, respectively, using TOPAS Academic 5. The backgrounds of the profiles were fitted using a Chebyshev polynomial with 13 coefficients, and the Stephens anisotropic peak function37 was used to account for the slight anisotropic line broadening. Two well-defined regions of electron density in the pore were revealed by difference Fourier analysis. In the pure CO2 experiment, these regions were assigned as CO2, and in the experiments using an equimolar CO2/N2 mixture, these regions were also assigned as CO2 rather than N2, due to both the similarity in position to the pure CO2 experiments, and also the shape of the electron density being more suited to a linear triatomic molecule as opposed to a small, more spherical, diatomic molecule. In all cases, the CO2 molecules were soft restrained rather than modelled as rigid bodies. The positions of these soft restrained atoms were refined, followed by a final refinement of the soft restrained framework, resulting in excellent fits and satisfactory structural models for the entire pressure range. Individual occupancies and isotropic temperature factors of the atoms in each CO2 molecule were made equal, and the values for each molecule were refined simultaneously.2.4. Preparation of the 25 wt% MFM-300(VIII)/PIM-1 MMM
A solution casting method was employed for fabrication of the MFM-300(VIII)/PIM-1 MMM. A quantity of 200 mg of MFM-300(VIII) powder was pre-dispersed in 5 mL of CHCl3 with sonication and stirring followed by addition of 600 mg of PIM-1 (ref. 38) in 10 mL of CHCl3. In order to aid affinity between the MOF particles and polymer matrix, 20 μL of dimethoxysilylpropyl modified poly(ethyleneimine) was added to the casting solution as a surface modifier and the mixture stirred for 24 h. The mixture was evaporated to 5–7 mL and cast on a PTFE substrate and the solvent evaporated at room temperature. The resulting free-standing MMM was dried in an oven at 120 °C for 12 h prior to the permeation measurements.2.5. Characterisation of the 25 wt% MFM-300(VIII)/PIM-1 MMM
The purity of MFM-300(VIII) was confirmed by powder X-ray diffraction before loading into the MMM. The membrane morphology was studied by scanning electron microscopy (SEM) using a Hitachi S-4800 Field-Emission Scanning Electron Microscope with Energy Dispersive X-ray (EDX) detector and cold cathode electron source. The membrane cross section was prepared via freeze fracturing using liquid N2. The sample was then coated with gold via sputtering using an Emitech coater. PXRD analysis was also carried out on a Philips X'Pert diffractometer to ensure retention of the crystallinity of the MOF once incorporated into the membrane.2.6. Measurements of gas permeation of the 25 wt% MFM-300(VIII)/PIM-1 MMM
Permeation was measured for pure gases (CO2, CH4, He, N2, H2) using the constant volume–variable pressure method.39 The recorded diffusion kinetics obeys Fick's first law of diffusion which relates the diffusive flux to the concentration under steady state. This postulates that a solute, in this case a gas, will move from an area of high concentration to an area of low concentration over a concentration gradient. This is described by the eqn (1),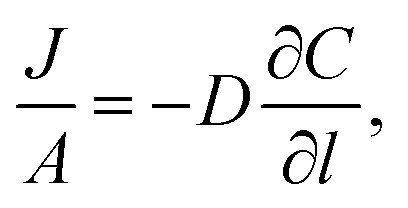 | (1) |
| C = S × p | (2) |
 | (3) |
As permeation can be considered a solubility–diffusion process, the permeability coefficient P can be expressed as a product of the average diffusion coefficient, D, and solubility coefficient, S as shown in eqn (4):
| P = D × S | (4) |
The diffusion coefficient, D, is a measure of the mobility of the gas between the upstream and downstream side of the membrane and is determined by the packing and motion of the polymer segments and MOF particles and by the size and shape of the gas molecules. The solubility coefficient, S, is determined by the condensability of the penetrating gas molecules, by interactions with the polymer and MOF particles.
The perm-selectivity αp of a given membrane for two gases (A and B) based upon single-component gas permeation measurements is given by eqn (5):
| αp = PA/PB | (5) |
3. Results and discussion
3.1. Construction of the new remote control gas dosing system
The new remote control gas dosing system is now fully operational on Beamline I11 (Fig. 1b). It offers many advantages over the previous manual system,26 including:• Complete programmability, leading to more efficient use of beam time;
• Much greater precision for gas loading, resulting in a higher reproducibility;
• Compatible with a number of toxic and corrosive gases;
• Safer operation, as users can operate it from outside of the X-ray hutch;
• Capability of mixing precisely up to 3 gases for sample loading.
Therefore, the development of this facility will enable new advanced crystallographic studies of crystalline porous materials to gain understanding of their function at the molecular level. A proof-of-concept study has been carried out on a VIII-based MOF to determine the preferred binding domains for CO2 molecules in the pore in the presence and absence of N2 gas. The study has yielded important evidence for the observed high CO2/N2 adsorption selectivity.
3.2. Structural analysis of single-component gas loaded MFM-300(VIII)
The complex MFM-300(VIII) [V2(OH)2(L)] (H4L = biphenyl-3,3′,5,5′-tetracarboxylic acid) was synthesised by hydrothermal reaction of VCl3 and H4L in slightly acidic (HCl) water at 210 °C.41 MFM-300(VIII), isolated as green microcrystalline powder, is isostructural to MFM-300(M) (M = Al, Ga, In),3,19,22 and comprises of chains of corner sharing [VO4(OH)2] octahedra, linked by mutually cis-μ2-OH groups, and further bridged by tetracarboxylate L4− ligands. This arrangement generates 1D square-shape pore channels in a ‘wine rack’ array. Desolvated MFM-300(VIII) displays a surface area of 1892 m2 g−1, a pore size of ca. 5.2 Å, and a total pore volume of 0.490 cm3 g−1 as determined from CO2 isotherm at 273 K, where the total uptake was recorded as 8.6 mmol g−1 at 1 bar. Desolvated MFM-300(VIII) shows an exceptionally high CO2 adsorption capacity of 6.0 mmol g−1 (26.4 wt%) at 298 K and 1.0 bar, comparable to the best-behaving materials such as MOF-74(Mg) or CPO-27(Mg) (26–27.5 wt%) under same conditions.42,43 In contrast, desolvated MFM-300(VIII) shows negligible N2 uptake, possibly due to the narrow pore window, leading to a high CO2/N2 selectivity of 81. We therefore sought to gain in-depth structural insights into the gas binding, and more importantly on competitive gas binding in this porous host.
In situ PXRD data confirm the absence of structural phase change of MFM-300(VIII) as a function of CO2 loading. Analysis of the bare MOF indicates the complete removal of solvent molecules from the pore. Difference Fourier analysis of the CO2-loaded structures revealed two well-defined regions of residual electron density within the pore of MFM-300(VIII): one situated above the hydroxyl group of the framework and the other located adjacent to this region (Fig. 2). Soft-restrained CO2 molecules were added to the structural model of bare MFM-300(VIII), and their positions, orientations and occupancies refined, revealing two distinct binding sites (A and B) across the entire pressure range. It is important to note that for clarity, half of the positionally disordered CO2 molecules are not shown in Fig. 2. CO2-A forms a hydrogen bond interaction with the μ2-OH group of the framework. CO2-B interacts with CO2-A via two separate dipole–dipole interactions resulting in distorted T-shape arrangement. When comparing these interactions with those found in dry ice at 150 K (ref. 44) (Fig. 3), there are many similarities, the most striking of which is the manner in which CO2-B ‘sandwiches’ itself between two molecules of CO2-A, maximising the number of dipole–dipole interactions between the molecules. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OA
OA![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) CB angle of interaction across the entire pressure range (128–139°) is also similar to CO′C′′ angle (132.35°) in solid CO2. The intermolecular distances for solid CO2, however, are shorter than those observed in MFM-300(VIII), with a OCOC(O)2 distance of 3.11 Å, compared to distances from 3.63(6)–3.89(4) Å found in MFM-300(VIII). This is most likely due to the solid state structure of CO2 being obtained at a much lower temperature than of CO2-loaded MFM-300(VIII). The combination of these interactions forms an infinite chiral network of CO2-A and CO2-B that propagates along the pore (Fig. 3). These structures vary significantly from that of MFM-300(Al)·3.2CO2 studied by PXRD at 273 K.3 In MFM-300(Al) the two CO2 sites align to create discrete T-shaped OCOC(O)2 units above each hydroxyl. These differences can be explained by the slightly narrower pore of MFM-300(Al) having more of a confinement effect than MFM-300(VIII) on the CO2 molecules.
CB angle of interaction across the entire pressure range (128–139°) is also similar to CO′C′′ angle (132.35°) in solid CO2. The intermolecular distances for solid CO2, however, are shorter than those observed in MFM-300(VIII), with a OCOC(O)2 distance of 3.11 Å, compared to distances from 3.63(6)–3.89(4) Å found in MFM-300(VIII). This is most likely due to the solid state structure of CO2 being obtained at a much lower temperature than of CO2-loaded MFM-300(VIII). The combination of these interactions forms an infinite chiral network of CO2-A and CO2-B that propagates along the pore (Fig. 3). These structures vary significantly from that of MFM-300(Al)·3.2CO2 studied by PXRD at 273 K.3 In MFM-300(Al) the two CO2 sites align to create discrete T-shaped OCOC(O)2 units above each hydroxyl. These differences can be explained by the slightly narrower pore of MFM-300(Al) having more of a confinement effect than MFM-300(VIII) on the CO2 molecules.
 | ||
| Fig. 2 Top: Difference Fourier maps of CO2-loaded MFM-300(VIII) from 100–1000 mbar viewed along the c-axis. Middle: Crystal structures of CO2-loaded MFM-300(VIII) from 100–1000 mbar viewed along the c-axis. Bottom: Detailed view of the interaction of CO2-A with the V–OH group and the intermolecular dipole interactions with CO2-B. In the case of the OHAOCO interaction, OAOCO distances are shown in brackets. For clarity, only half of the positionally disordered CO2 molecules have been shown. | ||
 | ||
| Fig. 3 (a) View of structure of MFM-300(VIII)·xCO2 along the a axis showing the infinite chiral network that propagates through the pore; (b) view of interactions between CO2-B and CO2-A in MFM-300(VIII)·xCO2; (c) view of the structure of CO2 in the solid state at 150 K. | ||
Throughout the 5 different loadings of CO2 [100 (I), 300 (II), 500 (III), 750 (IV) and 1000 (V) mbar], the position and orientation of CO2-A relative to the oxygen atom of the hydroxyl moiety remain consistent with the orientation of CO2-B changing only slightly over the pressure range. The distances for these interactions are summarised in Table 1. Inspection of the crystallographic occupancies of each site with respect to pressure (Fig. 2) shows that each site populates concurrently, with site A having a higher occupancy than site B at each pressure. The difference in site occupancy is more pronounced at low pressure where host–guest interaction is dominating the uptake. With increased pressure, the difference on site occupancy decreases. This suggests the presence of a coherent cooperativity between two sites, and implies a dependency of site B on the presence of site A. Combined occupancies of the two sites of CO2 atoms from these refinements were plotted as a function of pressure and compared to the CO2 adsorption isotherm of MFM-300(VIII) at 273 K (Fig. 4). The occupancy plot shows an excellent agreement with the isotherm, further validating the accuracy of the structural models, whilst also confirming the high accuracy of the remote control gas dosing system. Thus, these results highlight the dynamic nature of the adsorbed gases within the pore as the pressure changes, and suggest that the lowest energy crystallographic position for the guest molecules is affected by their crystallographic occupancy.
| Structure | CO2 pressure (mbar) | d 1 (Å) | d 2 (Å) | d 3 (Å) |
|---|---|---|---|---|
| a Indicates the partial pressure of CO2 in the equimolar CO2/N2 mixture. | ||||
| I | 100 | 3.33(1) | 3.69(7) | 3.88(6) |
| II | 300 | 3.36(1) | 3.63(6) | 3.90(5) |
| III | 500 | 3.38(1) | 3.63(6) | 3.89(5) |
| IV | 750 | 3.37(1) | 3.73(4) | 3.85(4) |
| V | 1000 | 3.35(1) | 3.67(4) | 3.88(4) |
| VI | 300a | 3.35(2) | 3.60(2) | 3.99(2) |
| VII | 500a | 3.34(2) | 3.73(4) | 3.90(4) |
| VIII | 1000a | 3.37(1) | 3.72(3) | 3.93(3) |
 | ||
| Fig. 4 (a) Plot of CO2 occupancies from single component CO2 adsorption experiments as a function of pressure (normalised to the site occupancy of the V center); (b) plot of combined CO2 occupancies from single component CO2 adsorption experiment as a function of pressure (blue squares) overlaid with the gravimetric CO2 isotherm of MFM-300(VIII) (red diamonds); (c) plot of combined CO2 occupancies from dual component N2/CO2 adsorption experiment as a function of pressure (blue squares) overlaid on gravimetric CO2 isotherm of MFM-300(VIII) (red diamonds). | ||
Similar difference Fourier analysis of N2-loaded MFM-300(VIII) revealed no specific regions of residual electron density in the pore, and refinement of the empty framework against the N2-loaded data revealed no apparent contribution from N2 molecules to the structure, consistent with the ultra-low N2 uptake in the corresponding isotherm.
3.3. Structural analysis of dual-component loading of an equimolar mixture of CO2/N2 in MFM-300(VIII)
As for the above structural refinements, difference Fourier analysis revealed two regions of high electron density and are attributed to CO2 molecules due to the similarity in position and shape to those in the single component CO2 refinements. The electron density was more elongated than spherical, suggesting the presence of the linear, triatomic, CO2 as opposed to the more spherical N2. The refinement of two soft-restrained CO2 molecules in these positions confirmed the presence of the same two binding sites (A and B) found in the single component CO2 structures (Fig. 5). No adsorption sites could be found for N2, most likely due to the poor uptake of N2 at 273 K in this material. Again, the positions of CO2-A and CO2-B across three loadings (VI, VII, VIII) remain consistent with those found in the single component CO2 structures. The CO2 occupancy plot from these refinements (Fig. 4c) again show excellent agreement with both the adsorption isotherm and the occupancy plot from the single component experiment, which shows that CO2 binding in MFM-300(VIII) does not change significantly in the presence of N2. Thus, this PXRD experiment on mixed gas loading affords direct structural insight into the competitive gas binding, underpinning the high adsorption selectivity of this MOF. | ||
| Fig. 5 Top: Difference Fourier maps of CO2/N2-loaded MFM-300(VIII) from 300–1000 mbar viewed along the c-axis. Middle: Structures of CO2/N2-loaded MFM-300(V) from dual component experiments from 300–1000 mbar viewed along the c-axis. Bottom: View of the interactions of CO2-A with the framework hydroxyl group and its two interactions with CO2-B. In the case of the OHAOCO interaction, OAOCO distances are shown in brackets. For clarity, only half of the positionally disordered CO2 molecules are shown. | ||
3.4. Analysis of gas permeation in MFM-300(VIII)/PIM-1 under dynamic conditions
The membrane was cast from solution with a surface modifier (Fig. 6). This allows fabrication of a uniform membrane with a homogenous distribution of MOF particles, and more significantly, without any macro-voids between the MOF particles and the polymer matrix as indicated by SEM analysis (Fig. 7). EDX elemental analysis also confirms the even distribution of the MOF throughout the membrane (Fig. 8). PIM-1 was used for this study due to its proven gas separation performance and inherent porosity.38 Single gas (CO2, N2, CH4, H2 and He) tests were performed on a 25 wt% MFM-300(VIII) loaded MMM in order to assess permeation performance (Table S3†). Addition of MFM-300(VIII) showed an improvement in CO2 permeation and ideal CO2/N2 selectivity over that of the bare PIM-1. The improvement in CO2 permeation in the MMM can be primarily attributed to the high chemical affinity shown of the 1D pore channels of MFM-300(VIII) and CO2 molecules as can be seen from the in situ PXRD studies described above. All permeation measurements were carried out at a range of temperatures (287–343 K) and a general increase in permeation was observed with rising temperature as expected, with the exception of CO2 which showed an initial increase in permeability between 287 K and 303 K followed by a decrease in permeability (Fig. 9). The initial increase in CO2 permeability between 287 K and 303 K, from 4450 Barrer to 4617 Barrer, could be explained by the increased speed of diffusion of the gas through the membrane, from 76 × 108 cm2 s−1 to 107 × 108 cm2 s−1 and although solubility decreases between these temperatures this does not offset the observed diffusion. The subsequent decrease in permeability seen at increasing temperatures can be explained by the decreased solubility of CO2 with the diffusion not significantly increasing to offset this due to the strong interactions between the CO2 molecules and the hydroxy groups present within the pores of MFM-300(VIII). This is further supported by the permeability trend shown with increasing temperature for other gases tested which have a much reduced interaction with the MOF host. | ||
| Fig. 6 Schematic representation of the preparation of the MMM derives from MFM-300(VIII) and PIM-1 with a photograph of the fabricated membrane and views by SEM and EDX. | ||
 | ||
| Fig. 7 SEM images obtained using a Hitachi S-4800 Field-Emission Scanning Electron Microscope at 3 kV of (a) cross-section of the fabricated MMM at 100 μm scale; (b) image of a single particle showing good affinity between the polymer matrix and the MOF particle. | ||
 | ||
| Fig. 8 EDX elemental maps showing a uniform distribution of the MFM-300(VIII) through the PIM-1 membrane. (a) SEM image of a cross-section of the membrane; (b) cross section elemental map of V in the membrane; (c) cross section elemental map of O in the membrane. | ||
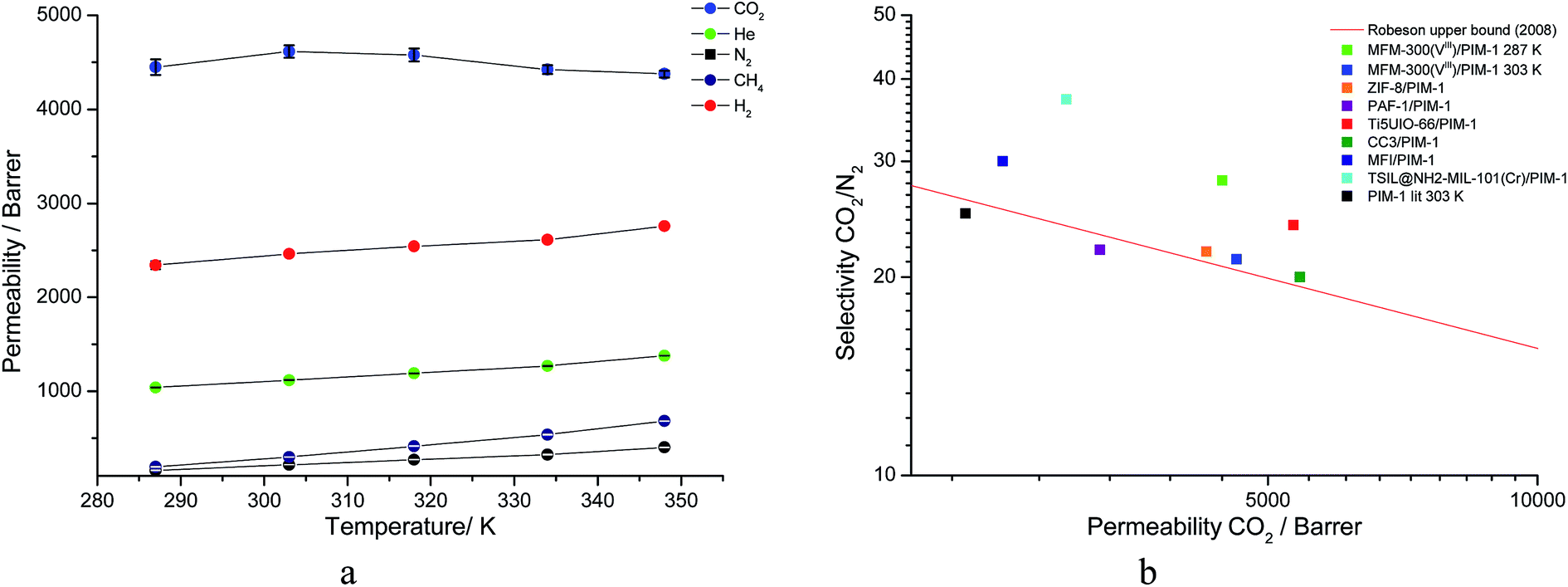 | ||
| Fig. 9 (a) Plot of change in gas permeability as a function of temperature. (b) Plot of the position of MFM-300(VIII)/PIM-1 at 287 K and 303 K in comparison to bare PIM-1 at 303 K (ref. 38) and other PIM-1 based MMMs for CO2/N2 selectivity. | ||
On the Robeson plot, it is evident that this MMM improves on the performance of the bare polymer PIM-1 and falls above the 2008 Robeson upper bound in terms of the ideal selectivity and CO2 permeability (Fig. 9). Moreover, MFM-300(VIII)/PIM-1 performs very competitively to other reported best-behaving PIM-1 based MMMs (Table 2). The new MMM also performs well when compared to membranes which have used other polymer supports. An example of this is a UiO-66-NH2/PEBA membrane fabricated by Shen et al.51 which showed a higher CO2/N2 selectivity of 66 but a much lower CO2 permeability of 87 Barrer compared to over 4000 Barrer for MFM-300(VIII)/PIM-1. Therefore, the dynamic experiments demonstrate and validate the potential of utilising this MFM-300(VIII) in CO2 separation, and the data may also be improved by applying nanosized MOF to increase the MOF surface per volume element and the interaction of MOF with the host polymer.
| Filler | Loading (wt%) | P CO2/Barrer | α (CO2/N2) | Operational conditions | Reference |
|---|---|---|---|---|---|
| a Volumetric loading for this sample as v%. | |||||
| MFM-300(VIII) | 25 | 4450 | 28.04 | 287 K, 1 bar | This work |
| MFM-300(VIII) | 25 | 4617 | 21.28 | 303 K, 1 bar | This work |
| ZIF-8 | 39 | 4270 | 21.89 | 295 K, 1 bar | 45 |
| PAF-1 | 30 | 3250 | 22 | 298 K, 1 bar | 46 |
| Ti5UIO-66 | 5 | 5340 | 24 | 298 K, 2 bar | 47 |
| CC3 | 30 | 5430 | 20 | 298 K, 1 bar | 48 |
| MFI | 23.5a | 2530 | 30 | 298 K, 1 bar | 49 |
| TSIL@NH2-MIL-101(Cr) | 5 | 2979 | 37.24 | 298 K, 3 bar | 50 |
4. Conclusions
In summary, a new remote control gas dosing system capable of millibar precision and fully automated operation has been successfully implemented on the powder diffraction Beamline I11 at Diamond Light Source. This system has shown unique in situ structural characterisation of MFM-300(VIII) as a function of loading of CO2, N2 and an equimolar mixture of CO2/N2. Rietveld structural analysis confirmed the presence of two distinct CO2 sites within the MOF pore with a hydrogen bond interaction between the framework μ-OH and CO2-A, and two further intermolecular dipole interactions between CO2-B and CO2-A. The positions of CO2-A and CO2-B were found to remain generally consistent across the entire pressure range. Upon dosing with an equimolar mixture of CO2 and N2, a similar network of CO2-A and CO2-B was observed in MFM-300(VIII). Occupancy plots from both the single and dual component experiments show excellent agreement with the adsorption isotherms, confirming the precision of the gas dosing system and the structural analysis. The fabrication of a MMM based upon PIM-1 doped with 25 wt% MFM-300(VIII) has been demonstrated. The inclusion of the MOF leads to an increased permeation to CO2 and an improvement of the ideal gas separation performance for CO2/N2 over the bare polymer. This increased CO2 permeation is likely due to the high chemical affinity of MFM-300(VIII) for CO2 as confirmed by in situ PXRD studies. Importantly, the newly formed MMM also falls above the Robeson 2008 upper bound for membrane performance at both 287 K and 303 K, demonstrating the potential of utilising this material in the application of CCS owing to its optimal compromise between selectivity and permeability.Acknowledgements
We thank EPSRC, ERC (Advanced Grant to MS), Diamond Light Source and University of Manchester for funding. We are especially grateful to Diamond for access to the Beamline I11. CGM is supported by a Diamond PhD studentship.References
- J. D. Figueroa, T. Fouta, S. Plasynski, H. McIlvried and R. D. Srivastavab, Int. J. Greenhouse Gas Control, 2008, 2, 9–20 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- M. H. Weston, W. Morris, P. W. Siu, W. J. Hoover, D. Cho, R. K. Richardson and O. K. Farha, Inorg. Chem., 2015, 54, 8162–8164 CrossRef CAS PubMed.
- Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- S. Ma and H. C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC.
- S. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schröder, Nat. Chem., 2015, 7, 121–129 CrossRef CAS PubMed.
- S. J. Geier, J. A. Mason, E. D. Bloch, W. L. Queen, M. R. Hudson, C. M. Brown and J. R. Long, Chem. Sci., 2013, 4, 2054–2061 RSC.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- A. N. Mlinar, B. K. Keitz, D. Gygi, E. D. Bloch, J. R. Long and A. T. Bell, ACS Catal., 2014, 4, 717–721 CrossRef CAS.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed.
- Z. Hu, B. J. Deiberta and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- J. L. C. Rowsell, E. C. Spencer, J. Eckert, J. A. K. Howard and O. M. Yaghi, Science, 2005, 309, 1350–1354 CrossRef CAS PubMed.
- J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2009, 131, 5516–5521 CrossRef CAS PubMed.
- S. Takamizawa, E. Nataka, T. Akatsuka, R. Miyake, Y. Kakizaki, H. Takeuchi, G. Maruta and S. Takeda, J. Am. Chem. Soc., 2010, 132, 3783–3792 CrossRef CAS PubMed.
- E. C. Spencer, J. A. K. Howard, G. J. McIntyre, J. L. C. Rowsell and O. M. Yaghi, Chem. Commun., 2006, 278–280 RSC.
- C. P. Krap, R. Newby, A. Dhakshinamoorthy, H. García, I. Cebula, T. L. Easun, M. Savage, J. E. Eyley, S. Gao, A. J. Blake, W. Lewis, P. H. Beton, M. R. Warren, D. R. Allan, M. D. Frogley, C. C. Tang, G. Cinque, S. Yang and M. Schröder, Inorg. Chem., 2016, 55, 1076–1088 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- Y. Yan, I. Telepeni, S. Yang, X. Lin, W. Kockelmann, A. Dailly, A. J. Blake, W. Lewis, G. S. Walker, D. R. Allan, S. A. Barnett, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 4092–4094 CrossRef CAS PubMed.
- M. Savage, I. da Silva, M. Johnson, J. H. Carter, R. Newby, M. Suyetin, E. Besley, P. Manuel, S. Rudić, A. N. Fitch, C. Murray, W. I. F. David, S. Yang and M. Schröder, J. Am. Chem. Soc., 2016, 138, 9119–9127 CrossRef CAS PubMed.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 3022–3030 CrossRef CAS PubMed.
- V. Bon, I. Senkovska, D. Wallacher, A. Heerwig, N. Klein, I. Zizak, R. Feyerherm, E. Dudzik and S. Kaskel, Microporous Mesoporous Mater., 2014, 188, 190–195 CrossRef CAS.
- J. E. Parker, J. Potter, S. P. Thompson, A. R. Lennie and C. C. Tang, Mater. Sci. Forum, 2012, 706–709, 1707–1712 CrossRef CAS.
- T. Mitra, R. S. Bhavsar, D. J. Adams, P. M. Budd and A. I. Cooper, Chem. Commun., 2016, 52, 5581 RSC.
- V. Nafisi and M.-B. Hägg, J. Membr. Sci., 2014, 459, 244–255 CrossRef CAS.
- S. Kulprathipanja, R. W. Neuzil and N. N. Li, US Pat., 4 740 219 (A), 1988.
- S. Kulprathipanja, E. W. Funk, S. S. Kulkarni and Y. A. Chang, US Pat., 4 735 193 (A), 1988 CrossRef CAS. See also: A. Singh and W. J. Koros, Ind. Eng. Chem. Res., 1996, 35, 1231–1234 CrossRef CAS; C. M. Zimmerman, A. Singh and W. J. Koros, J. Membr. Sci., 1997, 137, 145–154 CrossRef.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- G. Saracco, H. W. J. P. Neomagus, G. F. Versteeg and W. P. M. Van Swaaij, Chem. Eng. Sci., 1999, 54, 1997–2017 CrossRef CAS.
- M. G. Süer, N. Baç and L. Yilmaz, J. Membr. Sci., 1994, 91, 77–86 CrossRef.
- B. Zornoza, A. Martinez-Joaristi, P. Serra-Crespo, C. Tellez, J. Coronas, J. Gascon and F. Kapteijn, Chem. Commun., 2011, 47, 9522–9524 RSC.
- S. P. Thompson, J. E. Parker, J. Potter, T. P. Hill, A. Birt, T. M. Cobb, F. Yuan and C. C. Tang, Rev. Sci. Instrum., 2009, 80, 075107 CrossRef CAS PubMed.
- P. W. Stephens, J. Appl. Crystallogr., 1999, 32, 281–289 CrossRef CAS.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS.
- B. D. Freeman and H. Lin, in Springer Handbook of Materials Measurement Methods, ed. H. Czichos, T. Saito and L. Smith, Springer, New York City, 1st edn, 2006, ch. 7, pp. 371–387 Search PubMed.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- Z. Lu, H. G. W. Godfrey, I. da Silva, Y. Cheng, M. Savage, F. Tuna, E. J. L. McInnes, S. J. Teat, K. J. Gagnon, M. D. Frogley, P. Manuel, S. Rudić, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, Nat. Commun., 2017, 8, 14212 CrossRef CAS PubMed.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637–20640 CrossRef CAS PubMed.
- P. D. C. Dietzel, V. Besikiotisa and R. Bloma, J. Mater. Chem., 2009, 19, 7362–7370 RSC.
- A. Simon and K. Peters, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1980, B36, 2750–2751 CrossRef CAS.
- A. F. Bushell, M. P. Attfield, C. R. Mason, P. M. Budd, Y. Yampolskii, L. Starannikova, A. Rebrov, F. Bazzarelli, P. Bernardo, J. Carolus Jansen, M. Lanč, K. Friess, V. Shantarovich, V. Gustov and V. Isaeva, J. Membr. Sci., 2013, 427, 48–62 CrossRef CAS.
- C. H. Lau, K. Konstas, A. W. Thornton, A. C. Y. Liu, S. Mudie, D. F. Kennedy, S. C. Howard, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2015, 54, 2669–2673 CrossRef CAS PubMed.
- S. J. D. Smith, B. P. Ladewig, A. J. Hill, C. H. Lau and M. R. Hill, Sci. Rep., 2015, 5, 7823 CrossRef CAS PubMed.
- A. F. Bushell, P. M. Budd, M. P. Atteld, J. T. A. Jones, T. Hasell, A. I. Cooper, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Angew. Chem., Int. Ed., 2013, 52, 1253 CrossRef CAS PubMed.
- C. R. Mason, M. G. Buonomenna, G. Golemme, P. M. Budd, F. Galiano, A. Figoli, K. Friess and V. Hynek, Polymer, 2013, 54, 2222–2230 CrossRef CAS.
- J. Ma, Y. Ying, X. Guo, H. Huang, D. Liu and C. Zhong, J. Mater. Chem. A, 2016, 4, 7281–7288 CAS.
- J. Shen, G. Lui, K. Huang, Q. Li, K. Guan, Y. Li and W. Jin, J. Membr. Sci., 2016, 513, 155–165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1504685–1504693. See DOI: 10.1039/c6sc04343g |
| This journal is © The Royal Society of Chemistry 2017 |