
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective Narasaka–Heck cyclizations: synthesis of tetrasubstituted nitrogen-bearing stereocenters†
Nicholas J.
Race‡
a,
Adele
Faulkner‡
a,
Gabriele
Fumagalli
 a,
Takayuki
Yamauchi
a,
James S.
Scott
b,
Marie
Rydén-Landergren
c,
Hazel A.
Sparkes§
a and
John F.
Bower
*a
a,
Takayuki
Yamauchi
a,
James S.
Scott
b,
Marie
Rydén-Landergren
c,
Hazel A.
Sparkes§
a and
John F.
Bower
*a
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: john.bower@bris.ac.uk
bAstraZeneca, Alderley Park, Macclesfield, Cheshire SK10 4TG, UK
cAstraZeneca, Pepparedsleden 1, Mölndal, 43183, Sweden
First published on 24th November 2016
Abstract
The first examples of highly enantioselective Narasaka–Heck cyclizations are described. A SPINOL-derived P,N-ligand system enables Pd-catalyzed 5-exo cyclization of a range of oxime esters with sterically diverse trisubstituted alkenes to generate dihydropyrroles containing tetrasubstituted nitrogen-bearing stereocenters in 56 to 86% yield and 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 95:5 e.r. These processes are rare examples of reactions that proceed via enantioselective migratory insertion of alkenes into Pd–N bonds, and the first where trisubstituted alkenes are used to generate tetrasubstituted stereocenters with high enantioselectivity.
:10 to 95:5 e.r. These processes are rare examples of reactions that proceed via enantioselective migratory insertion of alkenes into Pd–N bonds, and the first where trisubstituted alkenes are used to generate tetrasubstituted stereocenters with high enantioselectivity.
Introduction
The intramolecular Heck reaction is a powerful method for the enantioselective construction of all-carbon quaternary stereocenters.1 Pioneering contributions from the Overman and Shibasaki groups2 have resulted in its establishment as a lynchpin reaction in the synthesis of a wide range of challenging natural products, most notably alkaloids.3 Within this class of compound, fully substituted nitrogen-bearing stereocenters are ubiquitous (Scheme 1A),4 and so related C–N bond formations via enantioselective aza-variants of the Heck reaction become appealing. First reported in 1999, the Narasaka–Heck cyclization of oxime esters with alkenes is the prototype aza-variant of the conventional Heck reaction, in so much as it incorporates the key steps of (a) N–O oxidative addition, (b) imino-palladation and (c) β-hydride elimination (Scheme 1B).5,6 Recently, related processes that use other classes of redox active N-donors have started to emerge.7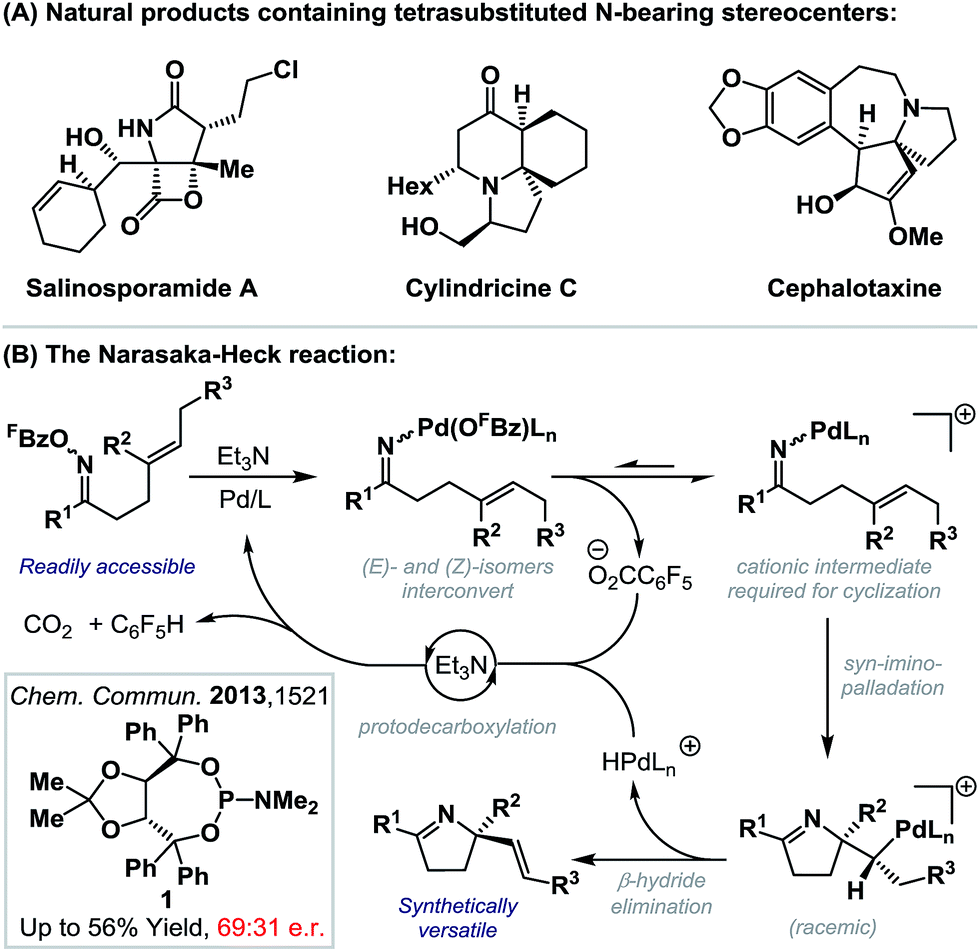 | ||
| Scheme 1 | ||
We have shown that the Narasaka reaction and related cascades are effective at generating tetrasubstituted stereocenters via cyclization onto a wide range of sterically encumbered alkenes.8c,d However, to date, highly enantioselective variants have remained elusive. In this report, we disclose the first examples of highly enantioselective Narasaka–Heck cyclizations, which provide efficient access to sterically congested tetrasubstituted stereocenters. These studies serve as proof-of-concept for enantioselective processes of this type and, in broader terms, provide rare examples of reactions that involve enantioselective migratory insertion of alkenes into N–Pd bonds.6,9–12 Indeed, to the best of our knowledge, this is only the third class of process where this step is used to generate tetrasubstituted nitrogen-bearing stereocenters with high enantioselectivity,9c,d and the first that achieves this using trisubstituted alkenes.
Results and discussion
Rendering Narasaka–Heck cyclizations enantioselective has proven especially challenging because processes that deliver racemic products exhibit prescriptive ligand requirements, with P(3,5-(CF3)2C6H3)3 emerging as by far the most efficient and general system to date.8a–d In earlier work we assayed a wide range of commercial mono- and bi-dentate chiral P-based systems, and established that TADDOL-phosphoramidate 1 can promote cyclization of 2a to 3a in 56% yield and 66:34 e.r. (Scheme 1B, box and Scheme 2).8c Evaluation of a range of non-commercial variants of 1 failed to deliver a system that offered appreciable additional benefits to yield or selectivity. Our attention therefore turned to P,N-based systems, inspired, in part, by their success in conventional cationic Heck reactions.1 Note that in the current scenario, entry to a cationic manifold is driven by facile, triethylammonium-mediated protodecarboxylation of the pentafluorobenzoate leaving group.8d The most promising early results were obtained using BINOL, SPINOL and H8-BINOL derived systems, as outlined in Scheme 2. We found that non-commercial ligands (Sa,S)-L-113 and (Sa,S)-L-314 provided (S)-3a with appreciable levels of enantioselectivity (92:8 and 85:15 e.r. respectively), but in low yield. However, (Sa,S)-L-215 offered the best balance between cyclization efficiency and selectivity, delivering the opposite (R)-enantiomer of 3a in 53% yield and 89:11 e.r.; this latter ligand system can be considered pseudo-diastereomeric with respect to (Sa,S)-L-1 and (Sa,S)-L-2, which accounts for the observed switch in enantioinduction.16
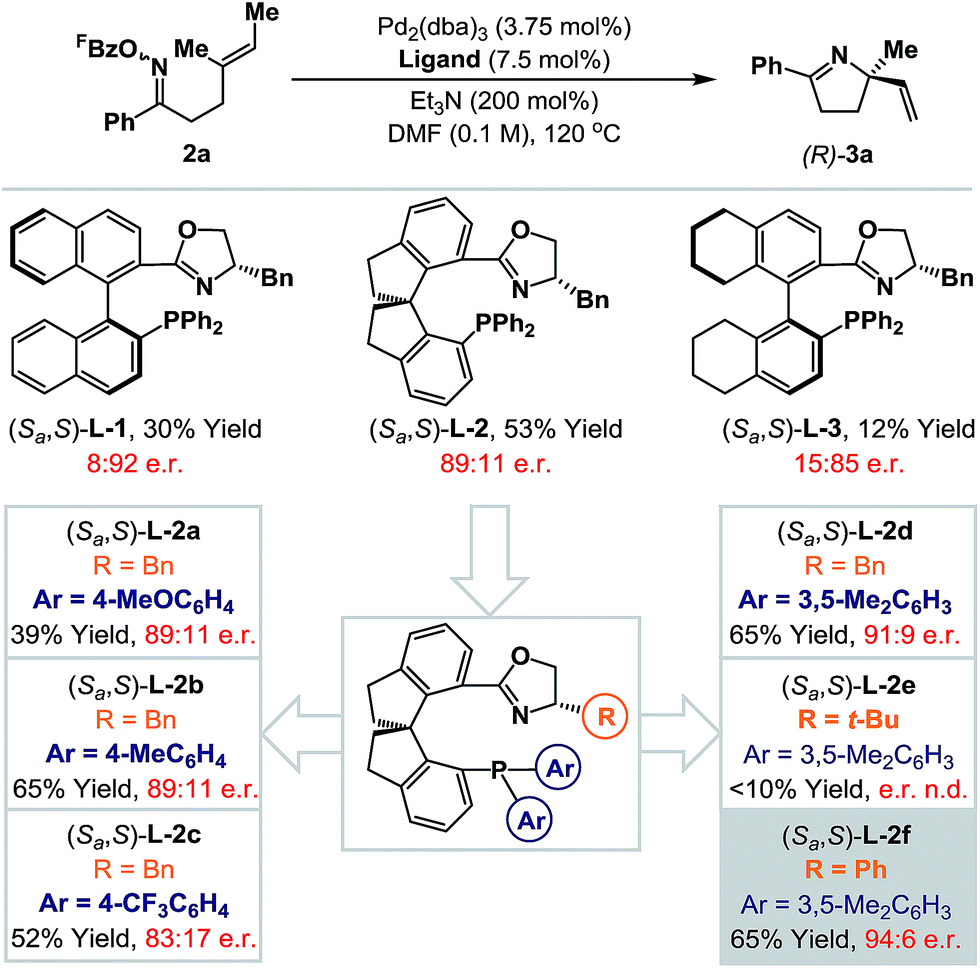 | ||
| Scheme 2 Development of an enantioselective protocol. | ||
(Sa,S)-L-2 is commercially available, but optimization of the oxazoline and phosphine aryl groups of this system necessitated the “in house” synthesis of a range of known non-commercial or novel analogues.17 Initially the phosphine aryl groups were varied ((Sa,S)-L-2a–d) and these studies revealed that replacement of the phenyl groups with 3,5-dimethylated variants ((Sa,S)-L-2d) offered a significant improvement in yield and a marginal enhancement in enantioselectivity for 3a. Notably, strongly electron-donating or -withdrawing groups at the para-position of the arene resulted either in lower yields or lower enantioselectivities (cf. (Sa,S)-L-2avs. (Sa,S)-L-2c). With a suitable phosphine aryl group established, attention turned to variations at the oxazoline portion. (Sa,S)-L-2e, in which a bulky tert-butyl group has replaced the benzyl moiety present in (Sa,S)-L-2d, was ineffective and generated 3a in low yield (<10%). However, replacement of the benzyl group with a phenyl substituent ((Sa,S)-L-2f) provided 3a in an increased e.r. of 94:6 and maintained cyclization efficiency at 65% yield. Although the improvements on moving from (Sa,S)-L-2a to (Sa,S)-L-2f may appear modest, they are significant, and this ligand confers approximately 10–20% enhancements for both yield and e.r. (vs. (Sa,S)-L-2) for additional examples discussed later. During the course of this work, the synthesis and application of (Sa,S)-L-2f to highly enantioselective reductions of 2-pyridyl cyclic imines was reported by Zhou and co-workers.18
With an optimal ligand system established, we evaluated initially its scope with respect to the alkene component (Table 1). A range of systems 2b–j, where Ar = phenyl or 2-naphthyl,19 cyclized to provide the targets in good to excellent yield and high enantioselectivity (91:9 to 95:5 e.r.). Notably, the system tolerates significant steric variation at R1 and R2, whilst maintaining cyclization efficiency and enantioselectivity. For example, cyclization of 2j, which possesses an iso-propyl substituent at R1, afforded 3j in 69% yield and 93:7 e.r. To achieve an optimal balance between cyclization efficiency and enantioselectivity, fine tuning of reaction temperature was required on a case-by-case basis. Control of substrate alkene geometry is crucial, as the alternate (Z)-isomer of 2c cyclized with considerably lower levels of enantioinduction.20 The absolute stereochemistry of cyclization products 3a–j was assigned on the basis of an X-ray structure of 3b and supporting VCD analysis of 3a and 3h (see the ESI†).21
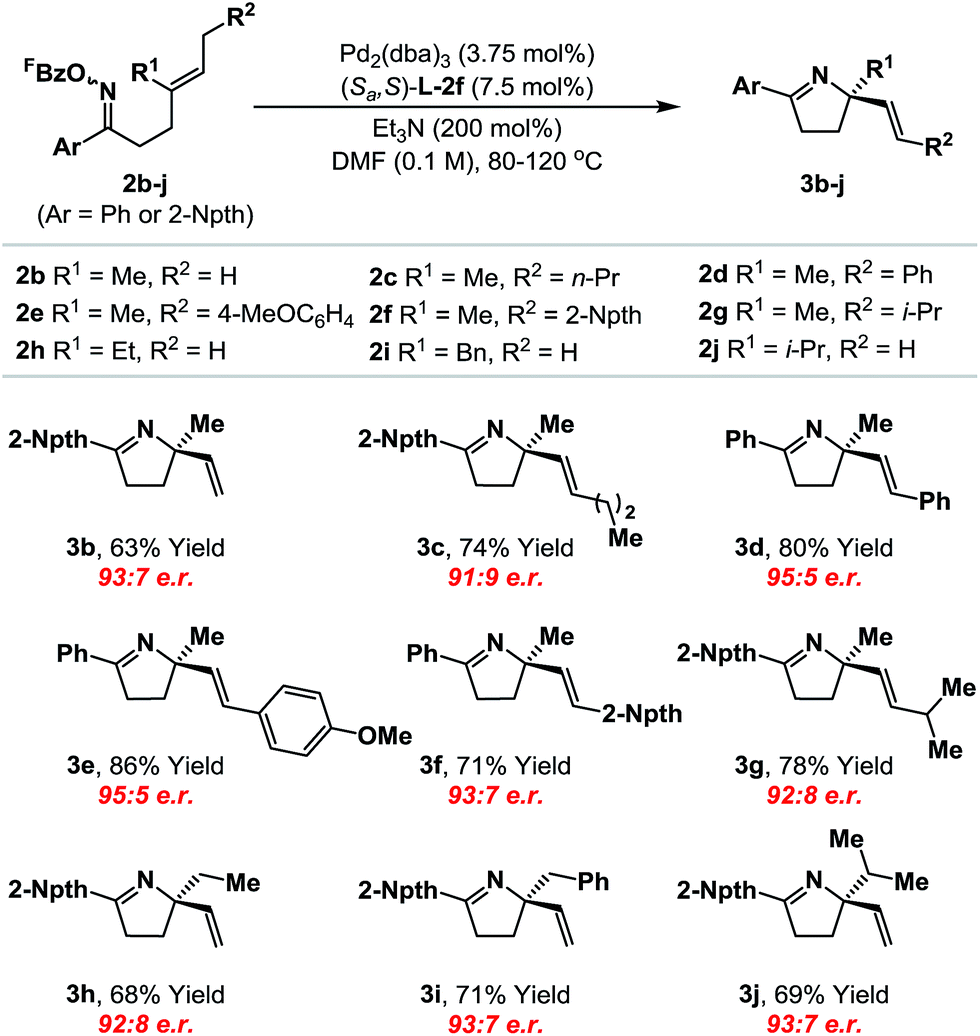
|
We have also conducted a preliminary evaluation of the scope of oxime ester moiety (Table 2). Systems 2k–n, which possess electron rich or poor aryl groups at R1, cyclized efficiently and with minimal variation in enantioinduction. The system can be extended to other, distinct classes of oxime ester. For example, cyclization of cyclopropyl and cyclohexyl derivatives 2o and 2p occurred efficiently to deliver the targets 3o and 3p with satisfactory levels of enantioselectivity. The stereochemical assignments of the products were made by analogy to 3b and were supported by VCD analysis of 3p (see the ESI†). Pertinent limitations of the oxime ester moiety in non-enantioselective Narasaka–Heck cyclizations have already been delineated in our earlier work.8a,22
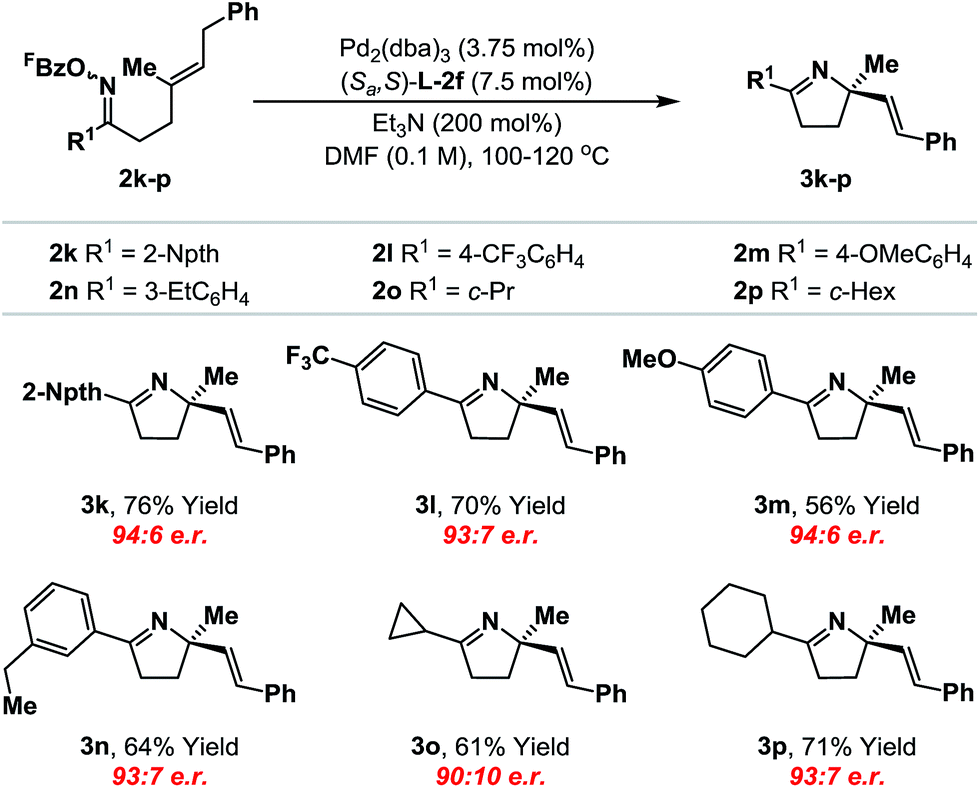
|
To rationalize the sense of enantioinduction in the processes described here, (Sa,S)-L-2f-ligated palladium complex 4 was synthesized and characterized by single crystal X-ray diffraction (see the ESI†), and this enabled the construction of a quadrant diagram (Scheme 3).21 The two xylyl groups of the phosphine provide little steric difference between quadrants I and III due to the similarity of the two N–Pd–P-aryl torsion angles (−115.5° and 124.6°). The oxazoline resides approximately perpendicular to the square plane of the complex (P–Pd–N–C(7) torsion angle = 80.7°), such that the phenyl substituent occupies quadrant II, and quadrant IV remains relatively unimpeded. Scheme 3B shows the conformations of the two diastereomeric complexes that lead to enantiodivergent iminopalladation during the conversion of 2a to 3a. The alkene likely coordinates trans to the phosphine, such that differentiation of its enantiotopic faces by the phenyl substituent of the oxazoline is facilitated. For diastereomer I, which leads to the major enantiomer (R)-3a, the terminal methyl group of the alkene occupies “free” quadrant IV and steric clashes are minimized. Minor enantiomer (S)-3a requires access to the indicated conformer of diastereomer II, where the alkene methyl substituent is placed in quadrant II and suffers unfavorable interactions with the oxazoline phenyl group. The increased enantioselectivity obtained with (Sa,S)-L-2fvs. (Sa,S)-L-2d is consistent with this model (APh = 3 vs. ABn = 1.8), as is the insensitivity of the system to increased substitution at R2 (cf.3bvs.3j). A key factor in the chemical efficiency of (Sa,S)-L-2f likely resides in the weak donor ability of the oxazoline nitrogen, which, in turn, should enhance σ-donation from the trans-imino group.23 This lowers the basicity of this moiety, such that competing protodepalladation is suppressed and cyclization efficiency is enhanced. A similar rationale was invoked for the success of P(3,5-(CF3)2C6H3)3 in our earlier work.8a–d In the present case, the structural features of the ligand backbone also play a key role, as highlighted by the studies outlined in Scheme 2.
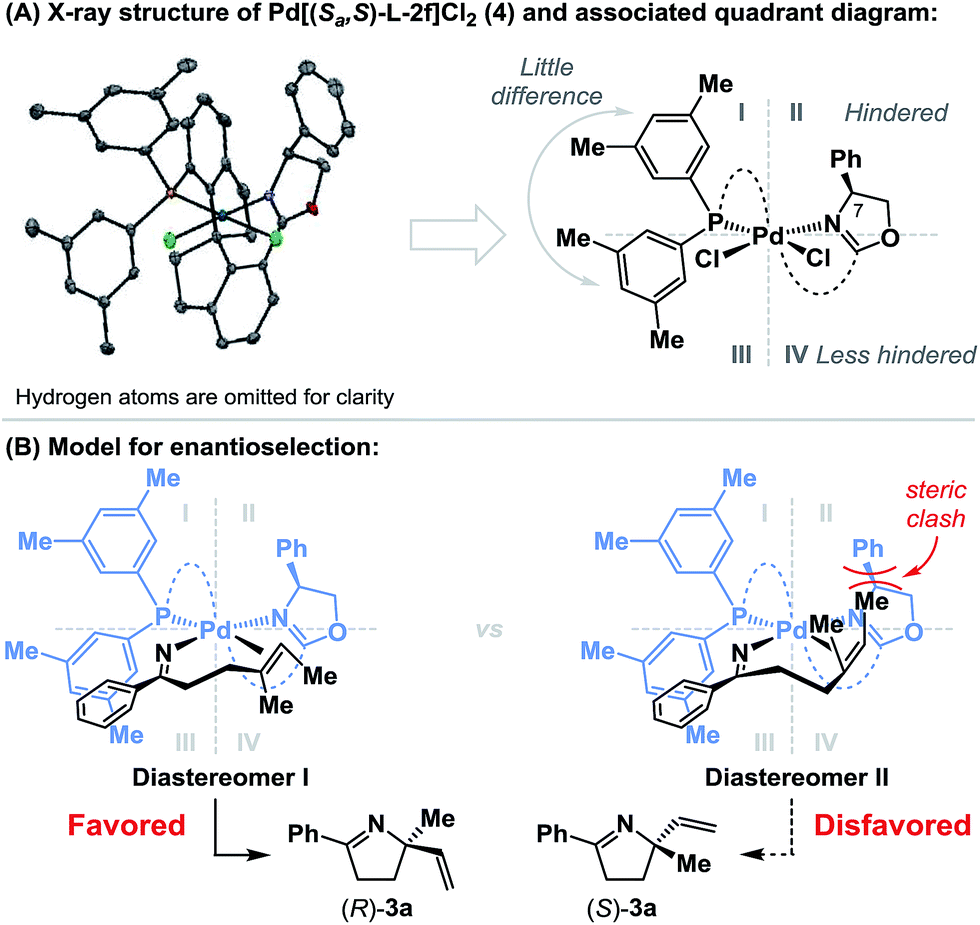 | ||
| Scheme 3 | ||
The heterocyclic products described here retain synthetically flexible imine and alkene moieties and this provides many opportunities for derivatization. Our preliminary focus has been upon reductive manipulations of the cyclization products (Scheme 4). Exhaustive hydrogenation of both the alkene and imine moieties of 3a (H2 (6 atm.), Pd/C, 4–6 days) generated efficiently acyclic target 5, which possesses a remote, tetrasubstituted stereocenter; this defines a flexible approach to this challenging class of substrates. Chemoselective reduction of the imine of 3j was achieved using DIBAL-H, and this occurred from the less hindered face to generate pyrrolidine 6 in 5:1 d.r. Related reductions of less sterically biased substrates proceeded with lower levels of diastereocontrol; efforts to address this issue will be a focus of future studies.24
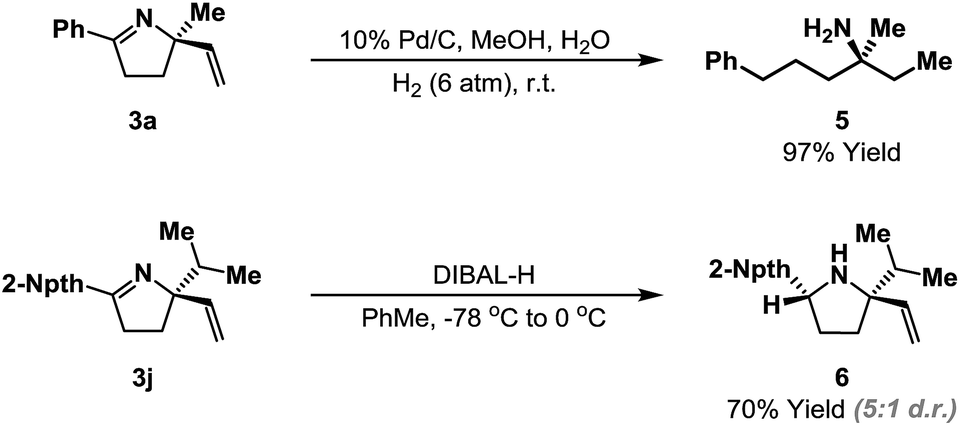 | ||
| Scheme 4 Reductive manipulations of the cyclization products. | ||
Conclusions
To conclude, we have outlined the identification and development of a P,N-based ligand system that promotes, for the first time, highly enantioselective Narasaka–Heck cyclizations. This provides access to challenging pyrrolidine derivatives that contain fully substituted nitrogen-bearing stereocenters and are key motifs in a wide range of alkaloid targets. The processes described here add to an emerging, yet rare class of reactions that proceed via enantioselective migratory insertion of alkenes into Pd–N bonds.9–12 Within this context, the current proof-of-concept study is unique in harnessing trisubstituted alkenes to generate tetrasubstituted stereocenters. Stereocontrolled manipulations of the cyclization products and the development of related enantioselective cyclizations and cascades are the focus of ongoing investigations in our laboratory.Acknowledgements
We thank the EPSRC (EP/J007455/1) and the European Research Council (ERC grant 639594 CatHet) for financial support. A. F. thanks AstraZeneca and N. J. R. thanks the Bristol Chemical Synthesis Doctoral Training Centre, funded by the EPSRC (EP/G036764/1), for a Ph.D. studentship. N. J. R. thanks the SCI for a postgraduate scholarship. J. F. B. is indebted to the Royal Society for a University Research Fellowship.Notes and references
- Selected reviews: (a) D. M. Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC; (b) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS; (c) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (d) M. Shibasaki, C. D. J. Boden and A. Kojima, Tetrahedron, 1997, 53, 7371 CrossRef CAS.
- (a) Y. Sato, M. Sodeoka and M. Shibasaki, J. Org. Chem., 1989, 54, 4738 CrossRef CAS; (b) N. E. Carpenter, D. J. Kucera and L. E. Overman, J. Org. Chem., 1989, 54, 5846 CrossRef CAS.
- Selected examples in synthesis: (a) A. B. Dounay, L. E. Overman and A. D. Wrobelski, J. Am. Chem. Soc., 2005, 127, 10186 CrossRef CAS PubMed; (b) A. D. Lebsack, J. T. Link, L. E. Overman and B. A. Stearns, J. Am. Chem. Soc., 2002, 124, 9008 CrossRef CAS PubMed; (c) M. E. Fox, C. Li, J. P. Marino Jr and L. E. Overman, J. Am. Chem. Soc., 1999, 121, 5467 CrossRef CAS; (d) T. Matsuura, L. E. Overman and D. J. Poon, J. Am. Chem. Soc., 1998, 120, 6500 CrossRef CAS.
- (a) Salinosporamide A: R. H. Felig, G. O. Buchanan, T. J. Mincer, C. A. Kauffman, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2003, 42, 355 CrossRef PubMed; (b) cylindricine C: C. P. Li and A. J. Blackman, Aust. J. Chem., 1994, 47, 1355 CrossRef CAS; (c) cephalotaxine: R. G. Powell, D. Weisleder, C. R. Smith Jr and I. A. Wolff, Tetrahedron Lett., 1969, 10, 4081 CrossRef.
- (a) H. Tsutsui and K. Narasaka, Chem. Lett., 1999, 28, 45 CrossRef; (b) H. Tsutsui, M. Kitamura and K. Narasaka, Bull. Chem. Soc. Jpn., 2002, 75, 1451 CrossRef CAS. For reviews, see: (c) M. Kitamura and K. Narasaka, Chem. Rec., 2002, 2, 268 CrossRef CAS PubMed; (d) K. Narasaka and M. Kitamura, Eur. J. Org. Chem., 2005, 4505 CrossRef CAS.
- For a review that encompasses processes involving migratory insertion of alkenes into N–Pd bonds, see: P. S. Hanley and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 8510 CrossRef CAS PubMed.
- (a) I. R. Hazelden, X. Ma, T. Langer and J. F. Bower, Angew. Chem., Int. Ed., 2016, 55, 11198 CrossRef CAS PubMed; (b) S. A. Shuler, G. Yin, S. B. Krause, C. M. Vesper and D. A Watson, J. Am. Chem. Soc., 2016, 138, 13830 CrossRef CAS PubMed.
- Aza-Heck reactions: (a) A. Faulkner and J. F. Bower, Angew. Chem., Int. Ed., 2012, 51, 1675 CrossRef CAS PubMed; (b) N. J. Race and J. F. Bower, Org. Lett., 2013, 15, 4616 CrossRef CAS PubMed; (c) A. Faulkner, J. S. Scott and J. F. Bower, Chem. Commun., 2013, 49, 1521 RSC; (d) A. Faulkner, J. S. Scott and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 7224 CrossRef CAS PubMed. For a copper-catalyzed variant, see: (e) A. Faulkner, N. J. Race, J. S. Scott and J. F. Bower, Chem. Sci., 2014, 5, 2416 RSC.
- Redox neutral 1,2-carboamination of alkenes: (a) D. Y. Mai and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 12157 CrossRef CAS PubMed; (b) B. A. Hopkins and J. P. Wolfe, Angew. Chem., Int. Ed., 2012, 51, 9886 CrossRef CAS PubMed; (c) B. A. Hopkins and J. P. Wolfe, Chem. Sci., 2014, 5, 4840 RSC; (d) Y. Miyazaki, N. Ohta, K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 3732 CrossRef CAS PubMed; (e) Z. J. Garlets, K. R. Parenti and J. P. Wolfe, Chem.–Eur. J., 2016, 22, 5919 CrossRef CAS PubMed.
- Oxidative 1,2-carboaminations of alkenes: (a) K.-T. Yip, M. Yang, K.-L. Law, N.-Y. Zhu and D. Yang, J. Am. Chem. Soc., 2006, 128, 3130 CrossRef CAS PubMed; (b) W. He, K.-T. Yip, N.-Y. Zhu and D. Yang, Org. Lett., 2009, 11, 5626 CrossRef CAS PubMed. For enantioselective copper catalyzed 1,2-carboaminations of alkenes, see: (c) T. W. Liwosz and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 2020 CrossRef CAS PubMed.
- 1,2-Diaminations of dienes and trienes: (a) H. Du, W. Yuan, B. Zhao and Y. Shi, J. Am. Chem. Soc., 2007, 129, 11688 CrossRef CAS PubMed; (b) L. Xu and Y. Shi, J. Org. Chem., 2008, 73, 749 CrossRef CAS PubMed.
- Aza-Wacker cyclizations: (a) R. I. McDonald, P. B. White, A. B. Weinstein, C. P. Tam and S. S. Stahl, Org. Lett., 2011, 13, 2830 CrossRef CAS PubMed; (b) A. B. Weinstein and S. S. Stahl, Angew. Chem., Int. Ed., 2012, 51, 11505 CrossRef CAS PubMed.
- (a) Y. Imai, W. Zhang, T. Kida, Y. Nakatsuji and I. Ikeda, Tetrahedron Lett., 1998, 39, 4343 CrossRef CAS; (b) M. Ogasawara, K. Yoshida and T. Hayashi, Heterocycles, 2000, 52, 195 CrossRef CAS; (c) M. Ogasawara, K. Yoshida, H. Kamei, K. Kato, Y. Uozumi and T. Hayashi, Tetrahedron: Asymmetry, 1998, 9, 1779 CrossRef CAS.
- Q. Li, P. Wan, S. Wang, Y. Zhuang, L. Li, Y. Zhou, Y. He, R. Cao, L. Qiu and Z. Zhou, Appl. Catal., A, 2013, 458, 201 CrossRef CAS.
- (a) S.-F. Zhu, J.-B. Xie, Y.-Z. Zhang, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886 CrossRef CAS PubMed; (b) S. Li, S.-F. Zhu, C.-M. Zhang, S. Song and Q.-L. Zhou, J. Am. Chem. Soc., 2008, 130, 8584 CrossRef CAS PubMed.
- (Ra,S)-L-2 provided (S)-3a in 74% yield and 75:25 e.r.
- Procedures: (a) V. B. Birman, A. L. Rheingold and K.-C. Lam, Tetrahedron: Asymmetry, 1999, 10, 125 CrossRef CAS; (b) Z. Li, X. Liang, F. Wu and B. Wan, Tetrahedron: Asymmetry, 2004, 15, 665 CrossRef CAS . See also ref. 15.
- C. Guo, D.-W. Sun, S. Yang, S.-J. Mao, X.-H. Xu, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2015, 137, 90 CrossRef CAS PubMed.
- Naphthyl systems were targeted in the hope of procuring crystalline products, such that absolute stereochemistry could be determined by X-ray diffraction.
- Using (Sa,S)-L-2, cyclization of (Z)-2c generated preferentially (S)-3c in 37% yield and 68:32 e.r. Under the current protocol, processes involving 1,2-disubstituted alkenes result in a mixture of dihydropyrrole and pyrrole products, resulting from low selectivity at the stage of β-hydride elimination.
- CCDC 1438659–1438660 contain the supplementary crystallographic data for this paper.†.
- Substrates where R1 = n-alkyl cyclize in low yield (<20%); a rationalization for the inefficiency of systems of this type has been provided in our earlier work.8a In specific, conformationally constrained cases Narasaka–Heck cyclizations can afford 6-membered rings.5c,d The present protocol does not address this issue and is not applicable to 6-ring cyclizations.
- M. Hatano, M. Yamanaka and K. Mikami, Eur. J. Org. Chem., 2003, 2552 CrossRef CAS.
- Intense efforts have been directed towards catalyst controlled manipulations of cyclic imines related to 3a–n because of the medicinal importance and economic value of 2-aryl pyrrolidines. Selected enantioselective methodologies: (a) F. Chen, Z. Ding, J. Qin, T. Wang, Y. He and Q.-H. Fan, Org. Lett., 2011, 13, 4348 CrossRef CAS PubMed; (b) M. Chang, W. Li, G. Hou and X. Zhang, Adv. Synth. Catal., 2010, 352, 3121 CrossRef CAS . See also ref. 18.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all compounds are provided. CCDC 1438659 and 1438660. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04466b |
| ‡ These authors contributed equally. |
| § Author to whom correspondence should be addressed regarding the X-ray structures of 3b and Pd[(Sa,S)-L-2f]Cl2 (4). |
| This journal is © The Royal Society of Chemistry 2017 |