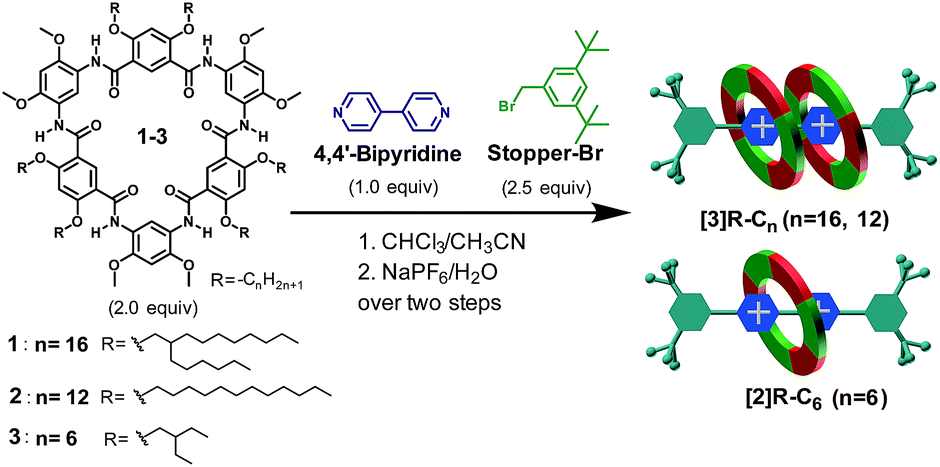
DOI:
10.1039/C6SC04714A
(Edge Article)
Chem. Sci., 2017,
8, 2091-2100
Macrocyclic shape-persistency of cyclo[6]aramide results in enhanced multipoint recognition for the highly efficient template-directed synthesis of rotaxanes†
Received
22nd October 2016
, Accepted 21st November 2016
First published on 22nd November 2016
Abstract
Examples of using two-dimensional shape-persistent macrocycles, i.e. those having noncollapsible and geometrically well-defined skeletons, for constructing mechanically interlocked molecules are scarce, which contrasts the many applications of these macrocycles in molecular recognition and functional self-assembly. Herein, we report the crucial role played by macrocyclic shape-persistency in enhancing multipoint recognition for the highly efficient template-directed synthesis of rotaxanes. Cyclo[6]aramides, with a near-planar conformation, are found to act as powerful hosts that bind bipyridinium salts with high affinities. This unique recognition module, composed of two macrocyclic molecules with one bipyridinium ion thread through the cavity, is observed both in the solid state and in solution, with unusually high binding constants ranging from ∼1013 M−2 to ∼1015 M−2 in acetone. The high efficacy of this recognition motif is embodied by the formation of compact [3]rotaxanes in excellent yields based on either a “click-capping” (91%) or “facile one-pot” (85%) approach, underscoring the great advantage of using H-bonded aromatic amide macrocycles for the highly efficient template-directed synthesis of mechanically interlocked structures. Furthermore, three cyclo[6]aramides bearing different peripheral chains 1–3 demonstrate high specificity in the synthesis of a [3]rotaxane from 1 and 2, and a [2]rotaxane from 3via a “facile one-pot” approach, in each case as the only isolated product. Analysis of the crystal structure of the [3]rotaxane reveals a highly compact binding mode that would be difficult to access using other macrocycles with a flexible backbone. Leveraging this unique recognition motif, resulting from the shape-persistency of these oligoamide macrocycles, in the template-directed synthesis of compact rotaxanes may open up new opportunities for the development of higher order interlocked molecules and artificial molecular machines.
Introduction
The development of mechanically interlocked molecules (MIMs) has invariably been accompanied by emerging recognition motifs and template-directed synthetic protocols.1 Among various factors, the combination of a macrocycle and a thread component with sufficient binding affinity constitutes a crucial determinant to create such motifs. In this regard, many elegant examples have been reported. These include the use of macrocyclic hosts such as crown ethers,2 tetracationic cyclophanes,3 calixarenes,4 cucurbiturils,5 cyclodextrins,6 and pillarenes7 to interact with the chosen thread components. However, uncovering simple and efficient recognition motifs still represents a grand challenge for the synthesis of MIMs, especially those involving non-metal coordination for extremely compact interlocked molecules, with high atom economy.
Thus far, most known examples of highly efficient, template-directed synthesis of [3]rotaxanes have relied on metal–ligand coordination.8 In all these cases, the “wheels” employed are based on macrocyclic molecules that are nonplanar and fairly flexible conformationally. In recent years, shape-persistent macrocycles, with noncollapsible and geometrically well-defined skeletons, such as phenylacetylene macrocycles,9 have attracted considerable attention due to their intriguing functions, including supramolecular gelation,10 channelized transportation,11 organic catalysis,12 molecular recognition13 and multifunctional self-assembly.14 Many shape-persistent macrocycles have well-defined surface topography and nanosized cavities with preorganized binding sites, and thus may exhibit enhanced complexation.15 Inspired by the pioneering work on synthesizing MIMs using flexible aromatic amide macrocycles16 or H-bonded oligoamide foldamers,17 we reasoned that a shape-persistent macrocycle, when serving as a wheel component for a MIM, may overcome the limit of known systems by engaging effectively in cooperative recognition interactions. However, introducing two-dimensional (2D) shape-persistent macrocycles into interlocked structures still presents a significant challenge. To the best of our knowledge, few examples of MIMs based on these macrocycles are known,18 especially [3]rotaxanes. In fact, there is only one report, based on a pentagonal cyanostar macrocycle, on forming [3]rotaxanes.18a
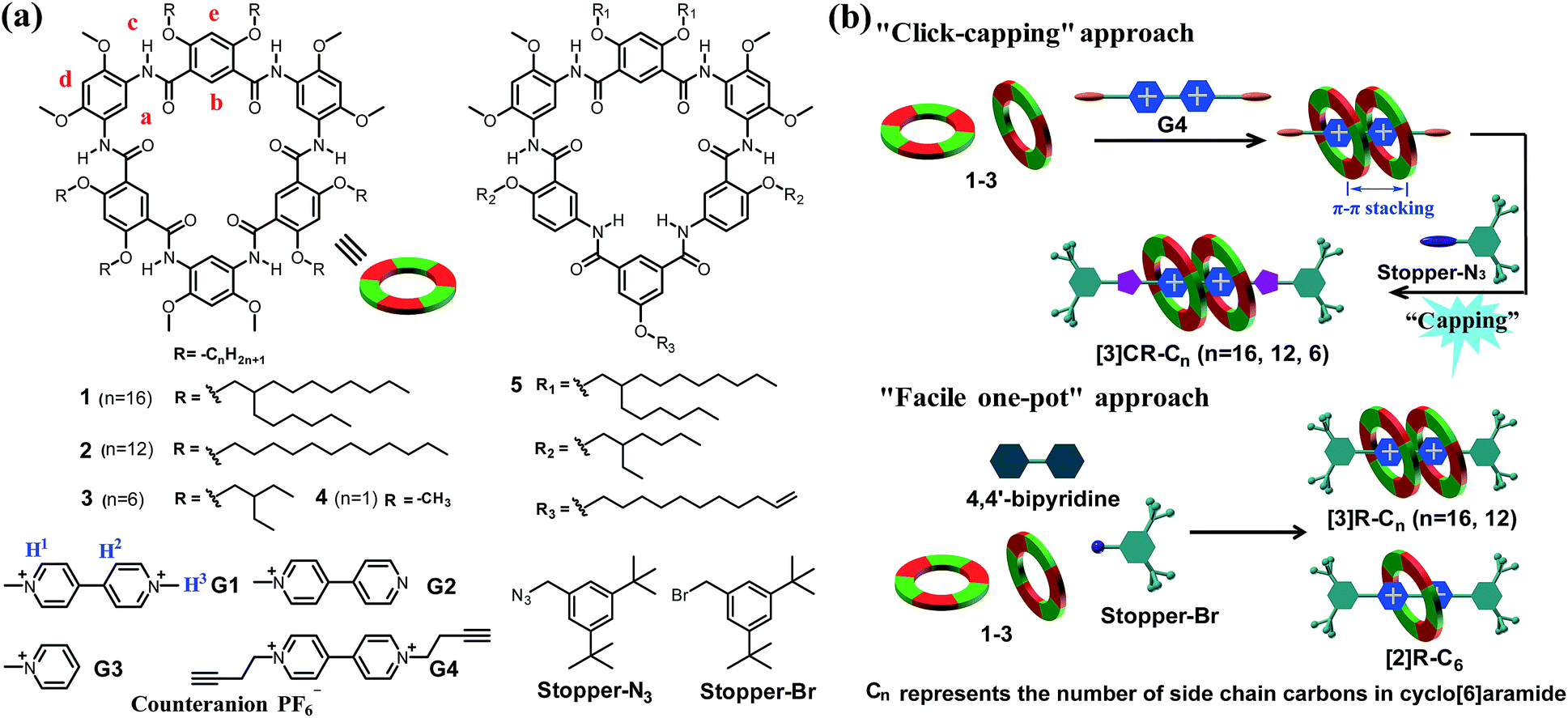
H-Bonded aromatic amide macrocycles19 are a large class of recently emerged 2D shape-persistent cyclic compounds featuring full amide linkages with rigid backbones enforced through intramolecular hydrogen bonds. Among them, cyclo[6]aramides, the smallest member of the aromatic oligoamide macrocycles20 with the amide carbonyl oxygens pointing inwards, are particularly noteworthy. They resemble a crown ether in their oxygen-rich cavities, but differ considerably in their conformational rigidity. In particular, the highly favorable intramolecular H-bond assisted macrocyclization strategy makes numerous geometrically well-defined macrocycles readily available, which facilitates their further applications. With a well-defined cavity of ca. 8 Å in diameter, large π-surfaces and precisely positioned binding sites, these macrocycles have displayed rich host–guest (H–G) chemistry. Our recent studies revealed that cyclo[6]aramides could bind organic cations and hydrogen bond donors.21 Especially notable is the complex consisting of such macrocycles and diquat, an isomer of paraquat, with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, in which a diquat molecule resides between two macrocyclic molecules rather than threads into their cavities due to electronic repulsion and steric hindrance.22 Paraquat is one of the most widely used guests for studying H–G interactions.23 Despite numerous reports on paraquat recognition, threading two rings on a single paraquat molecule with high binding affinity both in solution and in the solid state is still very difficult to achieve. The cryptand reported by Gibson and co-workers could form a 2:1 H–G complex only observed in the crystal structure and the highest binding constant for 1:1 stoichiometry achieved so far is 5.0 × 106 M−1 in acetone.24 A crown ether, bis-p-xylyl[26]crown-6, was used by Chiu and co-workers to form a [3]pseudorotaxane-like complex both in solution and in the solid state. However, only a very low binding constant (K1 = 700 M−1 and K2 = 60 M−1) was observed in acetonitrile.25 So far no successful examples have been reported on creating a tight 2:1 binding motif for constructing constrained [3]rotaxanes based on a single paraquat unit. Herein we report that cyclo[6]aramides, with their persistent shape and geometrically well-defined electron-rich cavity (Scheme 1a), could act as powerful hosts for the tight binding of bipyridinium salts in a 2:1 binding mode both in the solid state and in solution with exceedingly high binding constants. More importantly, this unique recognition motif has led to the template-directed synthesis of compact [3]rotaxanes in excellent yields based on either a “click-capping” or “facile one-pot” approach (Scheme 1b).
:1 stoichiometry, in which a diquat molecule resides between two macrocyclic molecules rather than threads into their cavities due to electronic repulsion and steric hindrance.22 Paraquat is one of the most widely used guests for studying H–G interactions.23 Despite numerous reports on paraquat recognition, threading two rings on a single paraquat molecule with high binding affinity both in solution and in the solid state is still very difficult to achieve. The cryptand reported by Gibson and co-workers could form a 2:1 H–G complex only observed in the crystal structure and the highest binding constant for 1:1 stoichiometry achieved so far is 5.0 × 106 M−1 in acetone.24 A crown ether, bis-p-xylyl[26]crown-6, was used by Chiu and co-workers to form a [3]pseudorotaxane-like complex both in solution and in the solid state. However, only a very low binding constant (K1 = 700 M−1 and K2 = 60 M−1) was observed in acetonitrile.25 So far no successful examples have been reported on creating a tight 2:1 binding motif for constructing constrained [3]rotaxanes based on a single paraquat unit. Herein we report that cyclo[6]aramides, with their persistent shape and geometrically well-defined electron-rich cavity (Scheme 1a), could act as powerful hosts for the tight binding of bipyridinium salts in a 2:1 binding mode both in the solid state and in solution with exceedingly high binding constants. More importantly, this unique recognition motif has led to the template-directed synthesis of compact [3]rotaxanes in excellent yields based on either a “click-capping” or “facile one-pot” approach (Scheme 1b).
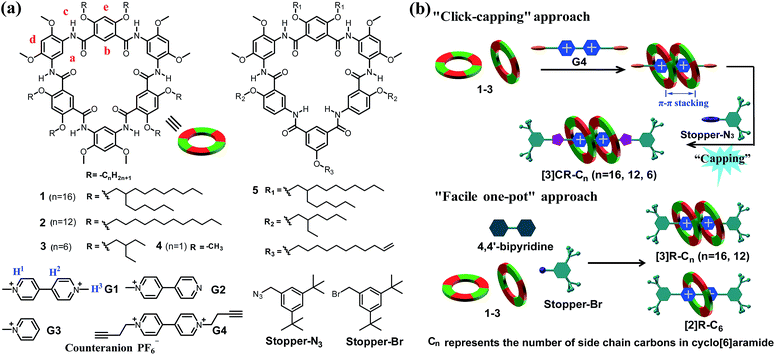 |
| | Scheme 1 (a) Chemical structures of cyclo[6]aramides 1–5, guests G1–G4, Stopper-N3 and Stopper-Br. (b) Schematic illustration of approaches for constructing compact [3]rotaxanes based on cyclo[6]aramides 1–3. | |
Results and discussion
Evidence for 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 cooperative host–guest complexation
:1 cooperative host–guest complexation
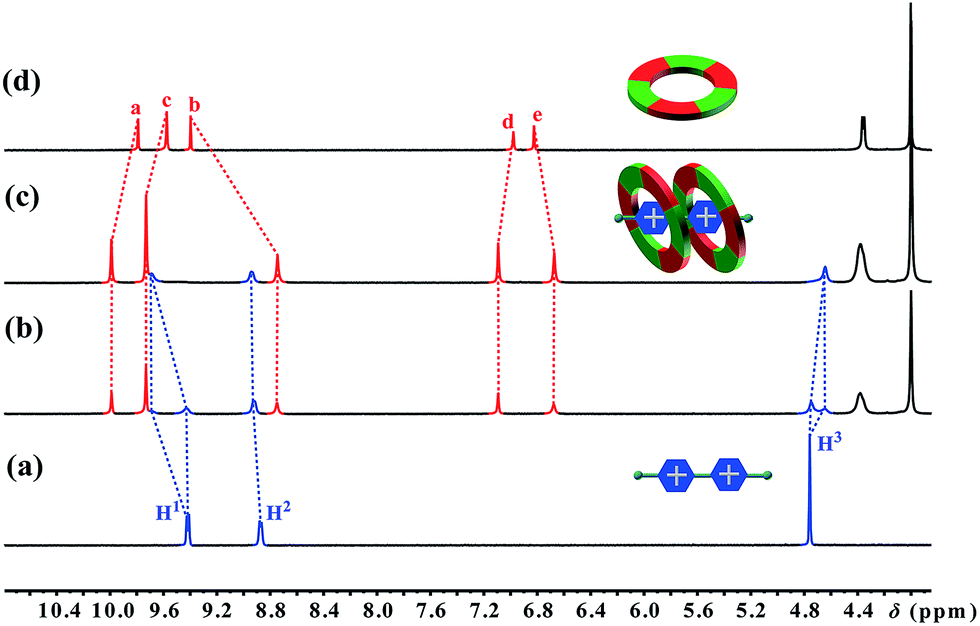
The first sign indicating the H–G interaction came from a color change upon adding paraquat G1 to a solution of cyclo[6]aramide 1 in acetone. An abrupt change from clear to light yellow was observed, indicating that a charge transfer (CT) interaction happened between 1 and G1. A CT band in the UV-vis absorption spectrum confirms the formation of the H-G inclusion complex (Fig. S69†). Then, the formation of the H–G complex was further explored using 1H NMR spectroscopy (Fig. 1). When 1.0 equiv. of 1 was added to a 2.0 mM solution of G1, two sets of signals from the bipyridinium ion were observed, which evolved into one set of signals with 2 equiv. of 1, indicating the slow exchange of the complex on the NMR time scale. Commensurate with this change is the appreciable shift for the aromatic and amide protons of the host upon guest binding. The binding event was supported with 2D nuclear overhauser effect spectroscopy (NOESY), which revealed through-space NOEs between bipyridinium protons and the internal aromatic protons Ha and Hb of G1–G4 (Fig. S55–S64†). Such through-space NOE contacts can transpire only if these two macrocycles are mutually parallel but orthogonal to G1. Two-dimensional diffusion ordered spectroscopic (2D-DOSY) analysis provided additional evidence for the formation of very stable complexes between 1 and G1–G4 (Fig. S65–S68†). For each complex, the protons of 1 and the guest have the same diffusion constant in solution. Furthermore, Job plots supplied information on the binding stoichiometry of 1 and G1–G4 in solution (Fig. S71–S78†). For example, in the case of the complex 12 ⊃ G1, the maximum change in absorbency was observed at 0.67, indicating a macrocycle-cation ratio of 2:1. Examining the 2:1 mixture of 1 and G1 using matrix-assisted laser ionization time of flight mass spectrometry (MALDI-TOF-MS) uncovered the peak with the highest intensity at m/z = 5005.219, corresponding to the complex [12 + G1 − PF6]+. The above results, taken in concert, clearly demonstrate the formation of a 2:1 complex 12 ⊃ G1. The same 2:1 stoichiometry was observed with guests G2 and G4, but G3 only shows 1:1 stoichiometry with 1 (Fig. S103–S109†). Another interesting observation made on complex 12 ⊃ G1 is its reversible redox-responsiveness, which was realized by the addition and removal of Zn powder (Fig. S143†).
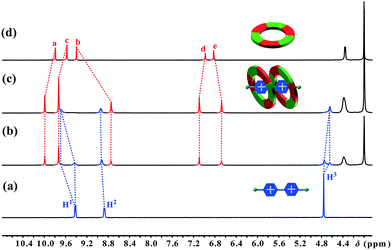 |
| | Fig. 1 Partial 1H NMR spectra (400 MHz, acetone-d6, 298 K) of (a) 2.0 mM G1, (b) 2.0 mM 1 and G1, (c) 4.0 mM 1 and 2.0 mM G1, and (d) 2.0 mM 1. | |
The binding constants K1 and K2 for the complexation of cyclo[6]aramide 1 with bipyridinium salts G1–G4 were obtained using UV-vis titration methods (Table 1). The UV-vis titration experiments revealed surprisingly high binding constants, K1 = 3.49 × 107 M−1 and K2 = 1.09 × 106 M−1, for complex 12 ⊃ G1. The binding of G1 with the second macrocycle is accompanied by a slightly negative cooperative effect (4K2/K1 < 1),26 which is probably caused by the remaining pyridinium unit that has become less electron deficient after the threading of the first macrocycle. To provide insight into the role of the second pyridinium moiety when the first site is occupied, G2, with one positive charge, was examined for binding to 1. Results from UV-vis titration show that, as compared to those of 12 ⊃ G1, the binding constants for G2 and 1 are drastically reduced to K1 = 3.62 × 104 M−1 and K2 = 2.28 × 104 M−1, which indicates the pivotal role played by the second positive charge of G1 for the high stability of complex 12 ⊃ G1. In addition, the positive cooperativity (4K2/K1 = 2.51) observed for 12 ⊃ G2, along with the similar Ka values of complex 1 ⊃ G3 to that of K1 for 12 ⊃ G1, suggests that additional inter-macrocycle π–π stacking may also assist the binding of the second macrocycle in the formation of 12 ⊃ G2. It is worth noting that G4 gives the highest binding constants for binding 1 (K1 = 1.68 × 108 M−1 and K2 = 2.47 × 107 M−1), which should greatly facilitate the synthesis of rotaxanes (Fig. S83–S95 and Table S1†). A similar trend in binding constants was observed in a competitive solvent (acetone-d6/DMSO-d6, 9/1, v/v) (Table 1, Fig. S94 and S95†). Additional infrared experiments on the complexes prepared from pyridinium salts and macrocycle 1 show that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency shift induced by complex formation is in the order of G4 > G1 > G2 > G3 (Fig. S110–S113 and Table S1†), which agrees well with the difference in the binding affinities of these guests with macrocycle 1. In order to demonstrate the crucial role played by shape persistency, cyclo[6]aramide 5, which is partially H-bonded and bears two rotatable amide groups, was synthesized. Since the depletion of partial intramolecular H-bonds results in free rotation of the two amide groups, the shape-persistency as observed in 1 should be attenuated to a significant extent. Indeed, the results from the binding experiments show that it binds G4 in a 2:1 binding mode (Fig. S81–S82†) with binding constants of K1 = 7.29 × 104 M−1 and K2 = 2.50 × 103 M−1 (Table 1 and Fig. S95 and S96†), which are four orders of magnitude lower than that of complex 12 ⊃ G4. This significantly reduced binding affinity as compared to 1 strongly suggests the importance of shape-persistency in the binding event. Despite the great advances made in the past decades, few recognition modules have shown such unusually strong binding in organic media.
O stretching frequency shift induced by complex formation is in the order of G4 > G1 > G2 > G3 (Fig. S110–S113 and Table S1†), which agrees well with the difference in the binding affinities of these guests with macrocycle 1. In order to demonstrate the crucial role played by shape persistency, cyclo[6]aramide 5, which is partially H-bonded and bears two rotatable amide groups, was synthesized. Since the depletion of partial intramolecular H-bonds results in free rotation of the two amide groups, the shape-persistency as observed in 1 should be attenuated to a significant extent. Indeed, the results from the binding experiments show that it binds G4 in a 2:1 binding mode (Fig. S81–S82†) with binding constants of K1 = 7.29 × 104 M−1 and K2 = 2.50 × 103 M−1 (Table 1 and Fig. S95 and S96†), which are four orders of magnitude lower than that of complex 12 ⊃ G4. This significantly reduced binding affinity as compared to 1 strongly suggests the importance of shape-persistency in the binding event. Despite the great advances made in the past decades, few recognition modules have shown such unusually strong binding in organic media.
Table 1 Binding constants of bipyridinium salts G1–G4 with cyclo[6]aramide 1a
| Complex |
Solvent |
Molar ratiob |
K
1 (M−1) |
K
2 (M−1) |
K
12 (M−2) |
Cooperativity α (4K2/K1)c |
|
The binding constant values were obtained through UV-vis titration experiments using a nonlinear fit curve calculated using Matlab 801. For each titration, at least 30 data points were collected. Typically, wavelength was monitored around the charge transfer absorption maxima for the complex formed, estimated errors for K1 and K2 = ±50%.
The stoichiometry was determined using Job plots based on NMR experiments.
The cooperativity with the interaction parameter a according to the equation α = 4K2/K1.
The binding constants were obtained using NMR titration experiments. For full details, see the ESI.
|
|
1
2 ⊃ G1 |
Acetone |
2:1 |
3.49 × 107 |
1.09 × 106 |
3.80 × 1013 |
0.12 |
|
1
2 ⊃ G1 |
Acetone/DMSO (9/1, v/v) |
2:1 |
3.29 × 106 |
3.72 × 105 |
1.22 × 1012 |
0.45 |
|
1
2 ⊃ G2 |
Acetone |
2:1 |
3.62 × 104 |
2.28 × 104 |
8.25 × 108 |
2.51 |
|
1 ⊃ G3 |
Acetone |
1:1 |
1.20 × 104 |
— |
— |
— |
|
1
2 ⊃ G4 |
Acetone |
2:1 |
1.68 × 108 |
2.47 × 107 |
4.15 × 1015 |
0.58 |
|
1
2 ⊃ G4 |
Acetone/DMSO (9/1, v/v) |
2:1 |
8.98 × 106 |
2.14 × 106 |
1.92 × 1013 |
0.95 |
|
5
2 ⊃ G4 |
Acetone-d6 |
2:1 |
[7.29 × 104]d |
[2.50 × 103]d |
[1.82 × 108]d |
0.13 |
X-ray crystal structure of [3]pseudorotaxane 32 ⊃ G1
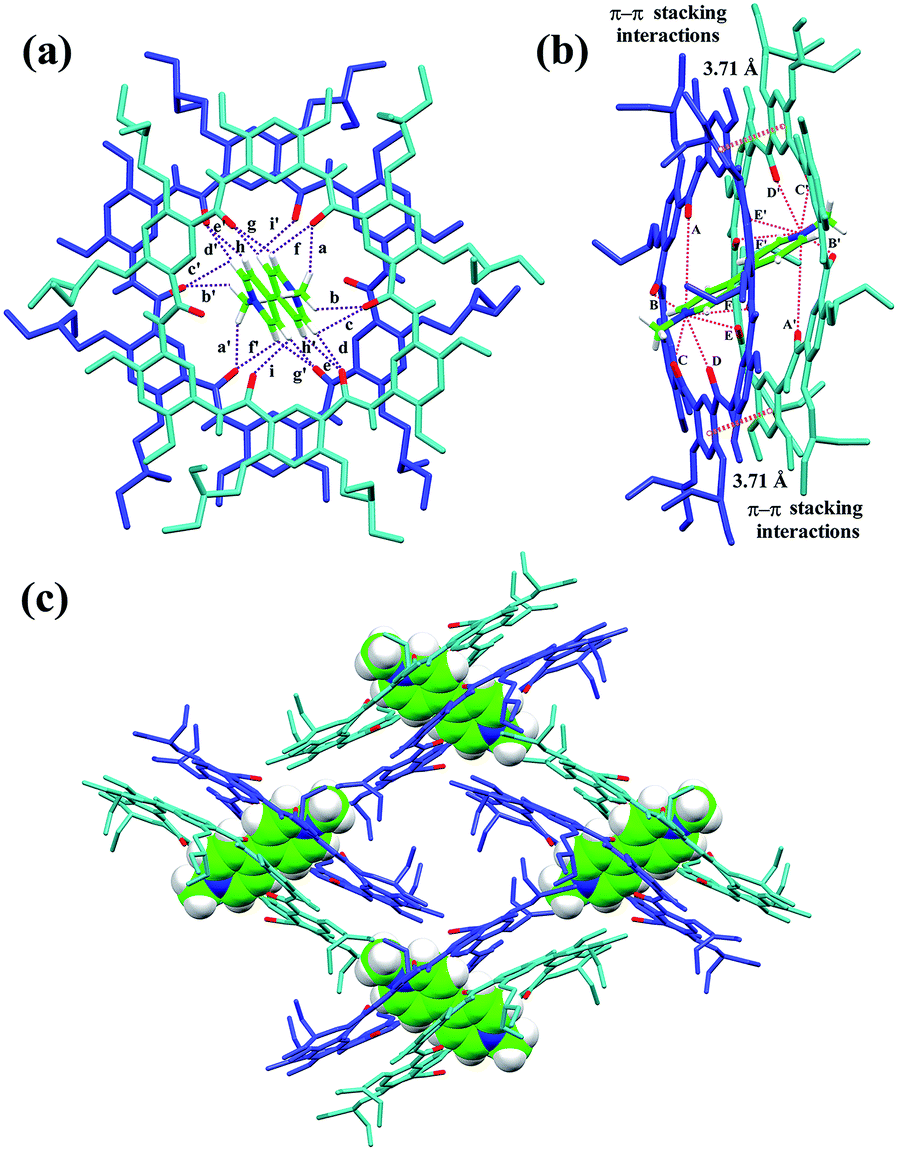
The slow evaporation of a solution containing 3 and G1 in a mixed solvent of acetone/CHCl3/methanol (10/1/0.3) afforded red crystals in 7 weeks. The analysis of the resulting solid state structure using single-crystal X-ray diffraction (XRD) experiments reveals that G1 is inserted into two macrocyclic molecules. In this complex, each of the pyridinium units is orientated orthogonally with respect to the two macrocycles and engages in multiple C–H⋯O H-bonding (Fig. 2a) and N+⋯O ion–dipole interactions that are reinforced by face to face π–π stacking interactions (3.71 Å) (Fig. 2b). These hydrogen bonds all have very short donor–acceptor distances ranging from 2.30 to 2.74 Å due to the rigid macrocyclic backbone constrained by intramolecular three-centre H-bonds. The length of the N+⋯O ion–dipole interactions varying from 3.32 to 4.73 Å is indicative of strong Coulomb interaction between the amide oxygen atoms and pyridinium cations. Therefore, the strong binding affinity between cyclo[6]aramide 1 and the bipyridinium guests is attributed to the result of the cooperative interplay of multipoint non-covalent forces. The geometrically well-defined and tightly packed solid-state structure of 32 ⊃ G1 is also observed which is stabilized by multiple van der Waals forces between the side chains of macrocycle 3 and G1 (Fig. 2c).
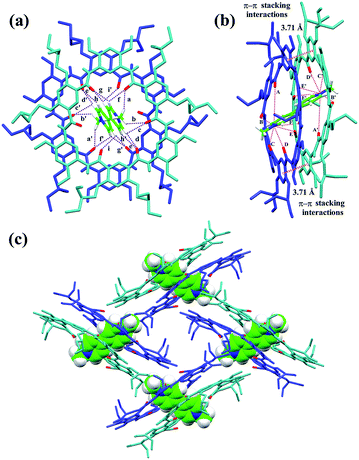 |
| | Fig. 2 X-ray crystal structure of complex 32 ⊃ G1 shown from (a) top and (b) side views. PF6− counterions and hydrogen atoms except the ones involved in hydrogen bonding were omitted for clarity. The dashed purple lines indicate the C–H⋯O hydrogen bonds (a–i) and (a′–i′), parameters are as follows, H⋯O distance (Å), C–H⋯O angles (deg): (a and a′) 2.63, 133.7; (b and b′) 2.64, 116.3; (c and c′) 2.66, 106.7; (d and d′) 2.55, 125.2; (e and e′) 2.64, 120.8; (f and f′) 2.42, 109.1; (g and g′) 2.30, 128.9; (h and h′) 2.74, 110.8; (i and i′) 2.65, 111.1. The dashed pink lines indicate the N+⋯O interactions (A–F) and (A′–F′), parameters are as follows, N+⋯O distance (Å): (A and A′) 4.73; (B and B′) 4.20; (C and C′) 3.35; (D and D′) 3.32; (E and E′) 4.81; (F and F′) 4.46. Face to face π-stacking parameters: centroid–centroid distance (Å), 3.71; ring plane–ring plane inclination (deg), 4.42. (c) Crystal packing structure of complex 32 ⊃ G1. G1 is shown in capped stick model or space filling model. Oxygen atoms in the cavity of the macrocycles are shown in red. PF6− counterions and hydrogen atoms have been removed for clarity. | |
“Click-capping” approach for the synthesis of rotaxanes
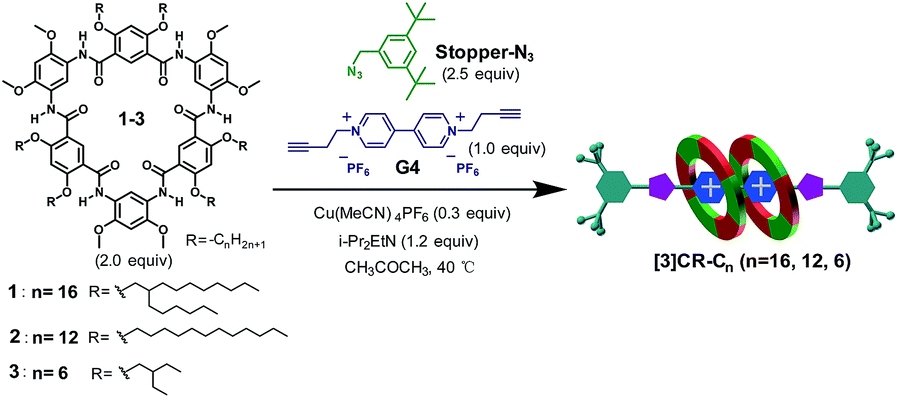
Click reactions, with their high efficiency and simplicity, are widely used for constructing MIMs.27 Buoyed by the observed tight binding, we anticipated that compact rotaxanes [3]CR-Cn (n = 16, 12 and 6) might be attained by tethering Stopper-N3 to the [3]pseudorotaxane formed from cyclo[6]aramides 1–3 and guest G4via a “click-capping” approach. Indeed, the templation reaction proceeded very well, leading to the specific formation of [3]rotaxanes (Table 2, entries 1–3). In the case of macrocycle 1, which is decorated peripherally with sterically crowded alkyl groups, the reaction offers [3]rotaxane [3]CR-C16 as the sole product in a yield of 86%. Using macrocycle 2, which tends to severely aggregate in solution due to the presence of linear alkyl chains,28 the yield of [3]rotaxane [3]CR-C12 was increased up to 91%. To the best of our knowledge, this is one of the rare examples of the synthesis of [3]rotaxanes in excellent yields reported hitherto using a non-metal coordination strategy.29 Due to its low solubility, macrocycle 3 forms [3]CR-C6 in a yield of 64%. Varying the solvent polarity and the ratios of reactants resulted in the controlled generation of [3]rotaxanes as the favourable product (Table 2, entries 4 and 5). To probe which binding mode (1:1 or 2:1) is more favourable for forming rotaxanes, a mixture of 1 equiv. of 1 and 1 equiv. of guest G4 was stirred in the presence of a catalyst. We speculated that the formation of [2]rotaxane should have dominated25 under this condition; however, besides [2]rotaxane in 34% yield, the yield of [3]rotaxane [3]CR-C16 still reached 36%, indicating that the recognition event favours 2:1 stoichiometry, rather than 1:1, during the binding process. The highly selective formation of [3]rotaxanes with cyclo[6]aramides originates from the combination of exceedingly high binding affinities and the compact self-assembly mode. These features are rarely observed in other similar H–G systems. In addition, face to face π–π stacking interactions between the two near-planar macrocycles as observed in the crystal structure of 32 ⊃ G1 could provide an additional driving force that contributes to the high efficiency. It is worth noting that the preparation of [3]rotaxanes with shape-persistent macrocycles as “wheels” in such a high yield (>90%) under non-metal coordination conditions is preceded by few examples and remains a formidable task.18a,30
Table 2 “Click-capping” approach for the synthesis of rotaxanes [3]CR-Cn
“Facile one-pot” approach for the synthesis of rotaxanes
The “facile one-pot” reaction, characterized by simply mixing and heating, and the absence of metallocatalysts, is another useful approach in the synthesis of rotaxanes with high yields.31 The synthesis of [3]rotaxanes [3]R-Cn (n = 16 and 12) or [2]rotaxane [2]R-C6 based on this method was achieved through mixing 2.0 equiv. of cyclo[6]aramide, 2.5 equiv. of Stopper-Br and 1.0 equiv. of 4,4′-bipyridine in CHCl3/CH3CN (1/1, v/v) (Table 3, entries 1–3). Highly efficient template-directed synthesis with macrocycle 1 or 2 was achieved with an excellent yield of 85%, which is rare in the known synthesis of [3]rotaxanes.32 Particularly noticeable was the formation of a [2]rotaxane as the only product when macrocycle 3, which bears short side chains, was used. This specificity in [2]rotaxane formation is rationalized according to the sparse dissolution of the macrocycle in the solution. Compound 3 alone was insoluble in CHCl3/CH3CN (1:1, v/v). However, gradual dissolution was observed to occur as the reaction progressed. This suggests a scanty concentration of macrocycle 3 in the reaction system as compared to that of macrocycle 1 or 2. Since the efficiency of forming [3]rotaxanes depends predominantly on the effective molar ratio (2:1) of the macrocycle and cationic guest in solution, the limited concentration of macrocyclic molecules with respect to that of the cationic axle tends to facilitate a binding process that favours a 1:1 binding mode, thereby leading to the specific formation of a [2]rotaxane.
Table 3 “Facile one-pot” approach for the synthesis of rotaxanes [3]R-Cn
Structural characterization of [2]/[3]rotaxanes
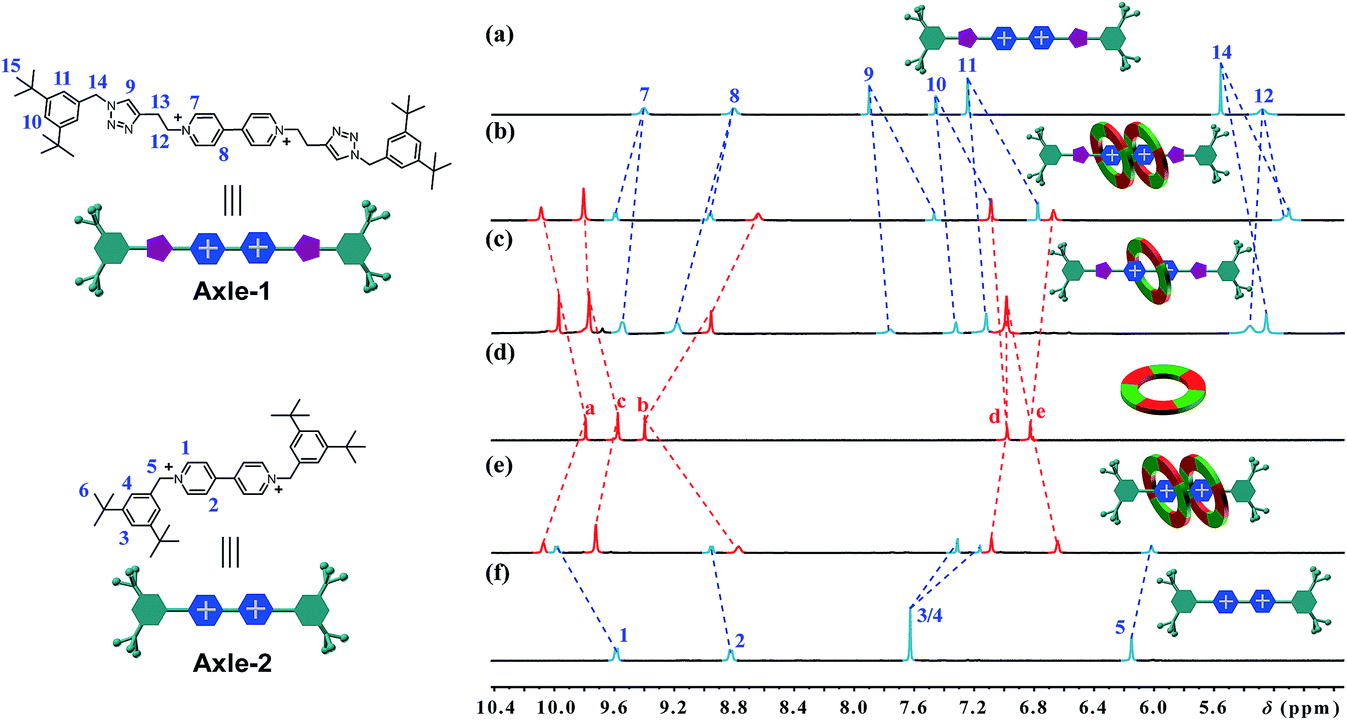
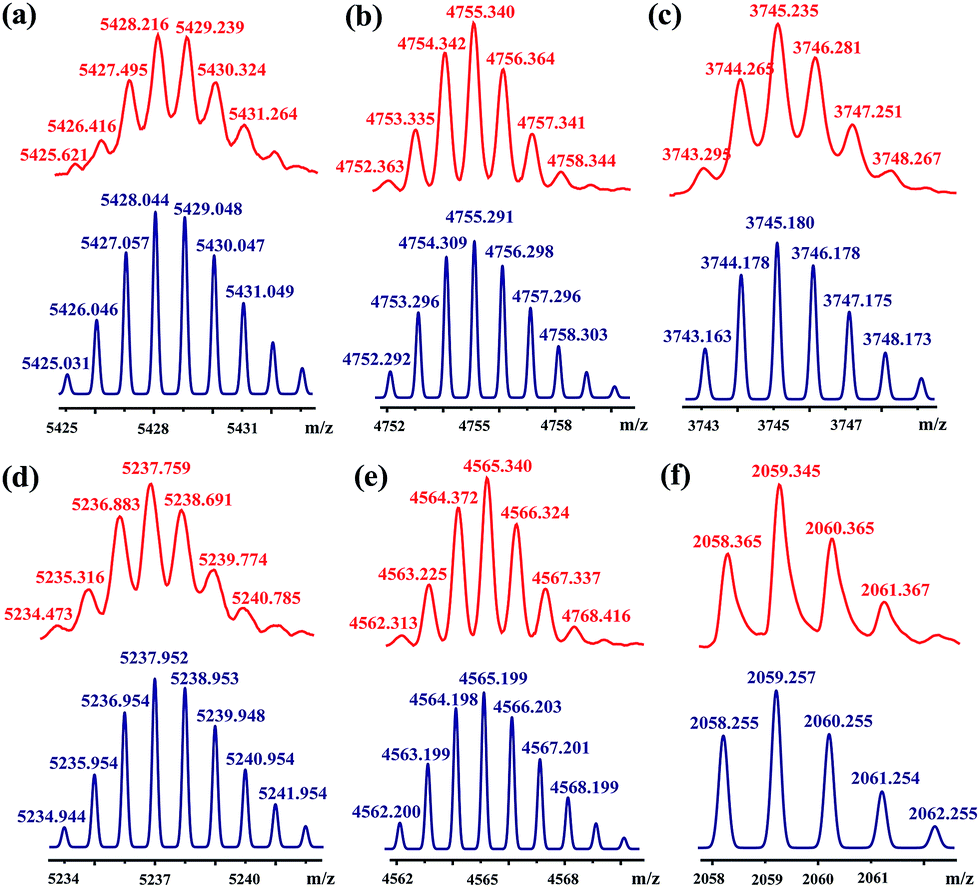
[3]Rotaxanes [3]CR-Cn (n = 6, 12 and 16) and [3]R-Cn (n = 12 and 16), and [2]rotaxanes [2]CR-C16 and [2]R-C6, were fully characterized using 1H and 13C NMR spectroscopy (Fig. S17–S32†) and mass spectrometry (Fig. S38–S44†). For example, the threaded structures of [3]CR-C16, [2]CR-C16 and [3]R-C16 are apparent by comparing their 1H NMR spectra (Fig. 3b, c and e) with those of the free threads Axle-1 and Axle-2 (Fig. 3a and f), and macrocycle 1 (Fig. 3d). The thread protons 7 and 8 in [3]R-C16 are shifted downfield due to C–H⋯O H-bonding. The shielding of protons 9, 10, 11 and 14 on Axle-1, along with the results from 2D NOESY, HSQC, HMBC, and DOSY NMR experiments, indicates the relative positions of the components and the formation of mechanical bonds (Fig. S118–S141†). MALDI-TOF-MS provided additional evidence for the formation of mechanically interlocked structures. In the mass spectra of [3]CR-C16, [3]CR-C12 and [3]CR-C6, the highest related peaks were observed at m/z = 5427.495, m/z = 4753.335 and m/z = 3744.265, which support the structural assignments for the formation of [3]rotaxanes (Fig. 4a–c). Furthermore, related peaks were also observed in the mass spectra of [3]R-C16, [3]R-C12 and [2]R-C6 at m/z = 5236.883, m/z = 4563.225 and m/z = 2058.365, pointing to the formation of a [3]rotaxane or [2]rotaxane (Fig. 4d–f). In all cases, all the peaks were isotopically resolved and fully agreed with their calculated theoretical distributions. It should be noted that all the rotaxanes are in red due to charge-transfer interactions between the macrocycles and axles, as indicated by the CT band observed in the UV-vis absorption spectra (Fig. S142†).
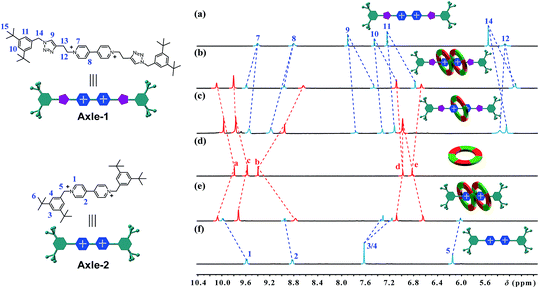 |
| | Fig. 3 Partial 1H NMR spectra of (a) Axle-1, (b) [3]CR-C16, (c) [2]CR-C16, (d) cyclo[6]aramide 1, (e) [3]R-C16 and (f) Axle-2 (400 MHz, acetone-d6, 298 K). The counterion is PF6−. | |
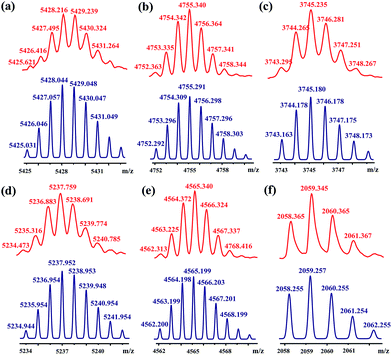 |
| | Fig. 4 MALDI-TOF-MS spectra of rotaxanes (a) [3]CR-C16, (b) [3]CR-C12, (c) [3]CR-C6, (d) [3]R-C16, (e) [3]R-C12 and (f) [2]R-C6 (top, red), and their simulated spectra (bottom, blue). | |
X-ray crystal structure of [3]CR-C6
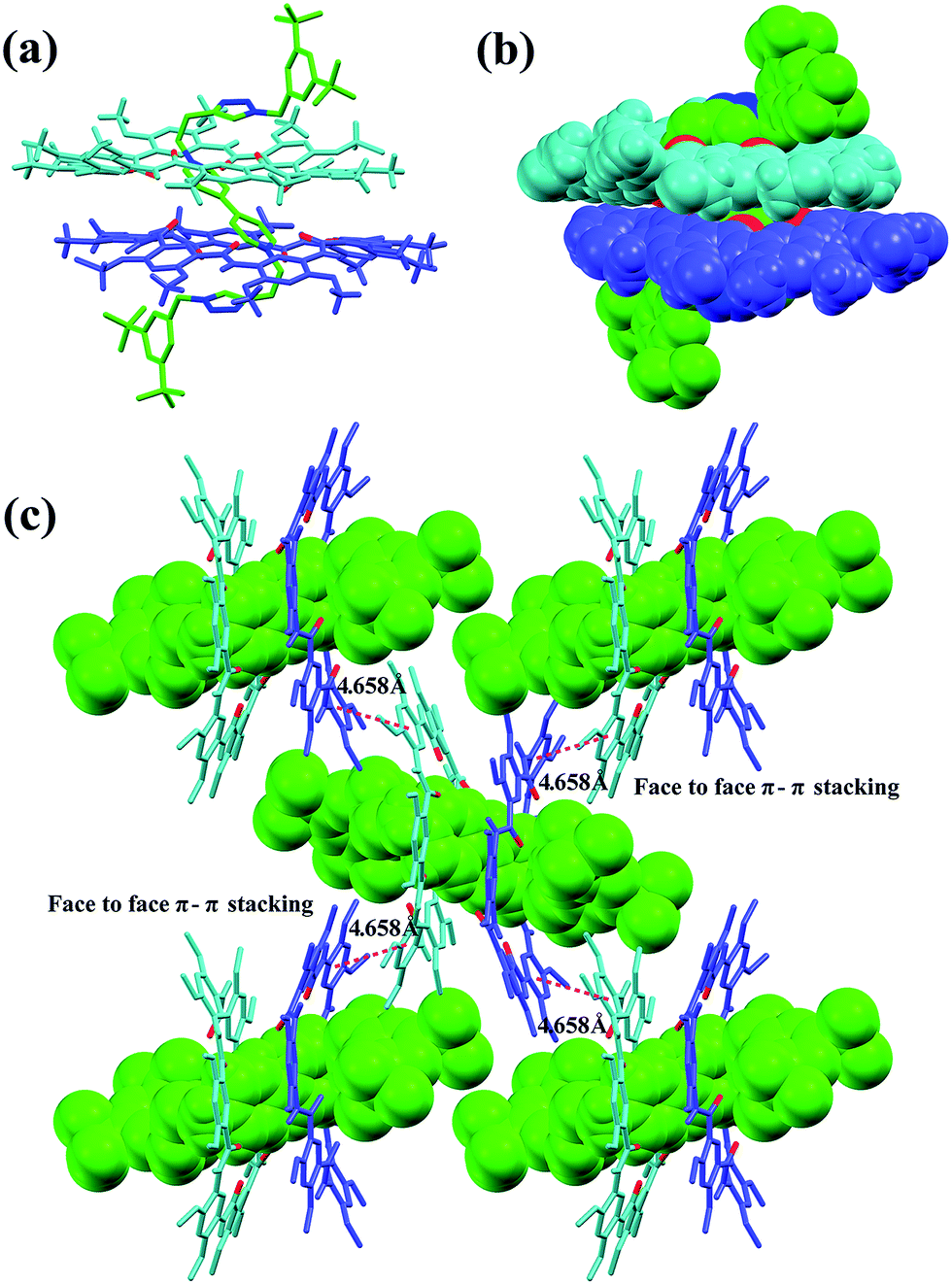
Single crystals of [3]rotaxane [3]CR-C6 were obtained by slowly diffusing methanol into an acetone solution containing [3]CR-C6 in about 8 weeks. The X-ray structure of [3]CR-C6 clearly shows that the thread Axle-1 penetrates through the cavities of the two neighboring macrocyclic molecules in a zigzag conformation (Fig. 5a and b). The mechanically interlocked structure is stabilized by twenty C–H⋯O H-bonds and twelve N+⋯O ion dipole interactions (Tables S4 and S5†). There are also a number of very weak face-to-face π–π stacking interactions (4.658 Å) between the macrocyclic molecules, resulting in a well-ordered and tightly packed solid-state structure (Fig. 5c). The numerous noncovalent forces revealed from the X-ray structure work cooperatively, leading to the surprisingly high stability of the [3]rotaxanes. In fact, when a mixture of Axle-1 and macrocycle 1 was heated under reflux for 3 hours in acetone-d6/DMSO-d6 (9:1, v/v), the 1H NMR spectra did not show any signs of the presence of rotaxanes, indicating the fact that threading does not occur (Fig. S117†), and thus it can be inferred that [3]rotaxane [3]CR-C6 is unlikely to experience a dethreading process under the conditions specified. The high stability is also demonstrated by the observation that complex 12 ⊃ G1 (or 12 ⊃ G4) and free macrocycle 1 were clearly seen for each of them on a TLC plate. Upon the addition of 10.0 equiv. of diethylamine (DEA) to [3]R-C16 or [2]R-C6 in acetone, only [2]R-C6 caused a color change from light yellow to blue, indicating the insensitivity of [3]R-C16 to redox responsiveness. Interestingly, trifluoroacetic acid (TFA) can reverse the redox process of [2]R-C6 (Fig. S144†).
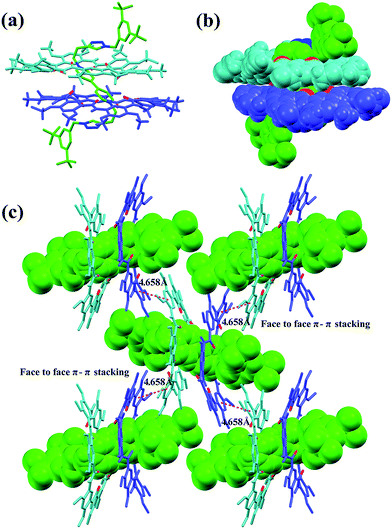 |
| | Fig. 5 X-ray crystal structure of [3]CR-C6 is shown in (a) capped stick model and (b) space-filling model. (c) Crystal packing structure of [3]CR-C6. Face to face π-stacking parameters are shown as dashed magenta lines: centroid–centroid distance (Å), 4.658; ring plane–ring plane inclination (deg), 2.96. Oxygen atoms in the cavity of the macrocycles are shown in red. PF6− counterions, hydrogen atoms and side chains have been removed for clarity. | |
Computational simulation of [3]R-C1
Since the growth of single crystals of the [3]rotaxane [3]R-Cn (n = 16, 12 and 6) synthesized according to the “facile one-pot” approach has proved to be extremely challenging, we resorted to molecular mechanics simulations to gain a better understanding of the noncovalent bonding interactions that direct rotaxane formation and stability. Further structural insights on [3]rotaxane [3]R-C1 were obtained through computational simulations based on the DFT method. Our computational study indicated that [3]rotaxane [3]R-C1 was built by assembling the two macrocyclic molecules of 4 in a near-planar conformation and the central motif Axle-1 in an interlocked orthogonal binding arrangement, which is in good agreement with the structure obtained from the single crystal of [3]CR-C6. Furthermore, multiple C–H⋯O H-bonds and N+⋯O ion–dipole interactions are also observed in the modelling structure which direct the [3]rotaxane formation (Fig. S153†). It is worth noting that there are no face to face π–π stacking interactions formed between the two macrocycles, which is the same as the observation from the crystal structure of [3]CR-C6.
Conclusions
In conclusion, we have demonstrated the unusually high binding affinity (Ka from ∼1013 M−2 to ∼1015 M−2 in acetone) of a novel threaded recognition motif comprising cyclo[6]aramides and bipyridinium salts in 2:1 (H:G) stoichiometry. The crystal structure of the [3]pseudorotaxane shows clear evidence of the high binding affinity, which is attributed as the result of the cooperative interplay of multipoint C–H⋯O H-bonds, N+⋯O ion–dipole interactions and π–π stacking interactions between the two neighboring macrocycles. Furthermore, the highly efficient synthesis of compact [3]rotaxanes achieved using either a “facile one-pot” or “click-capping” approach presents a rare example of constructing MIMs based on 2D shape-persistent macrocycles. The high efficiency of the formation of these rotaxanes highlights the unique advantage of macrocyclic shape-persistency, which results in enhanced multipoint recognition for the highly efficient synthesis of compact mechanically interlocked molecules. The concept of utilizing macrocyclic shape-persistency for boosting multipoint binding affinities for the template-directed synthesis of rotaxanes might be useful for the design of novel MIMs and the development of artificial molecular machines.
Experimental section
General experimental procedure for the “click-capping” approach for the synthesis of [3]CR-Cn (n = 16, 12 and 6)
A mixture of cyclo[6]aramide (2.0 equiv.), guest G4 (1.0 equiv.) and Cu(CH3CN)4PF6 (0.3 equiv.) was stirred in dry acetone at room temperature for 20 minutes under N2. Then a solution of Stopper-N3 (2.5 equiv.) and N,N-diisopropylethylamine (DIPEA) (1.2 equiv.) was injected. The mixture was further stirred at 40 °C for 24 h. The resulting solution was washed with 16% aqueous EDTA tetra-sodium saturated ammonia solution (2 × 50 mL). The organic layer was retained and the aqueous layer extracted twice with CH2Cl2 (2 × 50 mL). The organic extracts were combined and washed with water, dried over Na2SO4 and dried in vacuo. Removal of the solvent afforded a red solid and the crude material was purified using flash column chromatography with silica gel (CHCl3/CH3OH, 20:1) to yield the corresponding red solid. The detailed synthetic procedures and full characterization of new compounds are provided in the ESI.†
General experimental procedure for the “facile one-pot” approach for the synthesis of [3]R-C16, [3]R-C12 and [2]R-C6
A mixture of cyclo[6]aramide (2 equiv.), 3,5-di-tert-butylbenzyl bromide Stopper-Br (2.5 equiv.) and 4,4′-bipyridine (1 equiv.) was stirred in 6 mL of CHCl3/CH3CN (1/1, v/v) under N2 at 40 °C for 48 h. Removal of solvents afforded a pale red solid, which was dissolved in acetone/H2O, followed by the addition of saturated aqueous NaPF6. After stirring for 30 min, the organic solvent was then evaporated under reduced pressure. The precipitate was collected and washed with H2O. Then the crude material was purified using flash column chromatography with silica gel (CHCl3/CH3OH, 30:1, then CHCl3/CH3OH, 10:1) to yield the corresponding red solid. The detailed synthetic procedures and full characterization of new compounds are provided in the ESI.†
Single X-ray crystal data
Crystallographic data (excluding structure factors) for all the structures reported in this article have been deposited with the Cambridge Crystallographic Data Centre in CIF (see the ESI†). Crystal data for 32 ⊃ G1 (CCDC-1475246): C90H121N7O18PF6, M = 1733.91, monoclinic, space group P21/c, a = 22.630(8), b = 23.529(9), and c = 19.425(7) Å, α = 90, β = 112.979(5), and γ = 90°, V = 9522(6) Å3, Z = 4, T = 123(2) K, μ(Mo Kα) = 0.107 mm−1, 54751 reflections measured, 17399 unique (Rint = 0.0938) which were used in all calculations. The final wR(F2) was 0.2667 (all data). Crystal data for [3]CR-C6 (CCDC-1475247): C216H292N20O36P2F12, M = 4034.64, monoclinic, space group P21/n, a = 22.928(2), b = 25.6042(12), and c = 23.2131(15) Å, α = 90, β = 111.030(9), and γ = 90°, V = 12719.6(17) Å3, Z = 2, T = 150(2) K, μ(Mo Kα) = 0.089 mm−1, 52517 reflections measured, 23345 unique (Rint = 0.0624) which were used in all calculations. The final wR(F2) was 0.3386 (all data).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (21572143), Doctoral Program of the Ministry of Education of China (20130181110023), Open Project of State Key Laboratory of Supramolecular Structure and Materials (SKLSSM201629), and US National Science Foundation (CHE-1306326). The Comprehensive Training Platform of the Specialized Laboratory, College of Chemistry, Sichuan University, is acknowledged for NMR analysis.
Notes and references
-
(a) V. Balzani, M. Gómez-López and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS;
(b) G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571–586 CrossRef CAS PubMed;
(c) S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC;
(d) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed;
(e) C. Cheng, P. R. McGonigal, J. F. Stoddart and R. D. Astumian, ACS Nano, 2015, 9, 8672–8688 CrossRef CAS PubMed;
(f) E. R. Kay and D. A. Leigh, Angew. Chem., Int. Ed., 2015, 54, 10080–10088 CrossRef CAS PubMed;
(g) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
-
(a) C. Zhang, S. Li, J. Zhang, K. Zhu, N. Li and F. Huang, Org. Lett., 2007, 9, 5553–5556 CrossRef CAS PubMed;
(b) W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS PubMed;
(c) G. J. E. Davidson, S. Sharma and S. J. Loeb, Angew. Chem., Int. Ed., 2010, 49, 4938–4942 CrossRef CAS PubMed;
(d) Z.-J. Zhang, H.-Y. Zhang, H. Wang and Y. Liu, Angew. Chem., Int. Ed., 2011, 50, 10834–10838 CrossRef CAS PubMed;
(e) K. Zhu, V. N. Vukotic and S. J. Loeb, Angew. Chem., Int. Ed., 2012, 51, 2168–2172 CrossRef CAS PubMed;
(f) K.-D. Wu, Y.-H. Lin, C.-C. Lai and S.-H. Chiu, Org. Lett., 2014, 16, 1068–1071 CrossRef CAS PubMed;
(g) Z. Meng, J.-F. Xiang and C.-F. Chen, Chem. Sci., 2014, 5, 1520–1525 RSC;
(h) Y.-W. Wu, S.-T. Tung, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2015, 54, 11745–11749 CrossRef CAS PubMed.
-
(a) H. Li, A. C. Fahrenbach, S. K. Dey, S. Basu, A. Trabolsi, Z. Zhu, Y. Y. Botros and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8260–8265 CrossRef CAS PubMed;
(b) H. Li, Z. Zhu, A. C. Fahrenbach, B. M. Savoie, C. Ke, J. C. Barnes, J. Lei, Y.-L. Zhao, L. M. Lilley, T. J. Marks, M. A. Ratner and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 456–467 CrossRef CAS PubMed;
(c) Y. Wang, M. Frasconi, W.-G. Liu, J. Sun, Y. Wu, M. S. Nassar, Y. Y. Botros, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, ACS Cent. Sci., 2016, 2, 89–98 CrossRef CAS PubMed.
-
(a) A. Arduini, R. Ferdani, A. Pochini, A. Secchi and F. Ugozzoli, Angew. Chem., Int. Ed., 2000, 39, 3453–3456 CrossRef CAS;
(b) L. Wang, M. O. Vysotsky, A. Bogdan, M. Bolte and V. Böhmer, Science, 2004, 304, 1312–1314 CrossRef CAS PubMed;
(c) A. Arduini, F. Ciesa, M. Fragassi, A. Pochini and A. Secchi, Angew. Chem., Int. Ed., 2005, 44, 278–281 CrossRef CAS PubMed.
-
(a) J. W. Lee, Y. H. Ko, S.-H. Park, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 746–749 CrossRef CAS;
(b) R. Eelkema, K. Maeda, B. Odell and H. L. Anderson, J. Am. Chem. Soc., 2007, 129, 12384–12385 CrossRef CAS PubMed.
-
(a) E. J. F. Klotz, T. D. W. Claridge and H. L. Anderson, J. Am. Chem. Soc., 2006, 128, 15374–15375 CrossRef CAS PubMed;
(b) Y.-L. Zhao, W. R. Dichtel, A. Trabolsi, S. Saha, I. Aprahamian and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 11294–11296 CrossRef CAS PubMed;
(c) A. Hashidzume, H. Yamaguchi and A. Harada, Top. Curr. Chem., 2014, 354, 71–110 CrossRef PubMed.
-
(a) T. Ogoshi, T. Aoki, R. Shiga, R. Iizuka, S. Ueda, K. Demachi, D. Yamafuji, H. Kayama and T. Yamagishi, J. Am. Chem. Soc., 2012, 134, 20322–20325 CrossRef CAS PubMed;
(b) S. Dong, J. Yuan and F. Huang, Chem. Sci., 2014, 5, 247–252 RSC;
(c) G. Yu, D. Wu, Y. Li, Z. Zhang, L. Shao, J. Zhou, Q. Hu, G. Tang and F. Huang, Chem. Sci., 2016, 7, 3017–3024 RSC.
-
(a) V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed;
(b) J.-P. Collin, J. Frey, V. Heitz, J.-P. Sauvage, C. Tock and L. Allouche, J. Am. Chem. Soc., 2009, 131, 5609–5620 CrossRef CAS PubMed;
(c) S. M. Goldup, D. A. Leigh, R. T. McBurney, P. R. McGonigal and A. Plant, Chem. Sci., 2010, 1, 383–386 RSC;
(d) J. D. Crowley, S. M. Goldup, N. D. Gowans, D. A. Leigh, V. E. Ronaldson and A. M. Z. Slawin, J. Am. Chem. Soc., 2010, 132, 6243–6248 CrossRef CAS PubMed;
(e) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed;
(f) C. J. Campbell, D. A. Leigh, I. J. Vitorica-Yrezabal and S. L. Woltering, Angew. Chem., Int. Ed., 2014, 53, 13771–13774 CrossRef CAS PubMed.
-
(a) J. Zhang, D. J. Pesak, J. L. Ludwick and J. S. Moore, J. Am. Chem. Soc., 1994, 116, 4227–4239 CrossRef CAS;
(b) J. S. Moore, Acc. Chem. Res., 1997, 30, 402–413 CrossRef CAS;
(c) W. Zhang and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 4416–4439 CrossRef CAS PubMed.
-
(a) K. Balakrishnan, A. Datar, W. Zhang, X. Yang, T. Naddo, J. Huang, J. Zuo, M. Yen, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2006, 128, 6576–6577 CrossRef CAS PubMed;
(b) C. Ren, S. Xu, J. Xu, H. Chen and H. Zeng, Org. Lett., 2011, 13, 3840–3843 CrossRef CAS PubMed;
(c) J. Vollmeyer, S.-S. Jester, F. Eberhagen, T. Prangenberg, W. Mader and S. Höger, Chem. Commun., 2012, 48, 6547–6549 RSC.
-
(a) A. J. Helsel, A. L. Brown, K. Yamato, W. Feng, L. Yuan, A. J. Clements, S. V. Harding, G. Szabo, Z. Shao and B. Gong, J. Am. Chem. Soc., 2008, 130, 15784–15785 CrossRef CAS PubMed;
(b) X. Zhou, G. Liu, K. Yamato, Y. Shen, R. Cheng, X. Wei, W. Bai, Y. Gao, H. Li, Y. Liu, F. Liu, D. M. Czajkowsky, J. Wang, M. J. Dabney, Z. Cai, J. Hu, F. V. Bright, L. He, X. C. Zeng, Z. Shao and B. Gong, Nat. Commun., 2012, 3, 949 CrossRef PubMed;
(c) B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS PubMed;
(d) P. Xin, L. Zhang, P. Su, J.-L. Hou and Z.-T. Li, Chem. Commun., 2015, 51, 4819–4822 RSC;
(e) W. Si, P. Xin, Z.-T. Li and J.-L. Hou, Acc. Chem. Res., 2015, 48, 1612–1619 CrossRef CAS PubMed;
(f) Y. Huo and H. Zeng, Acc. Chem. Res., 2016, 49, 922–930 CrossRef CAS PubMed.
-
(a) H. Zhao, J. Shen, J. Guo, R. Ye and H. Zeng, Chem. Commun., 2013, 49, 2323–2325 RSC;
(b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
-
(a) A. R. Sanford, L. Yuan, W. Feng, K. Yamato, R. A. Flowers and B. Gong, Chem. Commun., 2005, 41, 4720–4722 RSC;
(b) P. S. Shirude, E. R. Gillies, S. Ladame, F. Godde, K. Shinya, I. Huc and S. Balasubramanian, J. Am. Chem. Soc., 2007, 129, 11890–11891 CrossRef CAS PubMed;
(c) Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS PubMed;
(d) B. Qin, C. Ren, R. Ye, C. Sun, K. Chiad, X. Chen, Z. Li, F. Xue, H. Su, G. A. Chass and H. Zeng, J. Am. Chem. Soc., 2010, 132, 9564–9566 CrossRef CAS PubMed;
(e) K. P. McDonald, Y. Hua, S. Lee and A. H. Flood, Chem. Commun., 2012, 48, 5065–5075 RSC;
(f) Y. Liu, J. Shen, C. Sun, C. Ren and H. Zeng, J. Am. Chem. Soc., 2015, 137, 12055–12063 CrossRef CAS PubMed;
(g) S. Lee, B. E. Hirsch, Y. Liu, J. R. Dobscha, D. W. Burke, S. L. Tait and A. H. Flood, Chem.–Eur. J., 2016, 22, 560–569 CrossRef CAS PubMed;
(h) B. Qiao, J. R. Anderson, M. Pink and A. H. Flood, Chem. Commun., 2016, 52, 8683–8686 RSC.
-
(a) A. S. Shetty, P. R. Fischer, K. F. Stork, P. W. Bohn and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 9409–9414 CrossRef CAS;
(b) S. Höger, D. L. Morrison and V. Enkelmann, J. Am. Chem. Soc., 2002, 124, 6734–6736 CrossRef;
(c) G.-B. Pan, X.-H. Cheng, S. Höger and W. Freyland, J. Am. Chem. Soc., 2006, 128, 4218–4219 CrossRef CAS PubMed;
(d) C. Ren, V. Maurizot, H. Zhao, J. Shen, F. Zhou, W. Q. Ong, Z. Du, K. Zhang, H. Su and H. Zeng, J. Am. Chem. Soc., 2011, 133, 13930–13933 CrossRef CAS PubMed;
(e) A. Singharoy, B. Venkatakrishnan, Y. Liu, C. G. Mayne, S. Lee, C.-H. Chen, A. Zlotnick, K. Schulten and A. H. Flood, J. Am. Chem. Soc., 2015, 137, 8810–8818 CrossRef CAS PubMed.
- Y. Liu, A. Singharoy, C. G. Mayne, A. Sengupta, K. Raghavachari, K. Schulten and A. H. Flood, J. Am. Chem. Soc., 2016, 138, 4843–4851 CrossRef CAS PubMed.
-
(a) A. S. Lane, D. A. Leigh and A. Murphy, J. Am. Chem. Soc., 1997, 119, 11092–11093 CrossRef CAS;
(b) E. M. Pérez, D. T. F. Dryden, D. A. Leigh, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 12210–12211 CrossRef PubMed;
(c) V. Aucagne, D. A. Leigh, J. S. Lock and A. R. Thomson, J. Am. Chem. Soc., 2006, 128, 1784–1785 CrossRef CAS PubMed;
(d) M. Alvarez-Pérez, S. M. Goldup, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem. Soc., 2008, 130, 1836–1838 CrossRef PubMed;
(e) R. Ahmed, A. Altieri, D. M. D'Souza, D. A. Leigh, K. M. Mullen, M. Papmeyer, A. M. Z. Slawin, J. K. Y. Wong and J. D. Woollins, J. Am. Chem. Soc., 2011, 133, 12304–12310 CrossRef CAS PubMed;
(f) A. Carlone, S. M. Goldup, N. Lebrasseur, D. A. Leigh and A. Wilson, J. Am. Chem. Soc., 2012, 134, 8321–8323 CrossRef CAS PubMed;
(g) M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235–240 CrossRef CAS PubMed.
-
(a) Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172–1175 CrossRef CAS PubMed;
(b) Y. Ferrand, Q. Gan, B. Kauffmann, H. Jiang and I. Huc, Angew. Chem., Int. Ed., 2011, 50, 7572–7575 CrossRef CAS PubMed;
(c) S. A. Denisov, Q. Gan, X. Wang, L. Scarpantonio, Y. Ferrand, B. Kauffmann, G. Jonusauskas, I. Huc and N. D. McClenaghan, Angew. Chem., Int. Ed., 2016, 55, 1328–1333 CrossRef CAS PubMed.
-
(a) S. Lee, C. H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed;
(b) C. Schweez, P. Shushkov, S. Grimme and S. Höger, Angew. Chem., Int. Ed., 2016, 55, 3328–3333 CrossRef CAS PubMed.
-
(a) Z.-T. Li, J.-L. Hou and C. Li, Acc. Chem. Res., 2008, 41, 1343–1353 CrossRef CAS PubMed;
(b) B. Gong, Acc. Chem. Res., 2008, 41, 1376–1386 CrossRef CAS PubMed;
(c) D. W. Zhang, X. Zhao, J. L. Hou and Z. T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed;
(d) H. Fu, Y. Liu and H. Zeng, Chem. Commun., 2013, 49, 4127–4144 RSC.
-
(a) L. Yuan, W. Feng, K. Yamato, A. R. Sanford, D. Xu, H. Guo and B. Gong, J. Am. Chem. Soc., 2004, 126, 11120–11121 CrossRef CAS PubMed;
(b) W. Feng, K. Yamato, L. Yang, J. S. Ferguson, L. Zhong, S. Zou, L. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2009, 131, 2629–2637 CrossRef CAS PubMed;
(c) Y. A. Yang, W. Feng and L. Yuan, Chem. J. Chin. Univ., 2011, 32, 1950–1961 CAS;
(d) L. Zhong, L. Chen, W. Feng, S. Zou, Y. Yang, N. Liu and L. Yuan, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 367–373 CrossRef CAS.
-
(a) J. Hu, L. Chen, Y. Ren, P. Deng, X. Li, Y. Wang, Y. Jia, J. Luo, X. Yang, W. Feng and L. Yuan, Org. Lett., 2013, 15, 4670–4673 CrossRef CAS PubMed;
(b) Y. He, M. Xu, R. Gao, X. Li, F. Li, X. Wu, D. Xu, H. Zeng and L. Yuan, Angew. Chem., Int. Ed., 2014, 53, 11834–11839 CrossRef CAS PubMed;
(c) J. Hu, L. Chen, J. Shen, J. Luo, P. Deng, Y. Ren, H. Zeng, W. Feng and L. Yuan, Chem. Commun., 2014, 50, 8024–8027 RSC;
(d) X. Li, B. Li, L. Chen, J. Hu, C. Wen, Q. Zheng, L. Wu, H. Zeng, B. Gong and L. Yuan, Angew. Chem., Int. Ed., 2015, 54, 11147–11152 CrossRef CAS PubMed;
(e) L. Chen, Z. Peng, S. Liu, X. Li, R. Chen, Y. Ren, W. Feng and L. Yuan, Org. Lett., 2015, 17, 5950–5953 CrossRef CAS PubMed.
- M. Xu, L. Chen, Y. Jia, L. Mao, W. Feng, Y. Ren and L. Yuan, Supramol. Chem., 2015, 27, 436–443 CrossRef CAS.
-
(a) M. Zhang, K. Zhu and F. Huang, Chem. Commun., 2010, 46, 8131–8141 RSC;
(b) Y. Han, Z. Meng, Y. X. Ma and C. F. Chen, Acc. Chem. Res., 2014, 47, 2026–2040 CrossRef CAS PubMed;
(c) Q.-H. Guo, L. Zhao and M.-X. Wang, Angew. Chem., Int. Ed., 2015, 54, 8386–8389 CrossRef CAS PubMed;
(d) F. Jia, Z. He, L.-P. Yang, Z.-S. Pan, M. Yi, R.-W. Jiang and W. Jiang, Chem. Sci., 2015, 6, 6731–6738 RSC.
-
(a) F. Huang, H. W. Gibson, W. S. Bryant, D. S. Nagvekar and F. R. Fronczek, J. Am. Chem. Soc., 2003, 125, 9367–9371 CrossRef CAS PubMed;
(b) F. Huang, K. A. Switek, L. N. Zakharov, F. R. Fronczek, C. Slebodnick, M. Lam, J. A. Golen, W. S. Bryant, P. E. Mason, A. L. Rheingold, M. Ashraf-Khorassani and H. W. Gibson, J. Org. Chem., 2005, 70, 3231–3241 CrossRef CAS PubMed.
- P.-N. Cheng, C.-F. Lin, Y.-H. Liu, C.-C. Lai, S.-M. Peng and S.-H. Chiu, Org. Lett., 2006, 8, 435–438 CrossRef CAS PubMed.
-
(a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC;
(b) E. N. W. Howe, M. Bhadbhade and P. Thordarson, J. Am. Chem. Soc., 2014, 136, 7505–7516 CrossRef CAS PubMed.
-
(a) V. Aucagne and D. A. Leigh, Org. Lett., 2006, 8, 4505–4507 CrossRef CAS PubMed;
(b) J. Winn, A. Pinczewska and S. M. Goldup, J. Am. Chem. Soc., 2013, 135, 13318–13321 CrossRef CAS PubMed;
(c) E. A. Neal and S. M. Goldup, Chem. Sci., 2015, 6, 2398–2404 RSC.
- Y. Yang, W. Feng, J. Hu, S. Zou, R. Gao, K. Yamato, M. Kline, Z. Cai, Y. Gao, Y. Wang, Y. Li, Y. Yang, L. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2011, 133, 18590–18593 CrossRef CAS PubMed.
- S. Li, M. Liu, J. Zhang, B. Zheng, X. Wen, N. Li and F. Huang, Eur. J. Org. Chem., 2008, 6128–6133 CrossRef CAS.
- B. Qiao, Y. Liu, S. Lee, M. Pink and A. H. Flood, Chem. Commun., 2016, 52, 13675–13678 RSC.
-
(a) A. Pun, D. A. Hanifi, G. Kiel, E. O'Brien and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 13119–13122 CrossRef CAS PubMed;
(b) C. Ke, N. L. Strutt, H. Li, X. Hou, K. J. Hartlieb, P. R. McGonigal, Z. Ma, J. Iehl, C. L. Stern, C. Cheng, Z. Zhu, N. A. Vermeulen, T. J. Meade, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 17019–17030 CrossRef CAS PubMed;
(c) X. Fu, Q. Zhang, S.-J. Rao, D.-H. Qu and H. Tian, Chem. Sci., 2016, 7, 1696–1701 RSC.
-
(a) E. A. Wilson, N. A. Vermeulen, P. R. McGonigal, A.-J. Avestro, A. A. Sarjeant, C. L. Stern and J. F. Stoddart, Chem. Commun., 2014, 50, 9665–9668 RSC;
(b) E. A. Neal and S. M. Goldup, Angew. Chem., Int. Ed., 2016, 55, 12488–12493 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further details of the synthesis, characterization, 1H and 2D NMR experiments and computational modelling. CCDC 1475246 and 1475247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04714a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Yi
Ren
a,
Chengyuan
Wang
a,
Lixin
Wu
b,
Wen
Feng
a,
Bing
Gong
c and
Lihua
Yuan
a,
Yi
Ren
a,
Chengyuan
Wang
a,
Lixin
Wu
b,
Wen
Feng
a,
Bing
Gong
c and
Lihua
Yuan