
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO/CO and NO/NO coupling at a hidden frustrated Lewis pair template†
Tongdao
Wang
a,
Long
Wang
a,
Constantin G.
Daniliuc
a,
Kamil
Samigullin
b,
Matthias
Wagner
b,
Gerald
Kehr
 a and
Gerhard
Erker
*a
a and
Gerhard
Erker
*a
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: erker@uni-muenster.de
bInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, 60438 Frankfurt am Main, Germany
First published on 4th January 2017
Abstract
N-Allyltetramethylpiperidine is readily isomerized to the corresponding enamine by treatment with catalytic amounts of B(C6F5)3. It adds HB(C6F5)2 at the nucleophilic enamine carbon atom to form a C/B Lewis adduct. This reacts with two molar equivalents of carbon monoxide by selective head to tail coupling to give a five-membered C2O2B heterocycle. In contrast the enamine/HB(C6F5)2 Lewis pair reacts with two molar equiv. of nitric oxide by head to head coupling. This reaction probably proceeds via equilibrium with the corresponding vicinal N/B Lewis pair. Most products were characterized by X-ray diffraction.
Introduction
Frustrated Lewis pairs (FLPs) have become well known for their ability of binding and/or activating a variety of small molecules.1,2 The reactivity of e.g. the ethylene-bridged P/B FLP 13 toward carbon monoxide is remarkable. Compound 1 adds both the phosphane Lewis base and the borane Lewis acid to the CO carbon atom to yield the five-membered heterocyclic carbonyl compound 2.4 This behavior of FLP 1 remotely resembles CO/metal coordination chemistry5 and in contrast to the conventional role of such P/B pairs as ambiphilic ligands.6 Compound 1 reacts analogously with nitric oxide (NO), again serving in a “pseudo-metal like” role, giving the persistent nitroxide radical 3 (see Scheme 1).7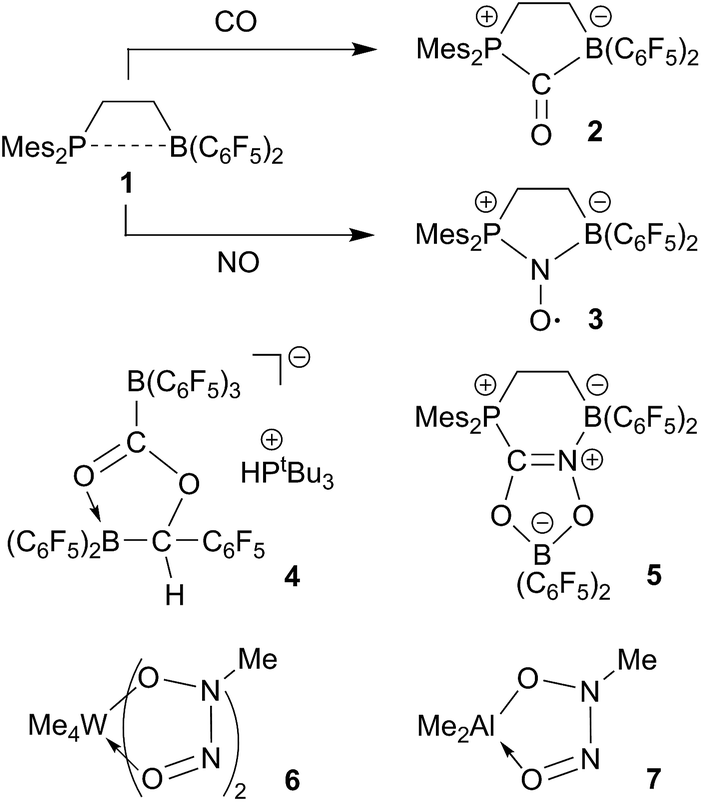 | ||
| Scheme 1 | ||
There are very few first cases where frustrated Lewis pairs induce coupling of two such element oxides. Stephan et al. had found the formation of 4 upon treatment of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of B(C6F5)3 and PtBu3 with CO/H2 (ref. 8) and we recently reported the first example of a CO/NO coupling reaction at the FLP 1 and elucidated the pathway of the unprecedented formation of product 5.9 It is also well known that a variety of alkyl or aryl transition metal and main group metal complexes, respectively, undergo reactions involving the head to head coupling of two nitric oxide (NO) molecules to give the respective O-metallated N-hydrocarbyl-N-nitroso-hydroxylaminato metal complexes.10–15 The tungsten and aluminum complexes 6 and 7 are representative examples. There have also been early reports about the reaction of Et3B with NO to apparently give a Et2B–ON(Et)NO type product.16
:1 mixture of B(C6F5)3 and PtBu3 with CO/H2 (ref. 8) and we recently reported the first example of a CO/NO coupling reaction at the FLP 1 and elucidated the pathway of the unprecedented formation of product 5.9 It is also well known that a variety of alkyl or aryl transition metal and main group metal complexes, respectively, undergo reactions involving the head to head coupling of two nitric oxide (NO) molecules to give the respective O-metallated N-hydrocarbyl-N-nitroso-hydroxylaminato metal complexes.10–15 The tungsten and aluminum complexes 6 and 7 are representative examples. There have also been early reports about the reaction of Et3B with NO to apparently give a Et2B–ON(Et)NO type product.16
We had previously shown that enamines can react with Piers' borane17 by addition of the Lewis acidic borane at the nucleophilic β-carbon atom. Nevertheless some of these systems were H2-activators, probably via dissociation of the Lewis pair adduct followed by in situ generation of the respective vicinal N/B FLP.18 We have now conveniently prepared an enamine/HB(C6F5)2 Lewis pair adduct containing the very bulky tetramethylpiperidinyl (TMP) moiety19 by an isomerization route starting from the respective allylamine precursor.20 The TMP-enamine/HB(C6F5)2 C/B adduct was shown to undergo CO/CO and NO/NO coupling reactions in rather differently oriented pathways. This opposing set of reactions will be described and discussed in this account.
Results and discussions
CO/CO coupling with the enamine/HB(C6F5)2 C/B addition product
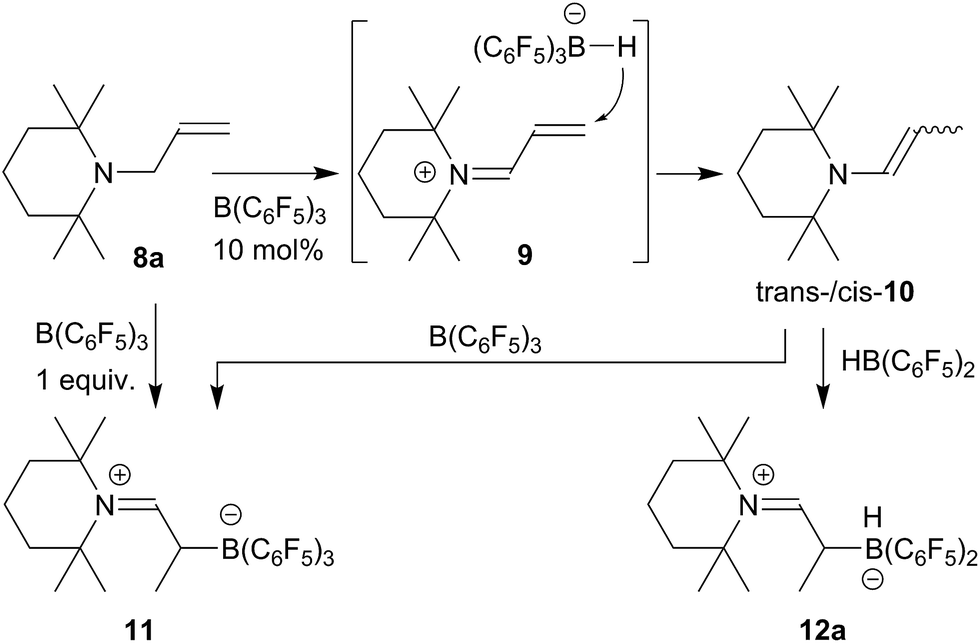
For this part of our study we started from N-allyl tetramethylpiperidine (8a) which was isomerized to the enamine 10 (trans/cis mixture of ∼10:8) by treatment with a catalytic quantity (10 mol%) of the strong boron Lewis acid B(C6F5)3 (toluene, r.t., 2 d).21 The trans-/cis-enamine mixture was isolated by distillation. Compound trans-10 shows an olefinic [N]–CH = 1H NMR resonance at δ 5.75 ppm (dq, 3JHH = 13.4 Hz, 4JHH = 1.6 Hz), whereas cis-10 features the respective olefinic resonance at δ 5.68 ppm (dq, 3JHH = 7.8 Hz, 4JHH = 1.8 Hz). We assume a reaction pathway (see Scheme 2) that involves hydride abstraction18,22 in the α-position to the amine to give the conjugated iminium/hydridoborate salt 9 followed by H− addition to the terminal ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 group.
CH2 group.
 | ||
| Scheme 2 | ||
The enamine isomers 10 rapidly add B(C6F5)3 to the enamine carbon atom18 (n-pentane, r.t., 30 min) to give the zwitterionic adduct 11 (isolated in 93% after workup). Compound 11 can also be obtained directly from the allylamine 8a by treatment with a stoichiometric quantity of B(C6F5)3 (toluene, r.t., 30 min); in this case in situ generation of the enamine is assumed. We isolated compound 11 in 81% from this stoichiometric reaction. Compound 11 shows a 11B NMR resonance at δ −11.9 ppm and a typical set of 19F NMR signals of o,p,m-fluorines of the three C6F5-groups at boron with Δδ19Fm,p = 4.5 ppm. The 1H/13C NMR iminium [N]CH– resonances were located at δ 8.74/δ 189.3 ppm, respectively. Compound 11 was also characterized by X-ray diffraction. The structure is depicted in the ESI.†
The reaction of the enamine isomer mixture 10 with Piers' borane [HB(C6F5)2]17 was carried out analogously (n-pentane, r.t., 30 min) and gave the zwitterionic addition product 12a as a white solid, isolated in 87% yield. Compound 12a was characterized by an X-ray crystal structure analysis (suitable single crystals were obtained at −35 °C from a dichloromethane solution covered with a n-pentane layer). In the structure (see Fig. 1) the bulky tetramethylpiperidide unit is part of the iminium functional group. It has the remaining saturated C2-unit attached, which features the –B(H)(C6F5)2 substituent bonded to carbon atom C2. The boron atom shows a pseudo-tetrahedral coordination geometry with a sum of heavy atom bond angles of ∑B1CCC = 335.6°. The structure of the B(C6F5)3/enamine adduct 11 is very similar (see the ESI†).
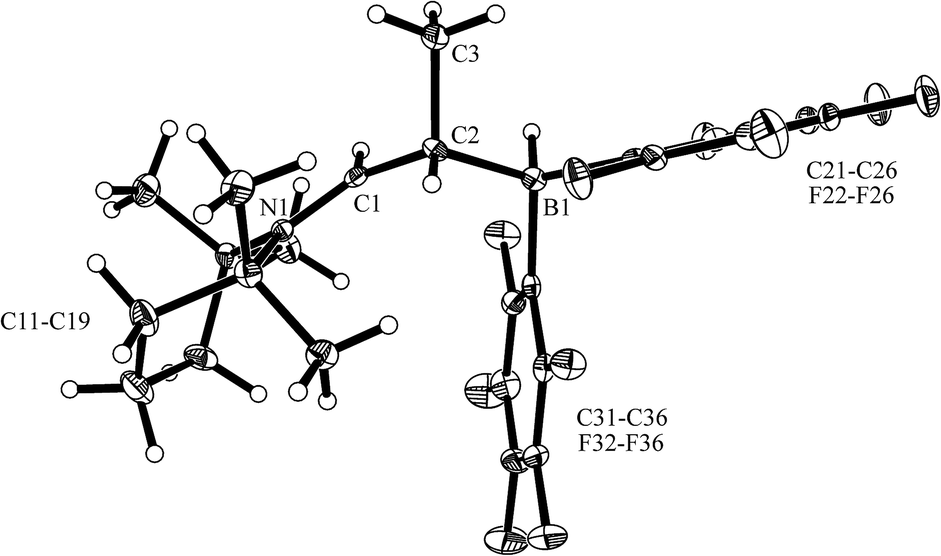 | ||
| Fig. 1 The molecular structure of the enamine/HB(C6F5)2 addition product 12a (thermal ellipsoids are shown at the 50% probability level). Selected bond lengths (Å) and angles (°): N1–C1 1.299(2), C1–C2 1.463(2), C2–C3 1.549(2), C2–B1 1.682(3), B1–C21 1.636(3), B1–C31 1.649(3); N1–C1–C2 131.8(2), C1–C2–C3 108.6(2), C1–C2–B1 106.2(1), N1–C1–C2–B1 125.4(2). | ||
In solution (CD2Cl2) compound 12a shows the 1H/13C NMR resonances of the [N]CH– iminium functionality at δ 8.12 (d, 3JHH = 13.1 Hz) and δ 189.2 ppm, respectively. The 11B NMR signal occurs at δ −20.0 ppm as a doublet (1JBH ∼ 93 Hz). The corresponding 1H NMR [B]–H resonance was located at 2.74 ppm as a broad 1:1:1:1 intensity quartet. Due to the chiral center (C2) we observe a 1:1 pair of o,p,m-19F NMR signals and a total of four tetramethylpiperidino 1H NMR methyl group signals.
We reacted the enamine/HB(C6F5)2 adduct 12a with carbon monoxide (CH2Cl2, r.t., 2.0 bar). The reaction was followed in situ by NMR spectroscopy. This showed the formation of the new product 13a (which contained two B(C6F5)2 units) and the boron free enamine 10. After 3 h reaction time workup eventually furnished the product 13a, isolated as a white solid in 43% yield. Since we needed two molar equiv. of HB(C6F5)2 for a complete conversion of 12a to the carbonylation product 13a, we reacted the precursor 12a with CO under similar conditions in the presence of an additional molar equivalent of Piers' borane. Workup after 3 h reaction time involving washing with CH2Cl2 gave the product 13a in 41% yield (for details see the ESI†).
The X-ray crystal structure analysis of 13a (Fig. 2, single crystals were obtained at −35 °C from a THF solution covered with a layer of n-pentane) revealed that a central five-membered heterocycle had been formed by head to tail coupling of two CO-molecules.8 The carbon atom of one of them (C4) now bears a hydrogen originating from a HB(C6F5)2 equivalent and the enamine derived substituent. The other CO equivalent is found C–O bonded between a pair of boron atoms. Both the boron atoms feature pseudotetrahedral coordination geometries (∑B1CCC = 344.4°, ∑B2CCC = 337.5°). The enamine derived residue shows an iminium functionality.
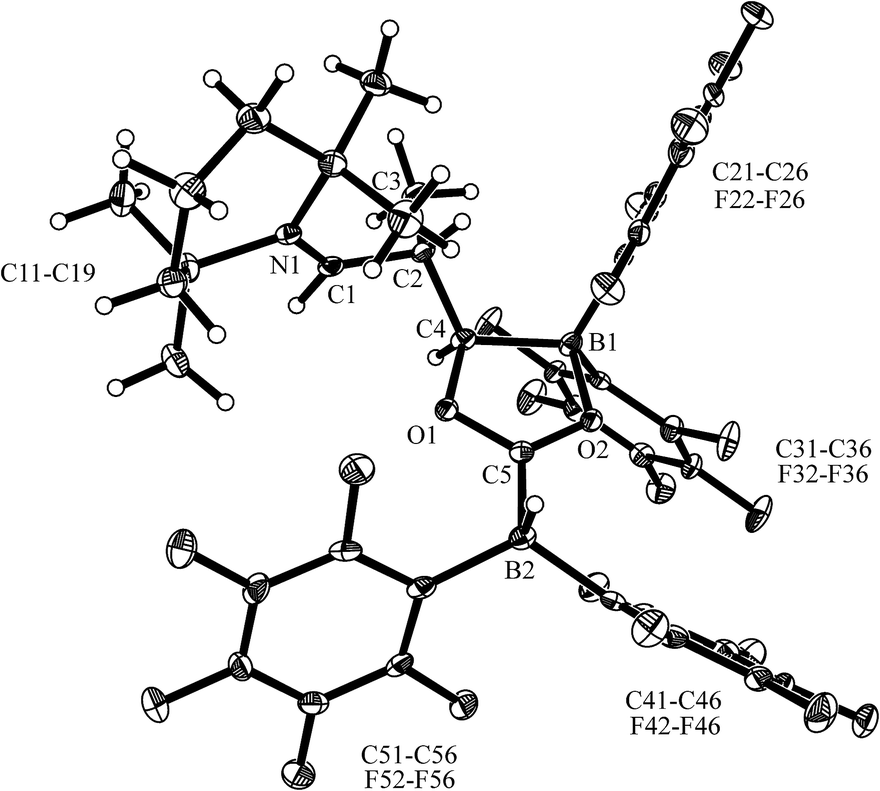 | ||
| Fig. 2 A projection of the carbonylation product 13a (thermal ellipsoids are shown at the 50% probability level). Selected bond lengths (Å) and angles (°): N1–C1 1.286(3), C1–C2 1.498(3), C4–B1 1.658(3), B1–O2 1.551(3), O2–C5 1.272(2), B2–C5 1.604(3), C5–O1 1.320(2), O1–C4 1.494(3); C4–O1–C5 111.5(2), B1–O2–C5 113.4(2). | ||
In solution (THF-d8) compound 13a features 1H/13C NMR resonances of the iminium moiety at δ 8.68 (3JHH = 10.4 Hz) and 184.5 ppm ([N]CH–), respectively. The central CO derived –O–C[B]–O– carbon atom shows a 13C NMR signal at δ 213.7 ppm (C5), whereas the [B]CH carbon atom (C4 in Fig. 2) shows a broad 13C NMR resonance δ 86.2 ppm.
Compound 13a shows a pair of 11B NMR signals at δ 2.6 and −26.1 ppm (1JBH = 85.8 Hz). Due to the C2–chirality center both the B(C6F5)2 units show the 19F NMR signals of pairs of diastereotopic C6F5 substituents.
We assume that the enamine/HB(C6F5)2 C/B-adduct becomes reversible under the applied carbonylation conditions and we, consequently, observe a reaction under frustrated Lewis pair (FLP) conditions.18,20,23,24 CO activation can occur by formation of Piers' borane carbonyl25,26 [OC–B(H)(C6F5)2] 14 as we had recently shown (we had actually isolated 14 at low temperature and characterized this borane carbonyl by X-ray diffraction). FLP addition of the enamine nucleophile followed by hydridoborate reduction of the acyl group in 15 would then lead to the intermediate 16 which could add to a second equivalent of the borane carbonyl to eventually yield the observed CO coupling product 13a (see Scheme 3).
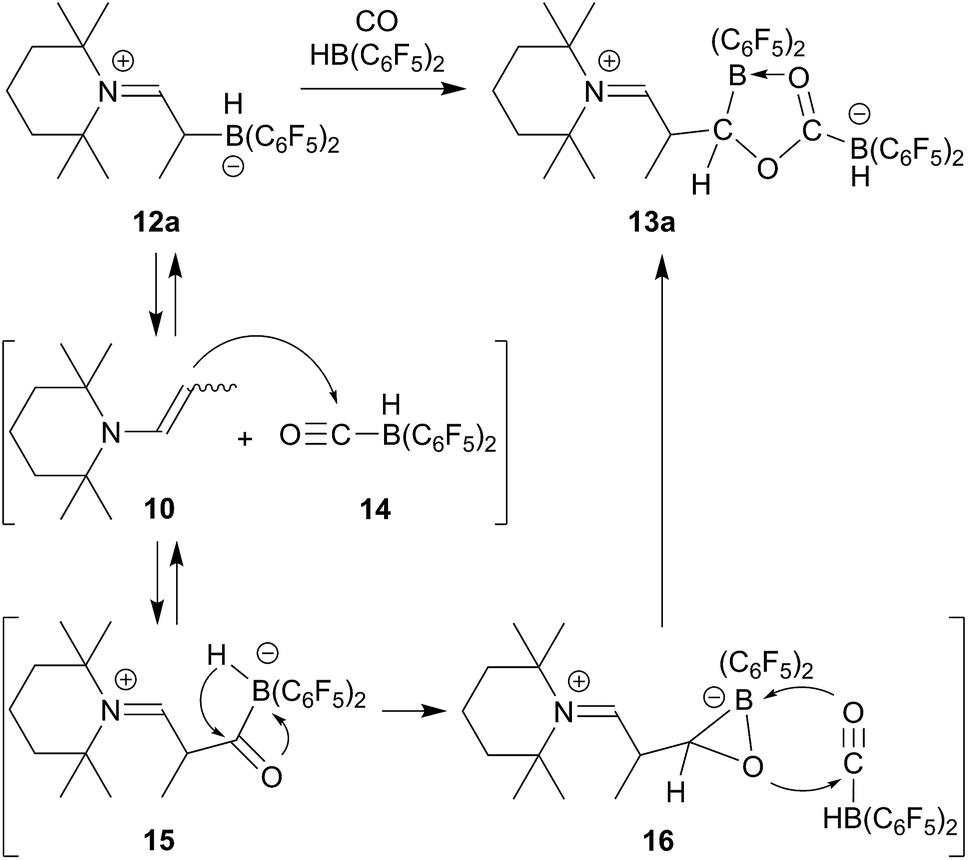 | ||
| Scheme 3 | ||
The reaction of the enamine/HB(C6F5)2 FLP 12a with CO proceeds similarly in the presence of one molar equiv. of the strong boron Lewis acid B(C6F5)3. The reaction was carried out at r.t. in dichloromethane with 2.0 bar carbon monoxide pressure. Workup after 3 h reaction time eventually gave the CO coupling product 13b as a white solid, isolated in 76% yield. In THF-d8 solution it shows a pair of 11B NMR signals at δ 2.6 and δ −17.7 ppm. We monitor a single set of o,p,m-C6F519F NMR resonances of the exocyclic B(C6F5)3 substituent and the corresponding resonances of a pair of diastereotopic C6F5 groups of the endocyclic B(C6F5)2 moiety. Compound 13b was also characterized by X-ray diffraction. Its structure is analogous to that of 13a (for details see the ESI†). We assume a similar reaction scheme for the formation of 13b as we had discussed it for 13a (see above) (Scheme 4).
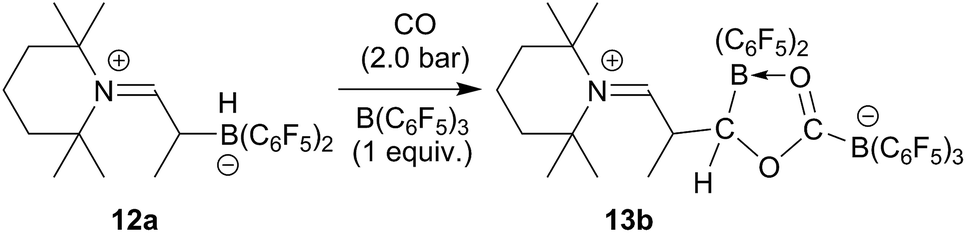 | ||
| Scheme 4 | ||
NO/NO coupling with the enamine/HB(C6F5)2 C/B Lewis pair
The enamine/HB(C6F5)2 adduct 12a reacted equally well with nitric oxide (NO). A solution of 12a in dichloromethane was exposed to a NO atmosphere (1.0 bar) for 3 h. Workup by removing the solvent in vacuo and washing of the residue with n-pentane gave the NO coupling product 17a as a pale yellow solid. It was isolated in 76% yield (see Scheme 5). Single crystals of compound 17a suited for the X-ray crystal structure analysis were obtained from a n-pentane solution at −35 °C (see Fig. 3). It shows that two NO molecules had been head to head coupled. This has formed the observed central planar five-membered N2O2B containing heterocycle. It contains a short N2N3 linkage and pairs of N–O and B–O bonds in the σ-bond length range. Nitrogen atom N2 has the amino-alkyl substituent attached to it. We note that this does not contain the iminium moiety of the starting material (12a) any more but instead features a saturated tert amine [N]–CH2– unit.
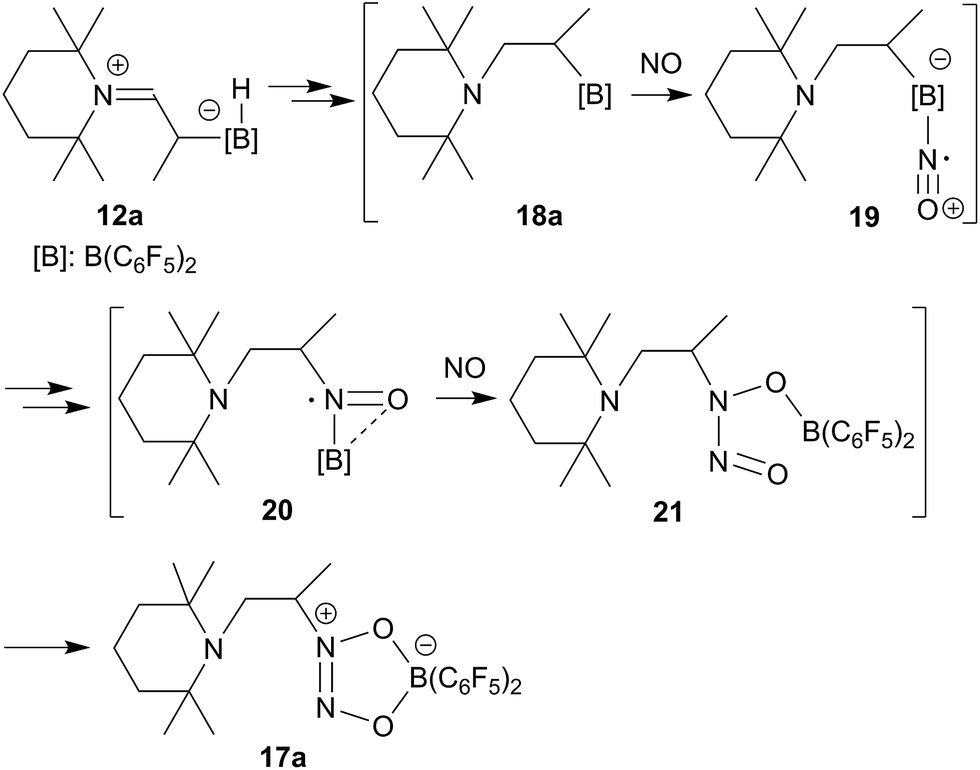 | ||
| Scheme 5 | ||
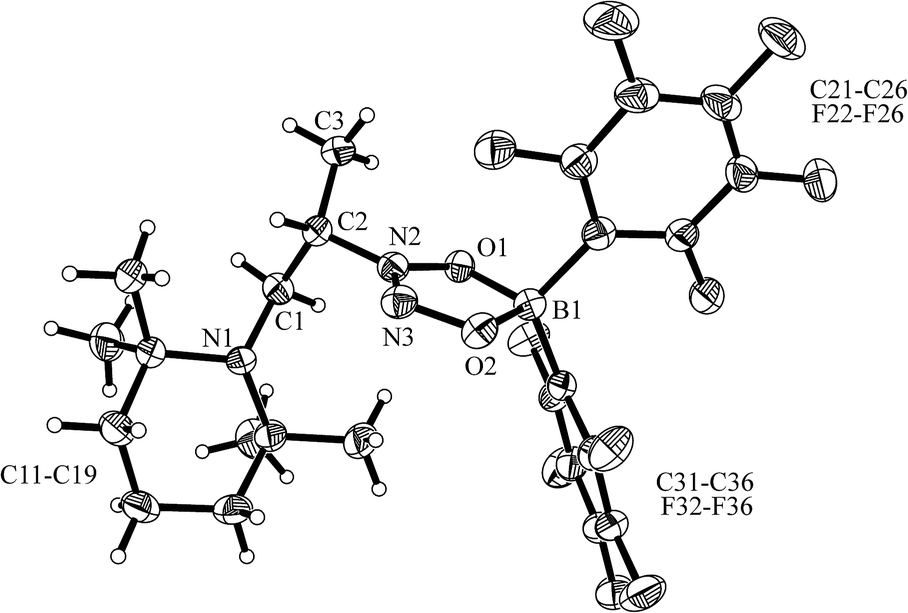 | ||
| Fig. 3 Molecular structure of the NO coupling product 17a (thermal ellipsoids are shown at the 30% probability level). Selected bond lengths (Å) and angles (°): N1–C1 1.459(3), N2–C2 1.473(3), N2–N3 1.256(3), N2–O1 1.344(2), N3–O2 1.333(3), O1–B1 1.527(3), O2–B1 1.538(3); N1–C1–C2 114.5(2), C2–N2–O1 119.2(2), O1–N2–N3 117.3(2), N2–O1–B1 105.6(2), N3–O2–B1 110.3(2), O1–B1–O2 97.6(2). | ||
In solution compound 17a shows the 1H NMR signals of the [N]–CH2–CH– section of the substituent at δ 2.72, 3.05 (CH2, 2JHH = 16.4 Hz, 3JHH = 9.5 Hz, 13C: δ 47.9) and δ 4.80 ppm (CH, 13C: δ 69.3), respectively. It features a 11B NMR resonance at δ +12.2 ppm and two sets of o,p,m-19F NMR signals of the pair of diastereotopic C6F5 substituents at boron.
From the structure of compound 17a (see Fig. 3), which contains the saturated [N]–CH2–CH– moiety, we must assume that the NO reaction started from the N/B FLP isomer 18a (see Scheme 5 and below).20,23,24 NO addition to boron would give the radical 19 which might have rearranged to its isomer 20.20,27 Trapping with a second equivalent of NO would then straightforwardly lead to 17a. We must stress that this pathway which is depicted in Scheme 5 provides a mere possible rationalization for the observed formation of the NO/NO coupling product; so far none of the alleged intermediates has been observed directly.
We have, however, obtained additional indirect evidence for the formation of the N/B FLP 18a, which is apparently in an endothermal equilibrium situation with 12a (a situation that had previously been observed with other enamine/RB(C6F5)2 borane addition products as well).18 In this case we added a slight excess of phenylacetylene to a solution of the enamine/HB(C6F5)2 adduct 12a in dichloromethane solution. The mixture was kept at r.t. for 1 h. Workup then gave the reaction product 22, which we isolated in 91% yield (see Scheme 6).
 | ||
| Scheme 6 | ||
The X-ray crystal structure analysis of 22 (see Fig. 4) revealed that we had apparently trapped the typical N/B FLP reaction product with the terminal acetylene.28 The alkyne had become deprotonated to give the ammonium cation section of 22 and the resulting alkynyl carbanion had become attached to the boron atom. In the zwitterionic compound a saturated –CH2–CH(CH3)– bridge is formed connecting the boron and nitrogen atoms. Consequently, we observe the 13C NMR acetylide signals at δ 111.9 and 98.4 ppm, respectively (11B: δ −17.3 ppm) and a broad NH 1H NMR resonance at δ 6.08 ppm. We assume that the adduct formation of the enamine 10 with Piers' borane to give 12a is reversible in solution. From this equilibrium there might be an (endothermic) pathway of hydroboration of the enamine double bond to generate the N/B FLP 18ain situ in an equilibrium situation. This we have not observed as such,18 but we assume that its typical N/B FLP reaction with the added phenylacetylene trapping reagent eventually led to the formation of the observed product 22 (see Scheme 6 and Fig. 4).
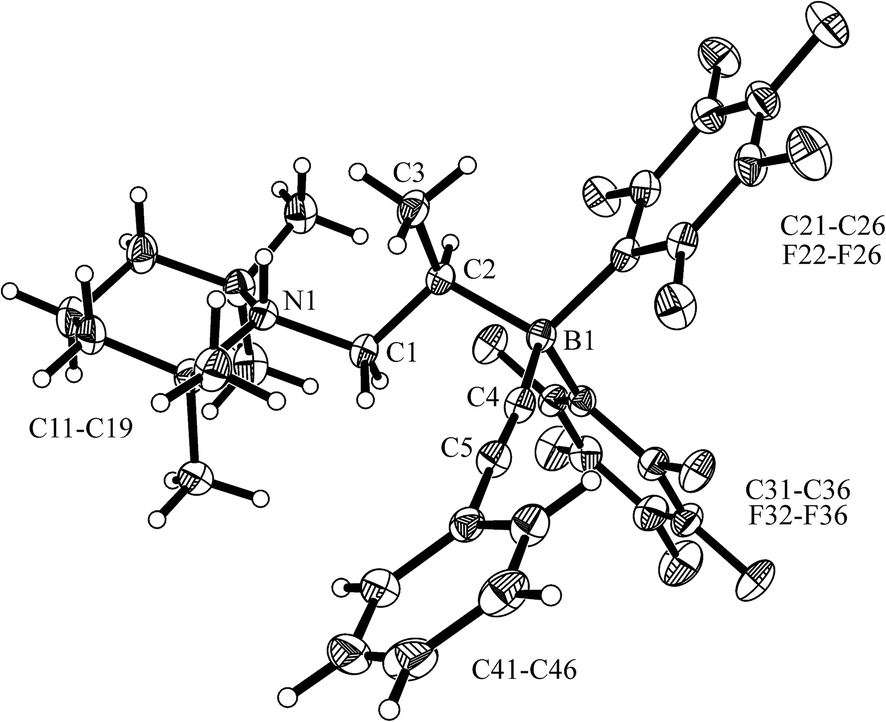 | ||
| Fig. 4 A projection of the molecular structure of compound 22 (thermal ellipsoids are shown at the 30% probability level). Selected bond lengths (Å) and angles (°): B1–C4 1.593(3), C4–C5 1.206(3), C5–C41 1.439(3), B1–C2 1.677(3), C1–C2 1.532(3), C1–N1 1.541(3); B1–C4–C5 174.3(2), C4–C5–C41 175.7(2), N1–C1–C2–B1 167.9(2), ∑N1CCC 343.5. | ||
This set the scene for the formation of the isopropyl substituted enamine/HB(C6F5)2 adduct 12b and its reaction with nitric oxide. We treated the substituted allyl amine precursor 8b20,29 with one molar equiv. of HB(C6F5)2 in n-pentane. This gave a yellow solution within 10 min. Workup after 30 min reaction time eventually gave the zwitterionic iminium/hydridoborate product 12b, which we isolated in 89% yield (see Scheme 7). The NMR spectra showed the presence of the iminium ion moiety (13C: δ 187.1 ppm), and a 11B NMR resonance at δ −21.1 ppm (d, 1JBH ∼ 92 Hz). We observed the 1H NMR signals of the [N]CH– proton at δ 8.40 ppm, the –CHMe2 isopropyl–H at δ 2.06 and the [B]H at ∼2.84 ppm (broad 1:1:1:1 quartet).
 | ||
| Scheme 7 | ||
We also did the analogous reaction of 8b with the isotopically labelled DB(C6F5)2 reagent. In the product we now find the deuterium atom scrambled over these two CH and the BH position listed above, which indicates that in this case the isomerization reaction of 8b to the respective enamine is probably taking place by means of hydroboration/retro-hydroboration (see Scheme 7, for further details see the ESI†).
Compound 12b reacted rapidly with nitric oxide. A solution of 12b in dichloromethane was stirred for 3 h at r.t. in a NO atmosphere (1.0 bar). Workup involving extraction with n-pentane eventually gave the NO coupling product 17b, which we isolated as a pale yellow solid in 69% yield (see Scheme 7). It was characterized by X-ray diffraction (see Fig. 5) and by NMR spectroscopy. The 1H NMR spectrum shows a total of six methyl group signals and an ABX type pattern of the [N]–CH2–CH– moiety [CH2: δ 2.95, 3.08 (2JHH = 16.3 Hz, 3JHH = 9.8 Hz), 4.44 (CH); 13C: δ 44.2, 78.9 ppm]. The 11B NMR resonance of compound 17b occurs at δ +12.6 ppm and we have observed two sets of o,p,m-19F NMR signals of the pair of diastereotopic C6F5 substituents at boron.
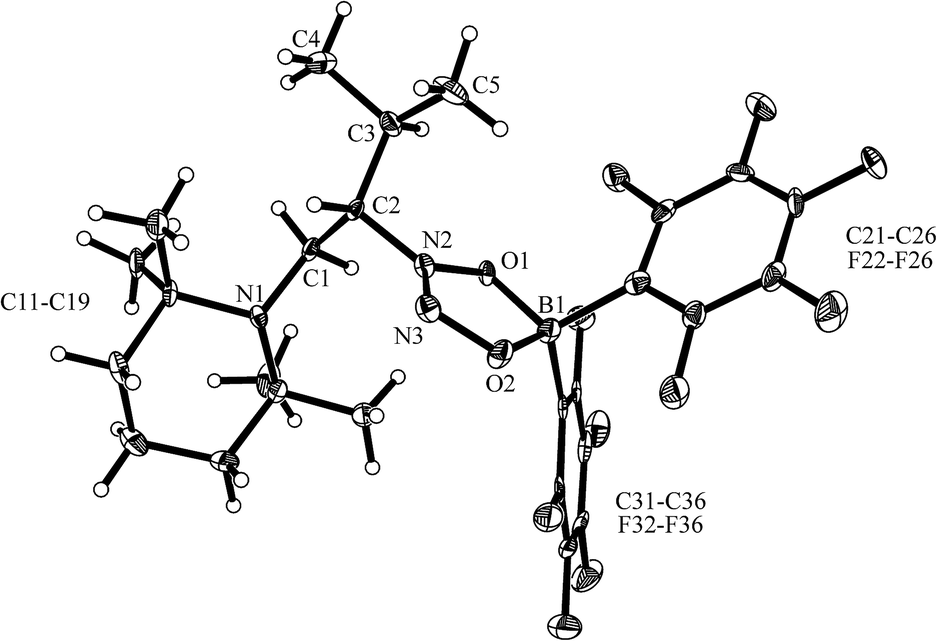 | ||
| Fig. 5 A view of the molecular structure of the NO coupling product 17b (thermal ellipsoids are shown at the 50% probability level). Selected bond lengths (Å) and angles (°): N1–C1 1.455(5), N2–C2 1.482(5), N2–N3 1.252(5), N2–O1 1.334(4), N3–O2 1.323(5), O1–B1 1.508(6), O2–B1 1.536(6); N1–C1–C2 113.5(4), C2–N2–O1 118.8(3), O1–N2–N3 117.4(4), N2–O1–B1 105.4(3), N3–O2–B1 109.4(3), O1–B1–O2 98.3(3). | ||
Conclusions
The HB(C6F5)2 adduct of the very bulky enamine 8a reacts very differently with the element oxides CO and NO.30 Its facile reaction with carbon monoxide results in a selective head to tail coupling of the CO molecule. Formally, the initial sequence might well be regarded as an addition reaction of a C/B frustrated Lewis pair31 to carbon monoxide. We assume a reaction pathway initiated by reversible HB(C6F5)2 cleavage from the adduct. It may then combine via nucleophilic enamine addition to Piers' borane carbonyl [(C6F5)2B(H)–CO 14], a reactive borane carbonyl that we had recently prepared and characterized. Hydridoborate carbonyl reduction and B/O addition to a second borane Lewis acid CO equivalent might then close the reaction cycle as it was shown in Scheme 3. It seems an essential consequence of this reaction that the enamine/HB(C6F5)2 adduct formation is reversible and that, consequently, the 10/HB(C6F5)2 ⇄ 12a system may function as a reactive C/B frustrated Lewis pair.The reaction of 12a with NO revealed another reaction type of the HB(C6F5)2/enamine C/B Lewis pair adduct. We here observe the formation of the product of selective NO/NO coupling. This is a typical reaction mode observed for many metal alkyls.10–15 Here we have probably found the analogous reaction of an alkyl–B(C6F5)2 functional group. Therefore, we assume that this reaction is initiated by the reaction of NO with the –CHR–B(C6F5)2 function of the in situ generated vicinal N/B Lewis pair, which may be formed by cleavage of the enamine/HB(C6F5)2 Lewis adduct followed by anti-Markovnikov hydroboration. The reaction then seems to follow the usual pathway, as it is often observed in the selective NO/NO coupling of metal hydrocarbyls,15 here to eventually give the respective boron based-ON(R)NO products 17. The NO coupling products1617 are topologically related to cupferron (24), a reagent that had frequently be used for chelate metal complexation.32–34 It is formed by nitrosation of N-phenyl-hydroxylamine (23) (Scheme 8). We will find out whether the boron based N-nitrosohydroxylaminato groups could potentially be used directly as reagents for metal complex formation.
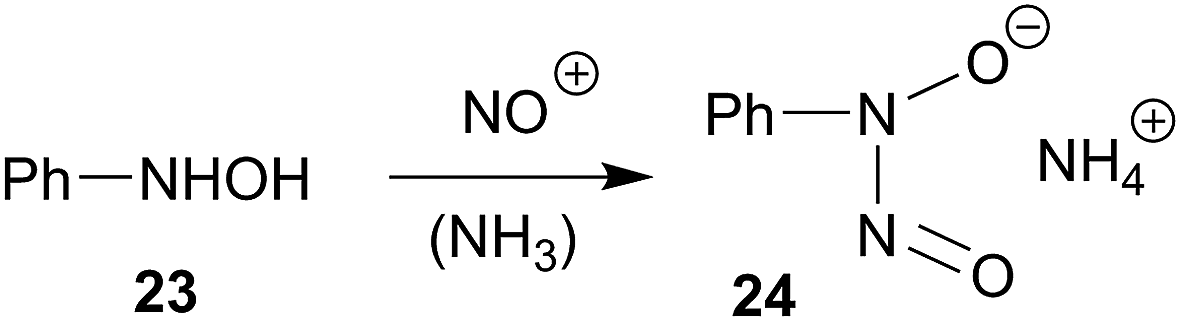 | ||
| Scheme 8 | ||
The dual reaction pathway of the enamine/HB(C6F5)2 Lewis pair 12 with CO and with NO indicates a remarkable variability of bulky Lewis acid/Lewis base combinations in small molecule chemistry.
Acknowledgements
Financial support from the European Research Council is gratefully acknowledged.Notes and references
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed.
- (a) Frustrated Lewis Pairs I, Uncovering and Understanding, Top. Curr. Chem., ed. G. Erker and D. W. Stephan, Heidelberg, 2013, vol. 332 Search PubMed; (b) Frustrated Lewis Pairs II, Expanding the Scope, Top. Curr. Chem., ed. G. Erker and D. W. Stephan, Heidelberg, 2013, vol. 334 Search PubMed.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC.
- M. Sajid, A. Lawzer, W. Dong, C. Rosorius, W. Sander, B. Schirmer, S. Grimme, C. G. Daniliuc, G. Kehr and G. Erker, J. Am. Chem. Soc., 2013, 135, 18567 CrossRef CAS PubMed.
- O. Ekkert, G. G. Miera, T. Wiegand, H. Eckert, B. Schirmer, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, S. Grimme, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 2657 RSC.
- (a) S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056 CrossRef CAS PubMed; (b) J. Grobe, K. Lütke-Brochtrup, B. Krebs, M. Läge, H.-H. Niemeyer and E.-U. Würthwein, Z. Naturforsch., B: J. Chem. Sci., 2006, 61, 882 CAS.
- (a) M. Sajid, A. Stute, A. J. P. Cardenas, B. J. Culotta, J. A. M. Hepperle, T. H. Warren, B. Schirmer, S. Grimme, A. Studer, C. G. Daniliuc, R. Fröhlich, J. L. Petersen, G. Kehr and G. Erker, J. Am. Chem. Soc., 2012, 134, 10156 CrossRef CAS PubMed; (b) M. Sajid, G. Kehr, T. Wiegand, H. Eckert, C. Schwickert, R. Pöttgen, A. J. P. Cardenas, T. H. Warren, R. Fröhlich, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 8882 CrossRef CAS PubMed; (c) J. C. M. Pereira, M. Sajid, G. Kehr, A. M. Wright, B. Schirmer, Z.-W. Qu, S. Grimme, G. Erker and P. C. Ford, J. Am. Chem. Soc., 2014, 136, 513 CrossRef CAS PubMed; (d) R. Liedtke, F. Scheidt, J. Ren, B. Schirmer, A. J. P. Cardenas, C. G. Daniliuc, H. Eckert, T. H. Warren, S. Grimme, G. Kehr and G. Erker, J. Am. Chem. Soc., 2014, 136, 9014 CrossRef CAS PubMed.
- R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 4974 CrossRef CAS PubMed.
- (a) K.-Y. Ye, G. Kehr, C. G. Daniliuc, L. Liu, S. Grimme and G. Erker, Angew. Chem., Int. Ed., 2016, 55, 9216 CrossRef CAS PubMed; (b) A description of an additional new example of this CO/NO coupling reaction was added to the ESI† of this paper.
- Tungsten: (a) S. R. Fletcher, A. Shortland, A. C. Skapski and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1972, 922 CAS; (b) S. R. Fletcher and A. C. Skapski, J. Organomet. Chem., 1973, 59, 299 CrossRef CAS; (c) A. Shortland and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1973, 872 RSC; (d) P. Legzdins, S. J. Rettig and L. Sánchez, Organometallics, 1988, 7, 2394 CrossRef CAS; (e) S. V. Evans, P. Legzdins, S. J. Rettig, L. Sánchez and J. Trotter, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, C51, 584 CrossRef CAS.
- Zirconium: (a) P. C. Wailes, H. Weigold and A. P. Bell, J. Organomet. Chem., 1972, 34, 155 CrossRef CAS; (b) A. R. Middleton and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1980, 1888 RSC; (c) C. J. Jones, J. A. McCleverty and A. S. Rothin, J. Chem. Soc., Dalton Trans., 1985, 405 RSC; (d) G. Fochi, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Chem. Soc., Dalton Trans., 1986, 445 RSC.
- Other transition metals: (a) J. D. Wilkins and M. G. B. Drew, J. Organomet. Chem., 1974, 69, 111 CrossRef CAS; (b) A. R. Middleton and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1981, 1898 RSC; (c) S. C. Puiu and T. H. Warren, Organometallics, 2003, 22, 3974 CrossRef CAS; (d) H. Lei, B. D. Ellis, C. Ni, F. Grandjean, G. J. Long and P. P. Power, Inorg. Chem., 2008, 47, 10250 CrossRef PubMed; (e) I. J. Casely, Y. Suh, J. W. Ziller and W. J. Evans, Organometallics, 2010, 29, 5209 CrossRef CAS; (f) S.-C. So, W.-M. Cheung, G.-C. Wang, E. K. Huang, M.-K. Lau, Q.-F. Zhang, H. H.-Y. Sung, I. D. Williams and W.-H. Leung, Organometallics, 2014, 33, 4497 CrossRef CAS.
- Main group metals: (a) E. Müller and H. Metzger, Chem. Ber., 1956, 89, 396 CrossRef; (b) I. S. Butler and M. L. Newbury, J. Coord. Chem., 1976, 6, 195 CrossRef; (c) S. Amirkhalili, A. J. Conway and J. D. Smith, J. Organomet. Chem., 1978, 149, 407 CrossRef CAS; (d) S. Amirkhalili, P. B. Hitchcock, J. D. Smith and J. G. Stamper, J. Chem. Soc., Dalton Trans., 1980, 2493 RSC.
- Carbanion equivalents: (a) M. J. Danzig, R. F. Martel and S. R. Riccitiello, J. Org. Chem., 1961, 65, 3327 CrossRef; (b) E. V. Arnold, L. K. Keefer and J. A. Hrabie, Tetrahedron Lett., 2000, 41, 8421 CrossRef CAS; (c) J. A. Hrabie, E. V. Arnold, M. L. Citro, C. George and L. K. Keefer, J. Org. Chem., 2000, 65, 5745 CrossRef CAS PubMed.
- Reviews: (a) J. A. McCleverty, Chem. Rev., 1979, 79, 53 CrossRef CAS; (b) J. A. Hrabie and L. K. Keefer, Chem. Rev., 2002, 102, 1135 CrossRef CAS PubMed.
- (a) S. J. Brois, Tetrahedron Lett., 1964, 345 CrossRef CAS; (b) See also: M. Inatome and L. Kuhn, Adv. Chem. Ser., 1964, 42, 183 CrossRef CAS.
- (a) D. J. Parks, R. E. von H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809 CrossRef CAS; (b) D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492 CrossRef CAS.
- (a) S. Schwendemann, R. Fröhlich, G. Kehr and G. Erker, Chem. Sci., 2011, 2, 1842 RSC; (b) S. Schwendemann, S. Oishi, S. Saito, R. Fröhlich, G. Kehr and G. Erker, Chem.–Asian J., 2013, 8, 212 CrossRef CAS PubMed.
- (a) D. M. Hodgson, C. D. Bray and N. D. Kindon, J. Am. Chem. Soc., 2004, 126, 6870 CrossRef CAS PubMed; (b) D. M. Hodgson, C. D. Bray, N. D. Kindon, N. J. Reynolds, S. J. Coote, J. M. Um and K. N. Houk, J. Org. Chem., 2009, 74, 1019 CrossRef CAS PubMed.
- T. Wang, G. Kehr, L. Liu, S. Grimme, C. D. Daniliuc and G. Erker, J. Am. Chem. Soc., 2016, 138, 4302 CrossRef CAS PubMed.
- (a) A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245 CrossRef CAS; (b) A. G. Massey, A. J. Park and F. G. A. Stone, Proc. Chem. Soc., London, 1963, 212 CAS.
- (a) F. Focante, P. Mercandelli, A. Sironi and L. Resconi, Coord. Chem. Rev., 2006, 250, 170 CrossRef CAS; (b) See also: H. Volz and H.-H. Kiltz, Liebigs Ann. Chem., 1971, 752, 86 CrossRef CAS.
- See for a comparison: (a) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 13559 CrossRef CAS PubMed; (b) K. Chernichenko, M. Nieger, M. Leskelä and T. Repo, Dalton Trans., 2012, 41, 9029 RSC; (c) K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M. Leskelä and T. Repo, Nat. Chem., 2013, 5, 718 CrossRef CAS PubMed; (d) K. Chernichenko, B. Kótai, I. Pápai, V. Zhivonitko, M. Nieger, M. Leskelä and T. Repo, Angew. Chem., Int. Ed., 2015, 54, 1749 CrossRef CAS PubMed; (e) M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef PubMed; (f) M.-A. Courtemanche, A. P. Pulis, É. Rochette, M.-A. Légaré, D. W. Stephan and F.-G. Fontaine, Chem. Commun., 2015, 51, 9797 RSC; (g) K. Chernichenko, M. Lindqvist, B. Kótai, M. Nieger, K. Sorochkina, I. Pápai and T. Repo, J. Am. Chem. Soc., 2016, 138, 4860 CrossRef CAS PubMed.
- (a) D. Winkelhaus, B. Neumann, H.-G. Stammler, R. J. F. Berger, Y. V. Vishnevskiy and N. W. Mitzel, Chem.–Eur. J., 2012, 18, 9312 CrossRef CAS PubMed; (b) L. A. Körte, S. Blomeyer, S. Heidemeyer, A. Mix, B. Neumann and N. W. Mitzel, Chem. Commun., 2016, 52, 9949 RSC.
- M. Sajid, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2014, 53, 1118 CrossRef CAS PubMed.
- See also: (a) A. G. Burg and H. I. Schlesinger, J. Am. Chem. Soc., 1937, 59, 780 CrossRef CAS; (b) T. P. Fehlner and W. S. Koski, J. Am. Chem. Soc., 1965, 87, 409 CrossRef CAS; (c) J. C. Carter, A. L. Moyé and G. W. Luther III, J. Am. Chem. Soc., 1974, 96, 3071 CrossRef CAS.
- See for a comparison: C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415 CrossRef CAS PubMed.
- (a) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396 CrossRef CAS PubMed; (b) C. M. Mömming, S. Frömel, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, J. Am. Chem. Soc., 2009, 131, 12280 CrossRef PubMed; (c) M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594 CrossRef CAS; (d) T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367 CrossRef CAS; (e) B.-H. Xu, K. Bussmann, R. Fröhlich, C. D. Daniliuc, J. G. Brandenburg, S. Grimme, G. Kehr and G. Erker, Organometallics, 2013, 32, 6745 CrossRef CAS.
- V. Sumerin, F. Schulz, M. Atsumi, C. Wang, M. Nieger, M. Leskelä, T. Repo, P. Pyykkö and B. Rieger, J. Am. Chem. Soc., 2008, 130, 14117 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625 RSC.
- (a) D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428 CrossRef CAS PubMed; (b) P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433 CrossRef CAS PubMed; (c) P. A. Chase, A. L. Gille, T. M. Gilbert and D. W. Stephan, Dalton Trans., 2009, 7179 RSC; (d) D. Holschumacher, C. Taouss, T. Bannenberg, C. G. Hrib, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2009, 6927 RSC; (e) M. Alcarazo, C. Gomez, S. Holle and R. Goddard, Angew. Chem., Int. Ed., 2010, 49, 5788 CrossRef CAS PubMed; (f) B. Ines, S. Holle, R. Goddard and M. Alcarazo, Angew. Chem., Int. Ed., 2010, 49, 8389 CrossRef CAS PubMed; (g) D. Holschumacher, T. Bannenberg, K. Ibrom, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2010, 39, 10590 RSC; (h) M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6422 CrossRef CAS; (i) E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2012, 18, 16938 CrossRef CAS PubMed.
- (a) C. S. Marvel and O. Kamm, J. Am. Chem. Soc., 1919, 41, 276 CrossRef CAS; (b) C. S. Marvel, Org. Synth., 1925, 4, 19 CrossRef; (c) D. van der Helm, L. L. Merritt Jr, R. Degeilh and C. H. MacGillavry, Acta Crystallogr., 1965, 18, 355 CrossRef CAS.
- See for a comparison: (a) K. G. Orrell, V. Šik and D. Stephenson, Magn. Reson. Chem., 1987, 25, 1007 CrossRef CAS; (b) D. Beaudoin and J. D. Wuest, Chem. Rev., 2016, 116, 258 CrossRef CAS PubMed.
- See also: (a) N. Xu, J. H. Christian, N. S. Dalal, E. G. Abucayon, C. Lingafelt, D. R. Powell and G. B. Richter-Addo, Dalton Trans., 2015, 44, 20121 RSC; (b) S. Kundu, S. C. E. Stieber, M. G. Ferrier, S. A. Kozimor, J. A. Bertke and T. H. Warren, Angew. Chem., Int. Ed., 2016, 55, 10321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and analytical details, spectral data and crystallographic data. CCDC 1506872–1506879. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04459j |
| This journal is © The Royal Society of Chemistry 2017 |