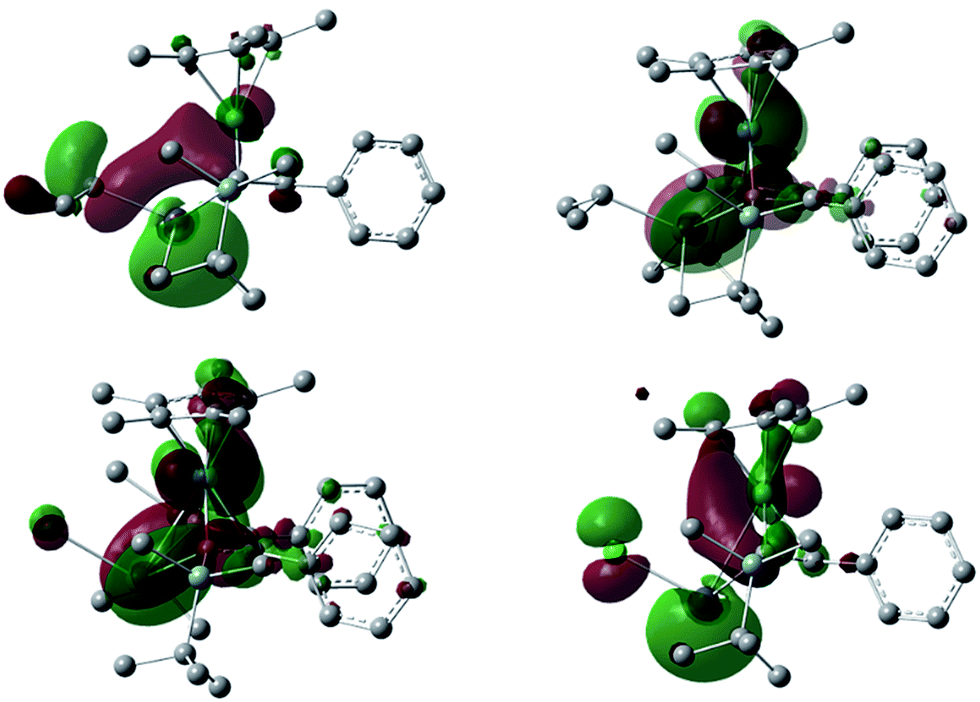DOI:
10.1039/C6SC04843A
(Edge Article)
Chem. Sci., 2017,
8, 3092-3097
Synthesis and reactivity of a ruthenocene-type complex bearing an aromatic π-ligand with the heaviest group 14 element†
Received
1st November 2016
, Accepted 11th February 2017
First published on 13th February 2017
Abstract
An anionic ruthenocene derived from a dilithioplumbole complex was prepared. In the complex, the plumbole ligand coordinates a ruthenium atom in an η5-fashion, similar to the cyclopentadienyl ligand in ferrocene. The ruthenocene that has the aromatic π-ligand with the heaviest group 14 element reacted with electrophiles to afford the plumbole complexes wherein the plumbole ligands show deviation from planarity, in contrast to the planar plumbole ring in the anionic ruthenocene. The bent angles of the plumbole ligands are dependent on the substituents on the lead atoms. Cyclic voltammetry measurements revealed that the plumbole complexes are oxidized more easily than the corresponding stannole complexes.
Introduction
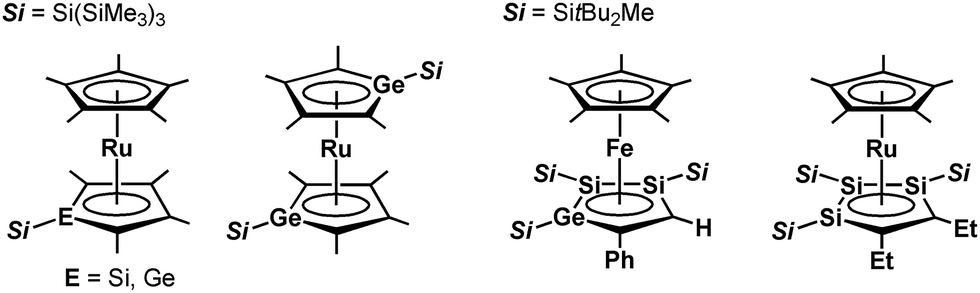
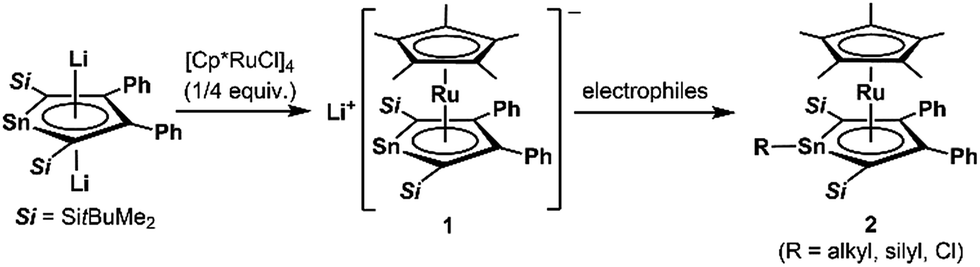
Since the discovery of ferrocene,1 the cyclopentadienyl anion (C5H5−; Cp−) has been one of the most widely-used ligands for transition-metal complexes, and the resulting ferrocene-type sandwich complexes have played important roles in many fields of chemistry, such as structural,2 synthetic,3 materials4 and supramolecular chemistry.5 To modify the properties of complexes bearing Cp ligands, the introduction of electron-donating, electron-withdrawing and sterically demanding groups to the skeletal carbon atoms of the cyclopentadienyl rings has been investigated, and indeed their properties and catalytic activities are effectively changed.6 Another possible method to change the properties of transition-metal complexes with Cp ligands is through the substitution of a skeletal carbon atom with a heavy group 14 atom: the utilization of group 14 metallole anions and dianions (heavy Cp anions and dianions)7 as ligands in the sandwich complexes. In fact, the synthesis of ferrocene-type transition-metal complexes bearing silicon8,9 and germanium analogs10,11 of heavy Cp anions has already been achieved (Fig. 1). Each of the ferrocene and ruthenocene analogs with the heavy Cps is oxidized more easily than the corresponding Cp and Cp*(C5Me5) derivatives, indicating that the heavy Cp ligands function as ligands that are more electron-donating than the Cp and Cp*. However, such derivatives were derived only from the silicon- and germanium-bearing ligands. Moreover, the substituents on the silicon and germanium atoms in the complexes are limited to silyl groups and therefore the effects of the substituents on the properties of the complexes remain elusive. In the course of our studies on the application of dilithiostannoles12 as ligands in transition-metal complexes,13 we have already reported on the synthesis of a lithium salt of anionic ruthenocene 1 bearing a stannole dianionic ligand,14 which enables the introduction of various substituents to the tin atoms (Scheme 1).15 The coordination mode of the resulting ruthenocene derivatives (2) is highly dependent on the substituent on the tin atom, and the complexes composed of η5-coordinating stannole ligands are oxidized more easily than the germole complex.10a These findings prompted us to apply plumbole anions and dianions16 as ligands, which would be envisaged to be more electron-donating than the tin analogs because a lead atom is the heaviest among the group 14 atoms. In terms of sandwich complexes bearing ligands containing the 6th row elements, a bismaferrocene and a dibismaferrocene have been reported.17 Although the X-ray characterization of the bismaferrocene and the dynamic behavior of the dibismaferrocene in solution were investigated, their electronic properties and reactivity were not discussed. Therefore, the synthesis, properties and reactivity of sandwich complexes with 6th row element-bearing ligands are of considerable fundamental importance. We report herein the synthesis of ruthenocene-type complexes, revealing a highly oxidizable nature due to the utilization of ligands having the heaviest group 14 element.
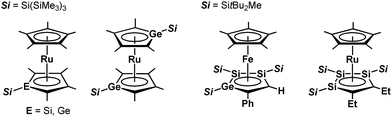 |
| | Fig. 1 Ferrocene- and ruthenocene-type sandwich complexes bearing ligands with heavy group 14 elements. | |
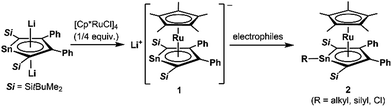 |
| | Scheme 1 Ruthenocene-type complexes bearing ligands with heavy group 14 elements. | |
Results and discussion
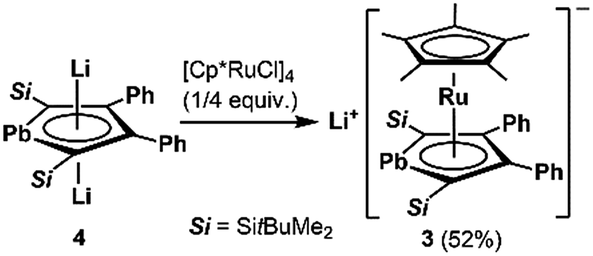
1. Synthesis and structure of the anionic ruthenocene 3
We succeeded in the synthesis of the key intermediate, the anionic ruthenium complex 3, by the reaction of silyl-substituted dilithioplumbole 416b with 0.25 equivalents of [Cp*RuCl]418 in THF (Scheme 2). After recrystallization of the crude product from THF and hexane, compound 3 was isolated as air- and moisture-sensitive red crystals in 52% yield. The 13C NMR signals assignable to the ring carbons of 3 (Cα: 145.60, Cβ: 121.66 ppm) are observed in a higher field compared to those of the starting material 4 (Cα: 213.47, Cβ: 159.38 ppm). A similar trend was found in the 207Pb NMR spectrum of 3: a signal appeared at a higher field (1552 ppm) compared to that of 4 (2573 ppm). According to the NMR calculations, all of these high-field shifts from compound 4 to 3 (Table 1) are mainly caused by a decrease in the paramagnetic contribution, indicating an increase in the electron density due to back-donation from the ruthenium moiety to the plumbole ring. The opposite case, which is a decrease in the electron density causing an increase in the paramagnetic contribution that results in magnetic deshielding, was already reported.19 Theoretical calculations on the relationship between the electron density on the carbon atoms and the paramagnetic terms in the 13C NMR chemical shifts are demonstrated in the ESI.†
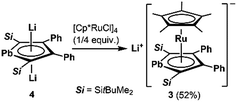 |
| | Scheme 2 Preparation of anionic ruthenocene 3 bearing a plumbole ligand. | |
Table 1
13C and 207Pb NMR chemical shifts (ppm) of 3 and 4
|
|
3
|
4
|
|
δ(13C) for Cα |
145.60 |
213.47 |
|
δ(13C) for Cβ |
121.66 |
159.38 |
|
δ(207Pb) |
1552 |
2573 |
X-ray diffraction analysis revealed that the plumbole ring in 3 has negligible C–C bond alternation, suggesting that the aromatic nature of the plumbole ring is retained, and 3 has a ferrocene-type sandwich structure (Fig. 2). The five-membered ring of the plumbole ring in 3 is almost planar with the sum of the internal angles being 539.4°, and the C–C bond distances within the plumbole ring are almost equal (1.425(5), 1.458(5) and 1.419(5) Å). The Pb–Ru distance in 3 is 2.8119(3) Å, which is slightly longer than those found in the compounds bearing Pb–Ru bonds (4 examples: 2.6668(5)–2.787(2) Å).20 The Ru–C distances in the plumbole ring are 2.200(4)–2.277(4) Å, which are similar to those found in the Cp* ligand (2.189(4)–2.252(4) Å) and those of decamethylruthenocene (2.171(4)–2.176(5) Å).21 These structural features strongly suggest that complex 3 should have a ferrocene-type sandwich type structure, where a lead atom in the ligand takes part for the first time in π-ligation and therefore the plumbole ligand coordinates the ruthenium atom in an η5-fashion. The HOMO of 3 reveals the interaction between the lead, ruthenium and carbon atoms of the plumbole ring, indicating that the original p-type lone pair of the dilithioplumbole interacts with the ruthenium and carbon atoms of the plumbole ring. The bonding interaction between the Pb and Ru atoms is also supported by its Wiberg Bond Index (WBI) of 0.42, which is larger than those of the Ru–C bonds in the Cp* ligand (0.34–0.35). MOs having interactions between the Ru atom and the plumbole ring are shown in the ESI.†
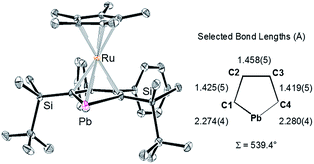 |
| | Fig. 2 ORTEP drawing of 3 (50% probability) and selected bond lengths of the plumbole ring. All hydrogen atoms and the cationic moiety are omitted for clarity. | |
The electrostatic potential surface of the HOMO in 3 reveals that the negative charge is mainly localized on the lead atom (Fig. 3), and therefore electrophilic functionalization would be envisaged to take place on the lead atom.
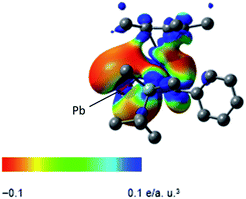 |
| | Fig. 3 Electrostatic potential surface of the HOMO for compound 3. | |
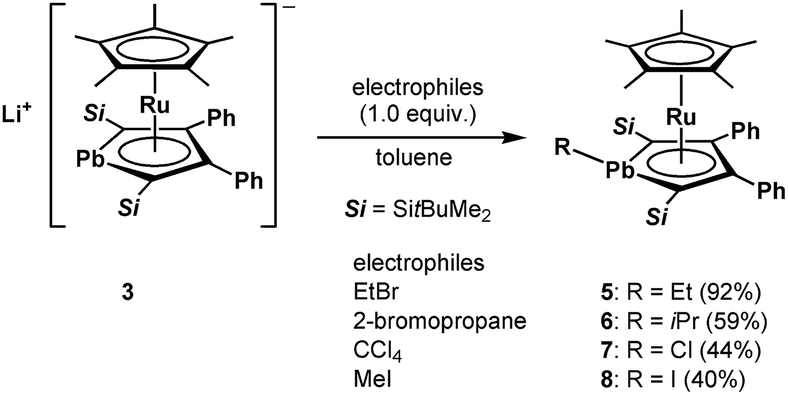
2. Reactions of the anionic ruthenocene 3
We achieved the synthesis of the ruthenium complexes 5–8 by the reactions of compound 3 with the corresponding electrophiles (Scheme 3). The formation of the halogenated compounds 7 and 8 was unexpected. However, heavy group 14 lithium species such as stannyllithiums are known to undergo halophilic reactions,22 and lithiation followed by chlorination by CCl4 is a well-known process for the preparation of chloro-derivatives.23 Accordingly, in the reaction of anionic ruthenocene 3 with CCl4, compound 3 attacks Cl to afford complex 7, and a further coupling reaction of 7 and the resulting CCl3 anion is suppressed due to steric reasons. In the reaction of anionic ruthenocene 3 with bromoalkanes and iodomethane, the first step would be a single electron transfer, as was reported in the reactions of stannyllithium with haloalkanes.24 However, we have already reported that the reaction of an anionic ruthenocene bearing a stannole ligand with iodomethane afforded the expected Me-bearing stannole complex.15 The introduction of an iodine atom to the lead atom is therefore characteristic of the reactivity of the plumbole-bearing anionic ruthenocene 3, even though the reason for this is still unclear.
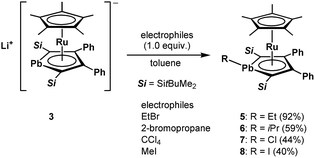 |
| | Scheme 3 Reactions of anionic ruthenocene 3 with electrophiles to afford complexes 5–8. | |
We found that the structures of the complexes are significantly different from that of 3 (Fig. 4). The plumbole rings of 6–8, which were characterized by X-ray diffraction analysis, deviate from planarity.25 The deviation from planarity is dependent on the substituent on the lead atom. The dihedral angles between the C4 and C1–Pb–C4 planes for the halogenated derivatives 7 and 8 (44.88(18) and 42.88(26)°, respectively) are larger than that for the alkyl derivative 6 (33.00(18)°). Although a similar trend was found in the corresponding tin complexes, the bent angles of the PbC4 rings are larger than those of the SnC4 rings: 44.9 vs. 41.2° for the chloro complexes, and 33.0 vs. 24.1° for the alkyl complexes.15 In parallel, the sum of the bond angles around the lead atom of 6 (313°) is larger than those for 7 and 8 (299 and 304°, respectively), indicating that pyramidalization around the lead atoms of 7 and 8 is larger than that in 6. In contrast, as the pyramidalization around the lead atoms becomes smaller, the C–C bond alternation in the plumbole rings becomes smaller (Table 2). The difference between the Cα–Cβ and Cβ–Cβ bond lengths is 0.045(6) Å in 6, while that in 8 is 0.070(10) Å. However, both of them are larger than that found in 3 (0.033 Å), which has a flatter plumbole ring.
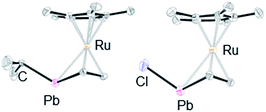 |
| | Fig. 4 ORTEP drawings of 6 (left) and 7 (right) (50% probability). All hydrogen atoms and the substituents on the plumbole carbon atoms are omitted for clarity. | |
Table 2 Comparison of the selected bond lengths (Å) and angles (°) of 3, 6, 7 and 8
|
|
3
|
6
|
7
|
8
|
| Pb–Cα |
2.274(4), 2.280(4) |
2.317(3), 2.363(3) |
2.385(5), 2.371(5) |
2.370(6), 2.375(6) |
| Cα–Cβ |
1.425(5), 1.419(5) |
1.421(5), 1.431(4) |
1.412(7), 1.408(7) |
1.408(8), 1.403(8) |
| Cβ–Cβ |
1.458(5) |
1.466(4) |
1.478(7) |
1.479(8) |
|
Σ (internal angle of the PbC4 ring) |
539.4 |
526.6 |
516.0 |
517.9 |
| Dihedral angle between the C4 and Cα–Pb–Cα planes |
6.9 |
33.0 |
44.9 |
42.9 |
The structural differences cause the following NMR characteristics. In the 13C NMR spectra, the signals of Cα of 5 (151.94 ppm) and 6 (149.76 ppm) were found in a field higher than those of 7 (176.20 ppm) and 8 (170.70 ppm). In the 13C NMR spectra of the stannole and plumbole derivatives,12,16,26 the chemical shifts for the Cα nuclei are highly dependent on the electronic states of the tin and lead atoms: the Cα nuclei of dilithiometalloles, which have dianionic character, resonate in a field lower (approximately 225 ppm for dilithioplumboles) than those for lithiometalloles (approximately 205 ppm for a lithioplumbole), bearing monoanionic character. The NMR signals for the Cα nuclei of 1,1-diphenylmetalloles are observed in a much higher field (for example, 150 ppm for hexaphenylplumbole).27
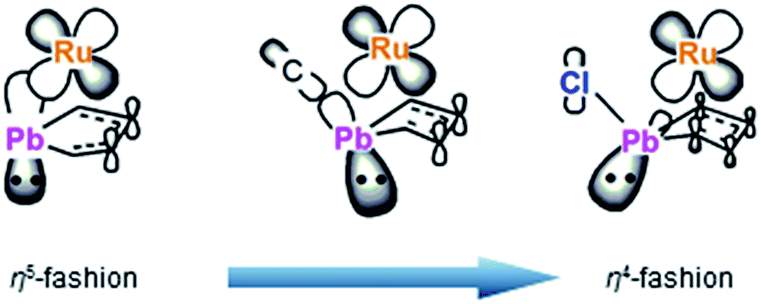
To understand the electronic structures of the plumbole–ruthenium complexes 5–8, we performed geometric optimization of complexes 6 and 7 as the representatives, to reproduce the X-ray characterized structures. We found differences in the electronic characteristics between alkyl-substituted 6 and halogen-substituted 7 (Fig. 5). The HOMO of 6 is mainly composed of the interaction between the Pb–C(iPr) σ-bond, the ruthenium and carbon atoms of the plumbole ring. However, as the contribution of the carbon atoms of the plumbole ring to the electronic delocalization is smaller than that found in the HOMO of 3, the plumbole ring of 6 deviates from planarity. On the other hand, the HOMO of 7 is mainly constituted by the Pb–C(plumbole) σ-bonds and the ruthenium moiety, which corresponds to the HOMO−1 of 6. The HOMO−1 of 7 is mainly composed of a σ-type lone pair localized on the lead atom and a ruthenium moiety. As a result, the bent angle of the plumbole ring in 7 is larger than that of 6. It is therefore concluded that the chloro-substituted Ru complex 7 can be regarded as a complex composed of a Cp*Ru cation and a plumbyl anion moiety, where the ruthenium is coordinated by a neutral butadiene moiety in an η4-fashion, while complex 6 has a coordination mode somewhere between the η4- and η5-fashion (Fig. 6). Such electronic states are consistent with those determined from the NMR analysis.
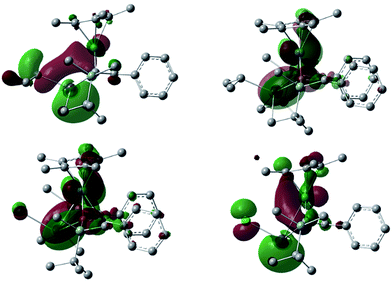 |
| | Fig. 5 The HOMO and HOMO−1 of the iPr-substituted complex 6 (above, left and right, respectively) and the HOMO and HOMO−1 of the chloro-substituted complex 7 (below, left and right, respectively) (isovalue = 0.03). All hydrogen atoms were omitted for clarity. | |
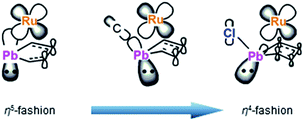 |
| | Fig. 6 Comparison of coordination modes in compounds 3, 6 and 7. | |
3. Physical properties of the plumbole ruthenium complexes
We next conducted cyclic voltammetry measurements for compounds 6 and 7 to investigate the electronic structures experimentally. An irreversible oxidation wave was observed (EOX = −0.01 V for 6, and EOX = 0.23 V for 7vs. Fc/Fc+), while decamethylruthenocene {(η5-Cp*)2Ru} exhibits an oxidation wave at 0.33 V (vs. Fc/Fc+) under similar conditions. It is therefore concluded that both the iPr-substituted complex 6 and chloro-substituted complex 7 are oxidized more easily than decamethylruthenocene. The Et-substituted stannole complex 9 is oxidized more easily than decamethylruthenocene, whereas the HOMO level of the chloro-substituted stannole 10 is lower than that of decamethylruthenocene (Fig. 7 and Table 3). Importantly, each of the plumbole complexes is oxidized more easily than the corresponding stannole complex, indicating that the introduction of a heavier atom increases the HOMO level of the resulting complex.15
 |
| | Fig. 7 Stannole complexes 9 and 10. | |
Table 3 Oxidative potentials of the plumbole and stannole complexes
|
|
6
|
7
|
9
|
10
|
| R |
iPr |
Cl |
Et |
Cl |
|
E
OX (E/V vs. Fc/Fc+) |
−0.01 |
0.23 |
0.04 |
0.48 |
Reflecting the differences in the electronic structures of 6 and 7, the color of the complexes 6 and 7 is different: complex 6 is red, while complex 7 is green. To investigate the origin of the color, we recorded the UV-Vis spectra of 6 and 7 (Fig. 8). The spectrum for 6 reveals the absorption maxima at 472 and 568 nm (ε 4700 and 650 mol cm L−1, respectively), while the absorption maxima of 7 are observed at 413 and 699 nm (ε 1600 and 300 mol cm L−1, respectively). Based on the TD-DFT calculations, the longest absorption maximum of 6 is assigned to the transition from the HOMO−1 to LUMO, while that of 7 is assignable to the transition from the HOMO to LUMO (see the ESI†). Both of the LUMOs are mainly composed of a ruthenium moiety, a σ* orbital of lead and the α-carbon atoms of the plumbole ring. As judged from the TD-DFT calculations of complexes 6 and 7, the weak, broad and longest absorptions mainly arise from d–d transitions.
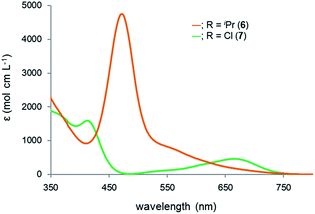 |
| | Fig. 8 UV-Vis absorption spectra of complexes 6 and 7 in hexane. | |
Conclusion
We succeeded in the synthesis of an anionic ruthenocene, where a plumbole ligand is coordinated in an η5-fashion, indicating that even a ligand containing a lead atom can reveal a coordination mode similar to that of the cyclopentadienyl ligand. The reactions of the anionic ruthenocene with electrophiles provided the corresponding ruthenium complexes that have bent plumbole moieties. The bent angles are dependent on the substituents on the lead atoms. The chloro-substituted complex 7 is regarded as a plumbyl anion-Cp*Ru+ complex, where a butadiene moiety coordinates the ruthenium, while the coordination mode of the plumbole ligand in the iPr-substituted complex 6 is considered as being somewhere between an η5- and η4-fashion. The plumbole complexes are oxidized more easily than the corresponding stannole complexes, indicating that the introduction of a heavier atom can increase the HOMO levels to tune the electronic properties of the complexes. The present work spotlights the importance of the introduction of heavy atoms to obtain desirable properties of materials and catalysts. The heavy ruthenocenes presented here would show unique catalytic activity derived from the strong electron-donating character of the plumbole ligands.
Acknowledgements
This paper is dedicated to Professor Shigeru Nagase on the occasion of his 70th birthday. This work was partially supported by the Grants-in-Aid for Scientific Research on Innovative Area “π-System Figuration, Control of Electron and Structural Dynamism for Innovative Functions” and Scientific Research (B) (No. 26102006 and 15H03774, respectively, for M.S.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. M.S. acknowledges a research grant from the Mitsubishi Foundation.
Notes and references
-
(a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS;
(b) G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef CAS.
-
(a) D. Schaarschmidt and H. Lang, Organometallics, 2013, 32, 5668–5704 CrossRef CAS;
(b) R. A. Musgrave, A. D. Russell and I. Manners, Organometallics, 2013, 32, 5654–5667 CrossRef CAS.
-
(a) Š. Toma and R. Šebesta, Synthesis, 2015, 47, 1683–1695 CrossRef;
(b) D.-Y. Zhu, P. Chen and J.-B. Xia, ChemCatChem, 2016, 8, 68–73 CrossRef CAS.
-
(a) I. Manners, Science, 2001, 294, 1664–1666 CrossRef CAS PubMed;
(b) R. Sun, L. Wang, H. Yu, Z.-u. Abdin, Y. Chen, J. Huang and R. Tong, Organometallics, 2014, 33, 4560–4573 CrossRef CAS;
(c) M. Saleem, H. Yu, L. Wang, A. Zain ul, H. Khalid, M. Akram, N. M. Abbasi and J. Huang, Anal. Chim. Acta, 2015, 876, 9–25 CrossRef CAS PubMed;
(d) S. O. Scottwell and J. D. Crowley, Chem. Commun., 2016, 52, 2451–2464 RSC.
-
(a) S. Deng, C. Schwarzmaier, M. Zabel, J. F. Nixon, M. Bodensteiner, E. V. Peresypkina, G. Balázs and M. Scheer, Eur. J. Inorg. Chem., 2011, 2991–3001 CrossRef CAS;
(b) L. Peng, A. Feng, M. Huo and J. Yuan, Chem. Commun., 2014, 50, 13005–13014 RSC;
(c) T. Fukino, H. Joo, Y. Hisada, M. Obana, H. Yamagishi, T. Hikima, M. Takata, N. Fujita and T. Aida, Science, 2014, 344, 499–504 CrossRef CAS PubMed;
(d) A. E. Kaifer, Acc. Chem. Res., 2014, 47, 2160–2167 CrossRef CAS PubMed;
(e) C. Heindl, E. V. Peresypkina, A. V. Virovets, V. Y. Komarov and M. Scheer, Dalton Trans., 2015, 44, 10245–10252 RSC.
-
(a) Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2011, 50, 10917–10921 CrossRef CAS PubMed;
(b) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846–11848 RSC;
(c) N. Umeda, K. Hirano, T. Satoh, N. Shibata, H. Sato and M. Miura, J. Org. Chem., 2011, 76, 13–24 CrossRef CAS PubMed;
(d) Y. Yamamoto, S. Mori and M. Shibuya, Chem.–Eur. J., 2013, 19, 12034–12041 CrossRef CAS PubMed;
(e) T. K. Hyster, D. M. Dalton and T. Rovis, Chem. Sci., 2015, 6, 254–258 RSC.
-
(a)
J. Dubac, C. Guérin and P. Meunier, in The Chemistry of Organic Silicon Compounds, John Wiley & Sons, Ltd, 2003, pp. 1961–2036 Search PubMed;
(b) M. Saito and M. Yoshioka, Coord. Chem. Rev., 2005, 249, 765–780 CrossRef CAS;
(c)
V. Y. Lee and A. Sekiguchi, in Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds, John Wiley and Sons, Chichester, 2010, pp. 335–414 Search PubMed;
(d) M. Saito, Coord. Chem. Rev., 2012, 256, 627–636 CrossRef CAS.
- W. P. Freeman, T. D. Tilley and A. L. Rheingold, J. Am. Chem. Soc., 1994, 116, 8428–8429 CrossRef CAS.
- H. Yasuda, V. Y. Lee and A. Sekiguchi, J. Am. Chem. Soc., 2009, 131, 9902–9903 CrossRef CAS PubMed.
-
(a) W. P. Freeman, T. D. Tilley, A. L. Rheingold and R. L. Ostrander, Angew. Chem., Int. Ed. Engl., 1993, 32, 1744–1745 CrossRef;
(b) W. P. Freeman, J. M. Dysard, T. D. Tilley and A. L. Rheingold, Organometallics, 2002, 21, 1734–1738 CrossRef CAS.
-
(a) V. Y. Lee, R. Kato, A. Sekiguchi, A. Krapp and G. Frenking, J. Am. Chem. Soc., 2007, 129, 10340–10341 CrossRef CAS PubMed;
(b) V. Y. Lee, R. Kato and A. Sekiguchi, Bull. Chem. Soc. Jpn., 2013, 86, 1466–1471 CrossRef CAS.
-
(a) M. Saito, R. Haga and M. Yoshioka, Chem. Commun., 2002, 1002–1003 RSC;
(b) M. Saito, R. Haga, M. Yoshioka, K. Ishimura and S. Nagase, Angew. Chem., Int. Ed., 2005, 44, 6553–6556 CrossRef CAS PubMed;
(c) R. Haga, M. Saito and M. Yoshioka, J. Am. Chem. Soc., 2006, 128, 4934–4935 CrossRef CAS PubMed;
(d) M. Saito, M. Shimosawa, M. Yoshioka, K. Ishimura and S. Nagase, Organometallics, 2006, 25, 2967–2971 CrossRef CAS;
(e) M. Saito, M. Shimosawa, M. Yoshioka, K. Ishimura and S. Nagase, Chem. Lett., 2006, 35, 940–941 CrossRef CAS;
(f) M. Saito, T. Kuwabara, C. Kambayashi, M. Yoshioka, K. Ishimura and S. Nagase, Chem. Lett., 2010, 39, 700–701 CrossRef CAS;
(g) T. Kuwabara, J.-D. Guo, S. Nagase, M. Minoura, R. H. Herber and M. Saito, Organometallics, 2014, 33, 2910–2913 CrossRef CAS.
-
(a) T. Kuwabara, M. Saito, J.-D. Guo and S. Nagase, Inorg. Chem., 2013, 52, 3585–3587 CrossRef CAS PubMed;
(b) T. Kuwabara, J. D. Guo, S. Nagase and M. Saito, Angew. Chem., Int. Ed., 2014, 53, 434–438 CrossRef CAS PubMed;
(c) T. Kuwabara and M. Saito, Organometallics, 2015, 34, 4202–4204 CrossRef CAS;
(d) T. Kuwabara, M. Nakada, J. Hamada, J. D. Guo, S. Nagase and M. Saito, J. Am. Chem. Soc., 2016, 138, 11378–11382 CrossRef CAS PubMed.
- T. Kuwabara, J.-D. Guo, S. Nagase, T. Sasamori, N. Tokitoh and M. Saito, J. Am. Chem. Soc., 2014, 136, 13059–13064 CrossRef CAS PubMed.
- T. Kuwabara, M. Nakada, J. D. Guo, S. Nagase and M. Saito, Dalton Trans., 2015, 44, 16266–16271 RSC.
-
(a) M. Saito, M. Sakaguchi, T. Tajima, K. Ishimura, S. Nagase and M. Hada, Science, 2010, 328, 339–342 CrossRef CAS PubMed;
(b) M. Saito, M. Nakada, T. Kuwabara and M. Minoura, Chem. Commun., 2015, 51, 4674–4676 RSC.
-
(a) A. J. Ashe, J. W. Kampf and S. M. Al-Taweel, J. Am. Chem. Soc., 1992, 114, 372–374 CrossRef CAS;
(b) A. J. Ashe, J. W. Kampf and S. M. Al-Taweel, Organometallics, 1992, 11, 1491–1496 CrossRef CAS;
(c) L. L. Lohr and A. J. Ashe, Organometallics, 1993, 12, 343–346 CrossRef CAS.
- P. J. Fagan, M. D. Ward and J. C. Calabrese, J. Am. Chem. Soc., 1989, 111, 1698–1719 CrossRef CAS.
- H. Nakatsuji, K. Kand, K. Endo and T. Yonezawa, J. Am. Chem. Soc., 1984, 106, 4653–4660 CrossRef CAS.
-
(a) N. C. Burton, C. J. Cardin, D. J. Cardin, B. Twamley and Y. Zubavichus, Organometallics, 1995, 14, 5708–5710 CrossRef CAS;
(b) M. P. Aarnts, D. J. Stufkens, A. Oskam, J. Fraanje and G. Kees, Inorg. Chim. Acta, 1997, 256, 93–105 CrossRef CAS;
(c) R. D. Adams, B. Captain and W. Fu, J. Organomet. Chem., 2003, 671, 158–165 CrossRef CAS;
(d) M. Shieh, Y.-Y. Chu, M.-H. Hsu, W.-M. Ke and C.-N. Lin, Inorg. Chem., 2011, 50, 565–575 CrossRef CAS PubMed.
- I. E. Zanin and M. Y. Antipin, Cryst. Rep., 2003, 48, 249–258 CrossRef CAS.
- H. Sato, N. Isono, I. Miyoshi and M. Mori, Tetrahedron, 1996, 52, 8143–8158 CrossRef CAS.
-
(a) C. Boga, E. Del Vecchio, L. Forlani and P. E. Todesco, J. Organomet. Chem., 2000, 601, 233–236 CrossRef CAS;
(b) M. A. P. Martins, D. J. Emmerich, C. M. P. Pereira, W. Cunico, M. Rossato, N. Zanatta and H. G. Bonacorso, Tetrahedron Lett., 2004, 45, 4935–4938 CrossRef CAS;
(c) K. Nakata and I. Shiina, Org. Biomol. Chem., 2011, 9, 7092–7096 RSC.
- M. Wakasa and T. Kugita, Organometallics, 1998, 17, 1913–1915 CrossRef CAS.
- Compound 6 was obtained as co-crystals with 7 with the ratio of 6 to 7 of 85/15. The origin of the chlorine may be LiCl that was contained in compound 3.
-
(a) R. Haga, M. Saito and M. Yoshioka, Eur. J. Inorg. Chem., 2007, 2007, 1297–1306 CrossRef;
(b) M. Saito, T. Kuwabara, K. Ishimura and S. Nagase, Bull. Chem. Soc. Jpn., 2010, 83, 825–827 CrossRef CAS.
- M. Saito, M. Sakaguchi, T. Tajima, K. Ishimura and S. Nagase, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 1068–1076 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, data for X-ray diffraction analysis of compounds 3, 6, 7 and 8, and details for theoretical calculations. CCDC 1507473–1507475, 1511941. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04843a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a