
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTo catalyze or not to catalyze: elucidation of the subtle differences between the hexameric capsules of pyrogallolarene and resorcinarene†
Qi
Zhang
a,
Lorenzo
Catti
a,
Ville R. I.
Kaila
c and
Konrad
Tiefenbacher
*ab
aDepartment of Chemistry, University of Basel, St. Johannsring 19, CH-4056 Basel, Switzerland. E-mail: konrad.tiefenbacher@unibas.ch
bDepartment of Biosystems Science and Engineering, ETH Zürich, Mattenstrasse 26, CH-4058 Basel, Switzerland. E-mail: tkonrad@ethz.ch
cDepartment of Chemistry, Technical University of Munich, Lichtenbergstrasse 4, 85747 Garching, Germany
First published on 15th November 2016
Abstract
The closely related, self-assembled resorcinarene and pyrogallolarene capsules display contrasting and puzzling encapsulation behaviors. Herein, we elucidate the reasons for these differences by combining experimental studies and DFT calculations. Furthermore, we report that, in contrast to the resorcinarene capsule, the pyrogallolarene derivative is not capable of catalyzing reactions with cationic transition states. The molecular mechanisms responsible for these observations are probed in detail.
The functional mimicking of natural enzymes has been a very fascinating but also challenging research topic for decades. Several supramolecular structures have been identified that are able to catalyze reactions inside their enzyme-like pockets.1 Nevertheless, neither the catalytic efficiency nor the selectivity of such systems, can usually compete with natural enzymes. Therefore, the refinement of known structures, as well as the development of new systems, is necessary. For the design of new artificial enzyme-like catalysts, it is of fundamental importance to understand the prerequisites for catalytic activity. We2 and others3 have shown that the hexameric resorcinarene capsule I,4 which self-assembles from six units of resorcinarene 1 and eight water molecules (Fig. 1), is an efficient catalyst for a variety of cationic reactions. Nevertheless, the reasons for the high catalytic efficiency remain unclear. To learn more about the pivotal requirements for the catalytic activity of hexamer I, we became interested in the structurally closely related pyrogallolarene hexamer II.5 It self-assembles from six units of pyrogallolarene 2. Surprisingly, hexamer II displays a different encapsulation behavior compared to I in chloroform solution. It was reported that hexamer I encapsulates both tertiary amines as well as alkylammonium species,4c,d,6 while II only binds tertiary amines.5e,h Very recently, the Cohen group disclosed that binding of ammonium salts in II can be observed to some extent in benzene solution.7 The high affinity of ammonium salts for I can be explained by strong cation–π interactions. However, the surprising exclusion of alkylammonium species from II in chloroform solution remained puzzling,4g especially since encapsulated tertiary amines were completely expelled after protonation by the addition of acid.5e,h This seemingly contradictory encapsulation behavior of II has remained a mystery for the last decade. We herein elucidate the reasons for these differences in chloroform solution. Importantly, we report that capsule II is catalytically completely incompetent in cationic reactions which are efficiently accelerated inside I and probe the molecular mechanisms responsible for these surprising observations.
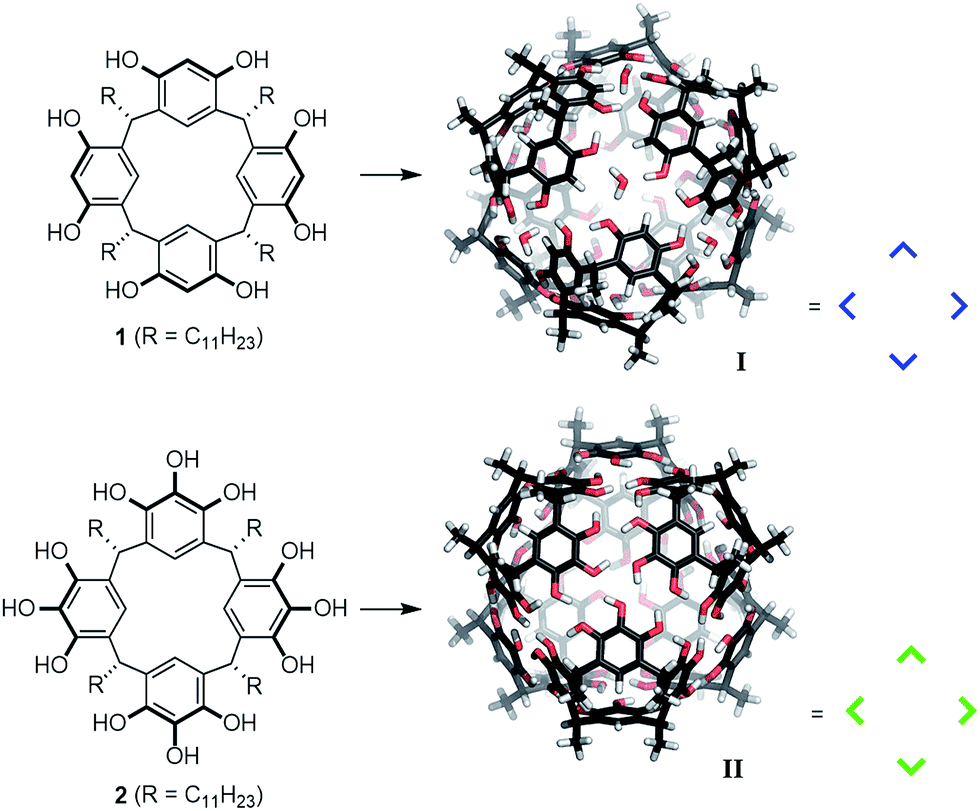 | ||
| Fig. 1 Structures of hexameric resorcinarene I and pyrogallolarene II capsules, optimized at the density functional theory (DFT: PBE-D3/def2-SVP) level and their respective building blocks 1 and 2. | ||
Our interest in capsule II started with the observation that, while I is an efficient catalyst for cationic reactions, hexamer II is catalytically incompetent in such reactions. The tail-to-head terpene cyclization of nerol (3)2c (Fig. 2) was investigated more closely with II. 1H-NMR analysis confirmed the formation of the host–guest complex 3@II (ESI-Fig. 14†). Therefore, the guest uptake cannot explain its catalytic inertness.
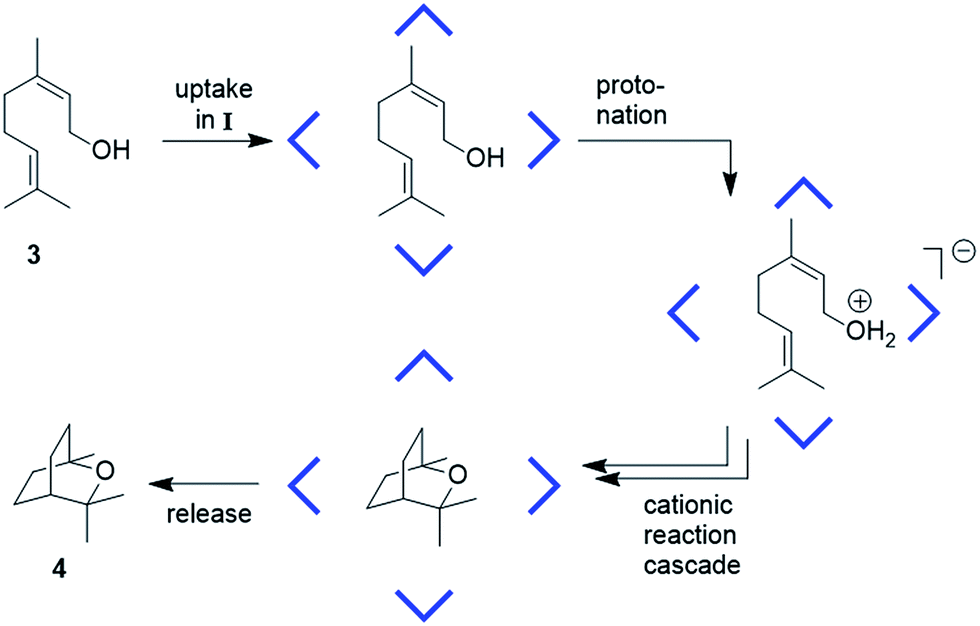 | ||
| Fig. 2 Schematic overview of the proposed mechanism for the cyclization of nerol (3) to eucalyptol (4), catalyzed inside capsule I (CDCl3, 30 °C).2c | ||
We first probed whether the absence of catalytic activity of II could arise from the reported resistance to bind alkyl ammonium salts,5e which may indicate a lack of cation–π stabilization inside the cavity. A cation–π stabilization of cationic transition states has been proposed as a possible reason for rate acceleration inside I,2c and may thus be a potential explanation for the observed catalytic differences. Interestingly, however, it was reported that tertiary amines bind well inside II.5e,h Recently, we demonstrated that a proton transfer from capsule I to the tertiary amines is responsible for their high affinity for I.2a This raised the question of whether the same is true inside II. Therefore, we investigated the encapsulation of differently sized tertiary amines inside II. The encapsulation of triethylamine (5a) in II is clearly evident in the 1H-NMR spectrum (ESI-Fig. 1†). The integral of the phenolic groups of II diminishes upon treatment with 5a, while a new broad peak emerges between 3 and 7 ppm. Careful integration (ESI-Schemes 1 and 2, Table 2†) reveals that it accounts for the diminished phenolic protons, as well as for the water signal, which is no longer visible as a separate peak. These observations are consistent with our previous findings for capsule I, and thus indicate the concomitant protonation of tertiary amines upon encapsulation in II.2a The integrity of the hexameric encapsulation complex was confirmed by DOSY spectroscopy (ESI-Fig. 2†). Therefore, the formation of smaller (dimeric, or monomeric) pyrogallolarene–cation complexes observed in methanol solution5c and in the solid state5g,8 can be excluded.
The size of the tertiary amine has a pronounced effect on protonation and encapsulation, as shown in Table 1a. The degree of protonation and encapsulation drops with the increasing size of the alkyl groups, a behavior that is consistent with a decreased cation–π interaction due to steric shielding. In the case of triethylamine (5a), the deprotonation degree of II is considerably lower than the encapsulation degree. Therefore, a [5H5]+@II−-complex, also observed in capsule I,2a is likely formed.
| (a) | |||
|---|---|---|---|
| R | Deprotonation degree of II | Encapsulation degree of 5 | |
a Precipitation occured (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex, ESI-Table 3). :1 complex, ESI-Table 3).
|
|||
| 5a | C2H5 | 29 ± 2% | 44 ± 2% |
| 5b | C3H7 | 19 ± 1% | 22 ± 1% |
| 5c | C4H9 | 12 ± 1% | 15 ± 1% |
| 5d | C6H13 | 10 ± 1% | 7 ± 1% |
These results indicate that capsule II encapsulates amines in their protonated form and therefore is able to stabilize cations inside its cavity. Indeed, our quantum chemical density functional theory (DFT) calculations also indicate that capsule II stabilizes cations as well as or even more strongly than capsule I (within ca. 4–12 kcal mol−1 for NEt4+; see ESI-Table 4†). This seems to contradict the previous observation that the ammonium salt Hex4NBr (6d+Br−) was rejected by capsule II.5e We therefore repeated the encapsulation studies not only with 6d+Br− but with differently sized ammonium salts (Table 1b). The small ammonium salt Et4NBr (6a+Br−) is indeed encapsulated to a considerable extent (see ESI-Fig. 5†). However, quantification was hampered by precipitation of a dimeric complex (see ESI-Table 3†). With the increasing size of the alkyl residues, encapsulation drops dramatically: only 4% encapsulation is observed in the case of Pr4NBr (6b+Br−), while the longer ammonium salts Bu4NBr (6c+Br−) and Hex4NBr (6d+Br−) do not show any degree of uptake inside II. This confirms the earlier observation that larger ammonium salts are rejected by II, but also demonstrated that the uptake of the smaller salts is possible. Nevertheless, there is a striking difference between the capsules, as I encapsulates salts (6a–6c+Br−) quantitatively and 6d+Br− to a large extent (see, ESI-Fig. 4†). This discrepancy is even more surprising considering that our DFT calculations suggest an even stronger stabilization of cations inside II relative to I. To elucidate the different behavior of I and II, we calculated the electrostatic potential (ESP) map of the capsules' inner surfaces. As shown in Fig. 3, the main difference between the two systems are high potential areas on the inner surface of I, which represent positively charged hydrogen atoms of the bound water molecules. These could potentially stabilize the anions of encapsulated ammonium salts via hydrogen bonds. Such a stabilization of anions inside II is lacking, and could therefore explain the dramatically weaker binding of ammonium salts. Our DFT optimizations of the capsules in presence of the ammonium salt 6a+Br− confirm this hypothesis: although capsule II stabilizes cations better than capsule I, capsule II binds the ammonium salt weaker (ca. 3–9 kcal mol−1, see ESI-Table 4†) than I.
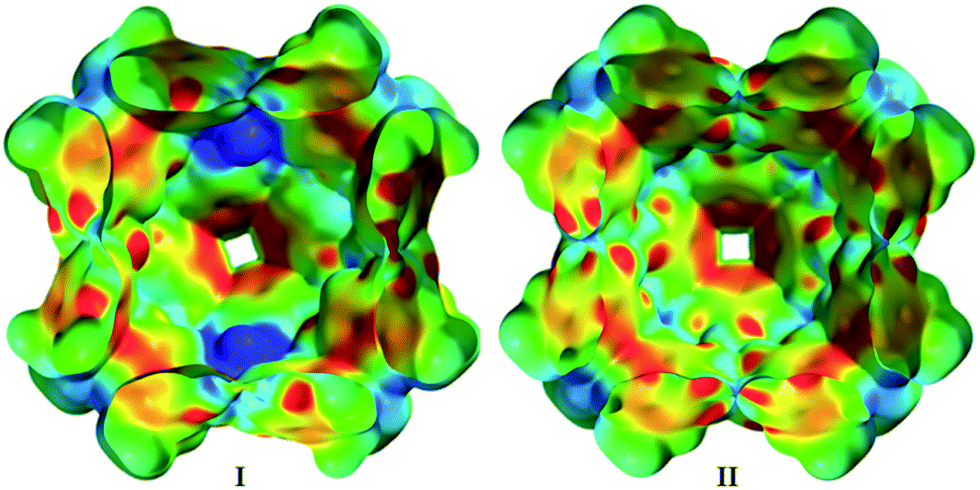 | ||
| Fig. 3 Comparison of the inner electrostatic potential (ESP) surfaces of capsules I and II (for details see ESI Chapter 6†). | ||
The energetically unfavorable encapsulation of anions inside II raises the question of whether the anion is encapsulated at all. Its presence would weaken the cation–π interactions, as has been shown in cyclophane hosts in organic solvents of low dielectric constants.9 Therefore the ammonium salt EtPr3N+MeSO3− (6e+MeSO3−) containing an organic anion, detectable by 1H and 13C-NMR spectroscopy, was investigated. 1H- and 13C-NMR investigations (ESI-Fig. 8–10†) confirm that the counterion is located outside of II (Fig. 4a). In capsule I, however, the ion pair is encapsulated (ESI-Fig. 11–13†). The minute uptake of 6+ by neutral II is likely achieved by an energetically unfavorable charge separation in CDCl3. This could explain the low encapsulation ratio of 6+ by II. If so, the addition of the large base 7 (Table 1), which cannot be encapsulated due to its size, should increase the uptake of ammonium species: it would deprotonate the capsule and form an ion pair with the bromide outside the capsule, as depicted in Fig. 4b. Indeed, upon the addition of 7, the encapsulation of ammonium species increases as anticipated (see also Table 1b). These findings solve the puzzling encapsulation behavior of capsule II and also explain the expulsion of encapsulated trihexylamine (5d) after HCl-addition5h (Fig. 4c). After the addition of HCl, the ion pair 5dH+Cl− is formed, which is rejected by capsule II.
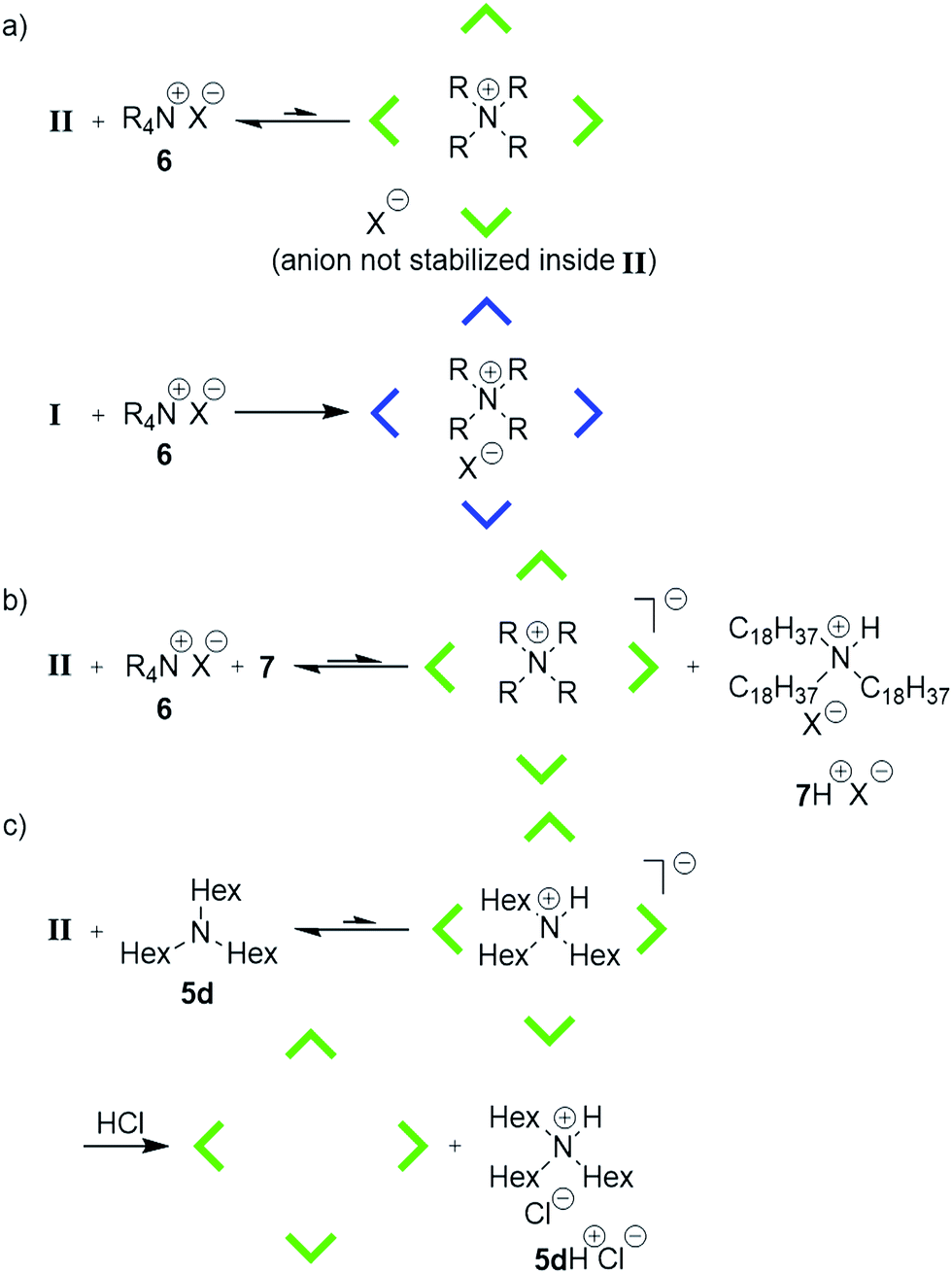 | ||
| Fig. 4 Schematic representation of encapsulation experiments of alkylammonium salts and amines with capsule I and II. (a) In capsule II, anions are not encapsulated; while capsule I encapsulated the ion pair. (b) Addition of a large base increases the uptake of ammonium species. (c) Tertiary amines are encapsulated in protonated form inside II. After addition of a strong acid, an ion pair is formed which resists encapsulation. | ||
The evidence presented clearly indicates that capsule II is able to stabilize cations inside its cavity due to cation–π interactions. Therefore, its catalytic incompetence originates from a different source. After successful substrate uptake, protonation is required for substrate activation (Fig. 2). Although II is able to protonate amines (Table 1a), the acidity of the system may be too low for activation of the alcohol substrate. Therefore, the acidity of hexamer II was determined analogously to I2a by a series of protonation experiments with amines of varying basicity. The pKa value of capsule II is approx. 9.5–10 (see ESI-Fig. 15 and 16;†ca. four pKa units higher than resorcinarene capsule I). These results are in excellent agreement with our DFT calculations at the PBE-D3/def2-SVP/ε = 4.81 level of theory, suggesting that the proton affinity of I is ca. 5 kcal mol−1 lower than that of II (ESI-Fig. 18†). The surprisingly low acidity of II may be a result of mesomeric destabilization of the phenolate (ESI-Fig. 17†). Our DFT calculations further show that the anionic defect can delocalize across several hydrogen bonds in I, while we observe a more localized defect in II, leading to a lower relative pKa in capsule I (ESI-Fig. 19†). Therefore, the low acidity of II is the likely cause of its catalytic incompetence which prevents activation of the substrate by proton transfer (see also Fig. 2). We tried to overcome this limitation by the addition of stronger external acids (see ESI 4.3†), but could not observe a difference to the background reaction, caused by the acid added. This result, however, is not too surprising, since an external acid forms an ion pair with the substrate, which will resist encapsulation (see also Fig. 4c).
The elucidation of the differences in these two systems allowed us to learn important lessons concerning catalytic activity in hydrogen-bond based molecular capsules. The identification of acidity as the crucial element of the catalytically active derivative I is essential for the design and construction of novel hydrogen bond-based supramolecular catalysts. These studies provided us with a first estimate on the required acidity for catalytic activity in such systems. Additionally, we demonstrated that externally added acid cannot function as a co-catalyst for capsule II since the substrate acid ion pairs formed are not encapsulated.
Conclusions
In conclusion, the differences between the closely related hexameric capsules I and II were elucidated for the first time. To the best of our knowledge this is the first study of two very closely related supramolecular host systems which completely differ in their catalytic activity. Evidence was presented showing that capsule II does not stabilize anions inside its cavity, as opposed to I. Therefore, alkyl ammonium salts are encapsulated only to a small extent with concomitant charge separation. Cations, nevertheless, are stabilized inside II even more strongly than inside Ivia cation–π interactions. The much lower acidity of capsule II was determined to be the cause of its catalytic incompetence. These findings are of great significance for future developments in the field of enzyme-like catalysis and have a profound impact on the design of new hydrogen bond-based catalytically active host systems.Acknowledgements
This project was supported by the Bayerischen Akademie der Wissenschaften (Junges Kolleg). V. R. I. K. is supported by grants from the Jane and Aatos Erkko Foundation and the German Research Foundation (DFG). We acknowledge the Leibniz Rechenzentrum (LRZ) and CSC – The IT Center for Science for computing time.Notes and references
- (a) D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed; (b) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 Search PubMed; (c) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (d) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 Search PubMed; (e) L. Marchetti and M. Levine, ACS Catal., 2011, 1, 1090–1118 Search PubMed; (f) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed; (g) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC; (h) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed; (i) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC; (j) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 Search PubMed.
- (a) Q. Zhang and K. Tiefenbacher, J. Am. Chem. Soc., 2013, 135, 16213–16219 Search PubMed; (b) L. Catti and K. Tiefenbacher, Chem. Commun., 2015, 51, 892–894 Search PubMed; (c) Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197–202 CrossRef CAS PubMed; (d) T. M. Bräuer, Q. Zhang and K. Tiefenbacher, Angew. Chem., Int. Ed., 2016, 55, 7698–7701 CrossRef PubMed; for reviews see (e) L. Catti, Q. Zhang and K. Tiefenbacher, Synthesis, 2016, 48, 313–328 Search PubMed; (f) L. Catti, Q. Zhang and K. Tiefenbacher, Chem.–Eur. J., 2016, 22, 9060–9066 CrossRef CAS PubMed.
- (a) G. Bianchini, G. L. Sorella, N. Canever, A. Scarso and G. Strukul, Chem. Commun., 2013, 49, 5322–5324 Search PubMed; (b) G. Bianchini, G. La Sorella, N. Canever, A. Scarso and G. Strukul, Chem. Commun., 2013, 49, 5322–5324 RSC; (c) T. Caneva, L. Sperni, G. Strukul and A. Scarso, RSC Adv., 2016, 6, 83505–83509 RSC; (d) S. Giust, G. La Sorella, L. Sperni, F. Fabris, G. Strukul and A. Scarso, Asian J. Org. Chem., 2015, 4, 217–220 Search PubMed; (e) G. La Sorella, L. Sperni, P. Ballester, G. Strukul and A. Scarso, Catal. Sci. Technol., 2016, 6, 6031–6036 RSC; (f) G. La Sorella, L. Sperni, G. Strukul and A. Scarso, ChemCatChem, 2015, 7, 291–296 Search PubMed; (g) G. La Sorella, L. Sperni, G. Strukul and A. Scarso, Adv. Synth. Catal., 2016, 358, 3443–3449 CrossRef CAS.
- For pioneering work see (a) L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS; (b) L. Avram and Y. Cohen, Org. Lett., 2002, 4, 4365–4368 CrossRef CAS PubMed; (c) L. Avram and Y. Cohen, J. Am. Chem. Soc., 2002, 124, 15148–15149 CrossRef CAS PubMed; (d) A. Shivanyuk and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7662–7665 Search PubMed; (e) A. Shivanyuk and J. J. Rebek, Chem. Commun., 2001, 2424–2425 Search PubMed; (f) A. Shivanyuk and J. Rebek, J. Am. Chem. Soc., 2003, 125, 3432–3433 CrossRef CAS PubMed; for a review see (g) L. Avram, Y. Cohen and J. Rebek Jr, Chem. Commun., 2011, 47, 5368–5375 Search PubMed; for recent studies see (h) S. Giust, G. La Sorella, L. Sperni, G. Strukul and A. Scarso, Chem. Commun., 2015, 51, 1658–1661 Search PubMed; (i) S. Giust, G. La Sorella, L. Sperni, F. Fabris, G. Strukul and A. Scarso, Asian J. Org. Chem., 2015, 4, 217–220 Search PubMed; (j) G. La Sorella, L. Sperni, G. Strukul and A. Scarso, ChemCatChem, 2015, 7, 291–296 Search PubMed.
- For pioneering work see (a) T. Gerkensmeier, W. Iwanek, C. Agena, R. Frohlich, S. Kotila, C. Nather and J. Mattay, Eur. J. Org. Chem., 1999, 2257–2262 CrossRef CAS; (b) J. L. Atwood, L. J. Barbour and A. Jerga, Chem. Commun., 2001, 2376–2377 Search PubMed; (c) A. Shivanyuk and J. Rebek, Chem. Commun., 2001, 2374–2375, 10.1039/b106793c; (d) J. L. Atwood, L. J. Barbour and A. Jerga, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4837–4841 CrossRef CAS PubMed; (e) L. Avram and Y. Cohen, J. Am. Chem. Soc., 2003, 125, 16180–16181 Search PubMed; (f) L. Avram and Y. Cohen, Org. Lett., 2003, 5, 3329–3332 Search PubMed; (g) A. Shivanyuk, J. C. Friese, S. Döring and J. Rebek, J. Org. Chem., 2003, 68, 6489–6496 Search PubMed; (h) L. Avram and Y. Cohen, J. Am. Chem. Soc., 2004, 126, 11556–11563 Search PubMed; for recent studies see (i) V. Guralnik, L. Avram and Y. Cohen, Org. Lett., 2014, 16, 5592–5595 Search PubMed; (j) L. Avram, A. Goldbourt and Y. Cohen, Angew. Chem., Int. Ed., 2016, 55, 904–907 CrossRef CAS PubMed.
- (a) M. Yamanaka, A. Shivanyuk and J. Rebek, J. Am. Chem. Soc., 2004, 126, 2939–2943 CrossRef CAS PubMed; (b) L. Avram and Y. Cohen, Org. Lett., 2003, 5, 1099–1102 CrossRef CAS PubMed.
- S. Yariv-Shoushan and Y. Cohen, Org. Lett., 2016, 18, 936–939 Search PubMed.
- (a) A. Ahman, M. Luostarinen, K. Rissanen and M. Nissinen, New J. Chem., 2007, 31, 169–177 Search PubMed; (b) N. K. Beyeh and K. Rissanen, Isr. J. Chem., 2011, 51, 769–780 CrossRef CAS.
- S. Bartoli and S. Roelens, J. Am. Chem. Soc., 1999, 121, 11908–11909 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04565k |
| This journal is © The Royal Society of Chemistry 2017 |