
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic activation of isocyanides: N-heterocyclic carbene-catalyzed enaminone synthesis from ketones†
Jungwon
Kim
 and
Soon Hyeok
Hong
*
and
Soon Hyeok
Hong
*
Department of Chemistry, College of Natural Science, Seoul National University, 1 Gwanak-ro, Seoul 08826, South Korea. E-mail: soonhong@snu.ac.kr
First published on 20th December 2016
Abstract
The first example of the use of an N-heterocyclic carbene (NHC) as an organocatalyst for the activation of isocyanides was demonstrated. On the basis of previous reports on the interaction between NHCs and isocyanides, we developed a catalytic cycle involving transient imidoyl intermediate. The reaction of ketones with isocyanides produced the corresponding enaminones with high efficiency. Control experiments suggested a novel role for the carbene in the activation of isocyanides, and a proton transfer process was found to be crucial for the generation of two activated species in the catalytic cycle. Various enaminones, some of which are not easily accessible by other methods, were synthesized in excellent yields. This study clearly demonstrates the potential of the nucleophilic activation of isocyanides in the expansion of their reactivity scope.
Introduction
Isocyanides are useful building blocks for the synthesis of various acyclic and cyclic compounds.1 Because the terminal carbon of isocyanide can act as both a nucleophile and the carbene center, control of its reactivity is key to developing novel transformations. In many cases, the isocyanide group can act as a mild nucleophile, meaning that it undergoes electrophilic activation in the presence of an electrophilic compound. In addition, various metal-based catalysts allow further transformations after the incorporation of the isocyanide motif into the starting substrate. Pd-catalyzed insertion reactions,2 Cu(Ag)-mediated electrophilic activation,3 and Cu-alkoxide systems4 for the generation of formimidate intermediates are well developed isocyanide activation strategies (Scheme 1A). Because of the mild nucleophilicity of the carbon atom of the isocyanide group, its utilization as a nucleophile has been limited to the use of highly electrophilic reaction partners, such as carbonyl compounds and metal catalysts. This has been a critical hurdle for achieving metal-free, non-Ugi-type transformations. In order to overcome this shortcoming, we envisioned a new approach where the isocyanide group is activated by a ‘nucleophilic moiety’ to transform the isocyanide into a highly basic intermediate. The reactions of isocyanides with external nucleophiles are relatively rare and most of them require highly reactive organometallic nucleophiles (Scheme 1B).5 The incorporation of milder nucleophiles into the isocyanide is possible only in the presence of an electrophilic group near the reactive center to trigger intramolecular cyclization reactions (Scheme 1B).6 To the best of our knowledge, the catalytic nucleophilic activation of isocyanides has not yet been reported.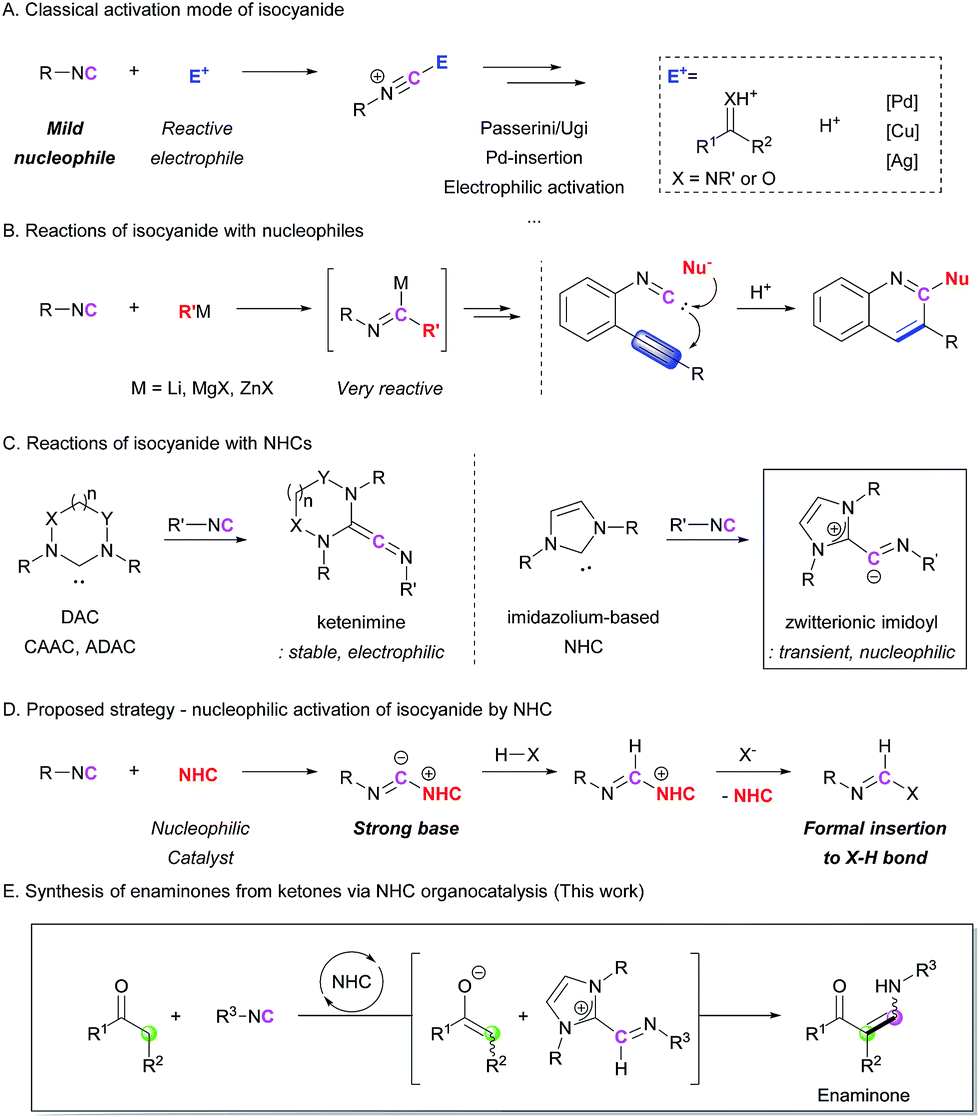 | ||
| Scheme 1 Strategies for isocyanide activation. | ||
We chose N-heterocyclic carbenes (NHCs) as potential nucleophilic activators.7 There have been several reports on the formation of ketenimines from the reaction between carbenes and isocyanides,8 such as diamidocarbene (DAC),8c,8d cyclic (alkyl)(amino)carbene (CAAC),8e and acyclic di(amino)carbene (ADAC)8e (Scheme 1C). Among them, a computational study on the formation of an imidoyl intermediate from the reaction between an imidazole-based NHC and an isocyanide, which proposed a zwitterionic imidoyl adduct as a favorable structure, attracted our attention (Scheme 1C).8c,8e Although this intermediate was not stable enough to be isolated, the transient zwitterionic imidoyl intermediate containing a localized anion charge on the terminal carbon of the isocyanide structure could be highly reactive.8e On the basis of this report, we envisioned the possibility to enhance the nucleophilicity (basicity) of the terminal carbon atom of the isocyanide group by conversion into an imidoyl intermediate (Scheme 1D). This intermediate would abstract a proton from an unactivated substrate, generating activated nucleophilic and electrophilic species. Thus, formal X–H insertion without the aid of a metal catalyst could be achieved. To realize this strategy, we first attempted the reaction between isocyanide and ketone derivatives, which can form an enaminone as the final product (Scheme 1E). The proposed methodology provides an unprecedented synthetic route to enaminones via C–C bond formation. Although various synthetic strategies have been developed for the preparation of enaminones,9 the limited scope and relatively low availability of reagents are significant drawbacks to these procedures. Herein, we report a novel organocatalytic enaminone synthesis from isocyanides and ketones with excellent functional group tolerance. This methodology is applicable to the preparation of a wide range of enaminones via the first catalytic activation of isocyanides by an NHC.
Results and discussion
At the outset, the reaction between acetophenone (1a) and 4-methylphenyl isocyanide (2a) was conducted, and in our first attempt the desired (Z)-enaminone (3a) was isolated in 19% yield (Scheme S1†). The exclusive (Z)-selectivity of 3a can be explained by intramolecular hydrogen bonding, which was confirmed by 1H NMR spectroscopy.10 To further increase the reactivity, several reaction parameters were varied (Tables 1 and S1–S4†). Among the several solvents examined, DMA afforded the highest yields of 3aa (entries 1–4). Next, various bases were evaluated, showing that replacing a strong base (NaOtBu) with a relatively weak one (K2CO3) did not significantly affect the reactivity (entry 5 and Table S1†). Interestingly, the use of IMes as an organocatalyst was critical to obtain the desired product, and the reaction did not proceed at all in the presence of other related imidazolium salts (entries 6 and 7, Table S3†). Screening of the amount of NHC salt, base, and solvent and increasing the reaction temperature resulted in further enhancement of the reaction efficiency (entries 8–10). The reaction time could be reduced to 6 h without significant decrease in reactivity (entry 11). The reaction without NHC salt did not generate the desired product (entry 12) or showed lower reactivity (entry 13), proving that this transformation cannot be catalyzed only by a base.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | NHC salt (mol%) | Base (mol%) | Solvent (M) | T | X | Yield (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), NHC salt, base, and solvent for 24 h, including the 5 min induction period. Yields determined by GC using mesitylene as an internal standard. b 6 h, without induction time. c Isolated yields. IMesHCl = 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride. IPrHCl = 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride. IAdHBF4 = 1,3-bis(1-adamantyl)imidazolium tetrafluoroborate. DMA = N,N-dimethylacetamide. N. R. = no reaction. | ||||||
| 1 | IMesHCl (20) | NaOtBu (30) | 1,4-Dioxane (0.25) | 50 | 1.0 | 30 |
| 2 | IMesHCl (20) | NaOtBu (30) | MeCN (0.25) | 50 | 1.0 | 19 |
| 3 | IMesHCl (20) | NaOtBu (30) | Toluene (0.25) | 50 | 1.0 | 36 |
| 4 | IMesHCl (20) | NaOtBu (30) | DMA (0.25) | 50 | 1.0 | 43 |
| 5 | IMesHCl (20) | K2CO3 (30) | DMA (0.25) | 50 | 1.0 | 45 |
| 6 | IPrHCl (20) | K2CO3 (30) | DMA (0.25) | 50 | 1.0 | N. R. |
| 7 | IAdHBF4 (20) | K2CO3 (30) | DMA (0.25) | 50 | 1.0 | N. R. |
| 8 | IMesHCl (15) | K2CO3 (20) | DMA (0.25) | 50 | 1.0 | 50 |
| 9 | IMesHCl (15) | K2CO3 (20) | DMA (0.125) | 80 | 1.0 | 73 |
| 10 | IMesHCl (15) | K2CO3 (20) | DMA (0.125) | 80 | 1.5 | 92 |
| 11b | IMesHCl (15) | K2CO3 (20) | DMA (0.083) | 80 | 1.5 | 93 (86)c |
| 12 | None | K2CO3 (20) | DMA (0.125) | 80 | 1.0 | N. R. |
| 13b | None | NaOtBu (20) | DMA (0.083) | 80 | 1.5 | 13c |
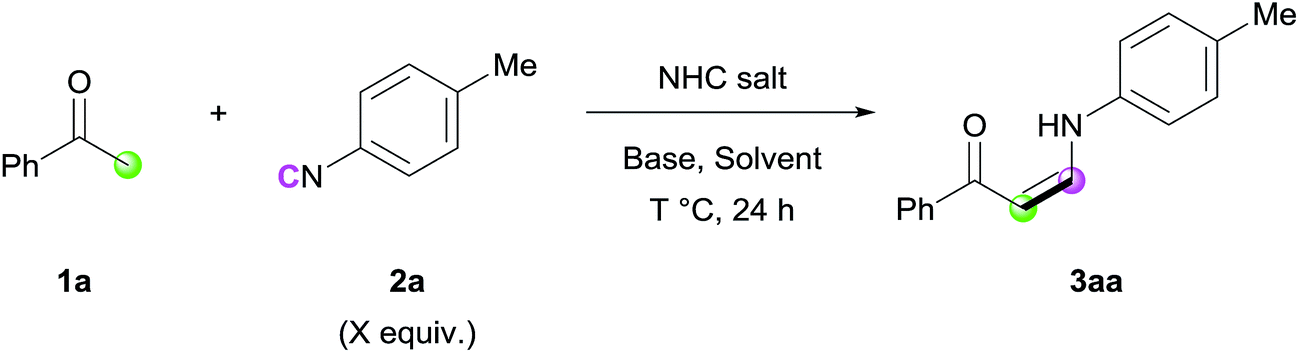
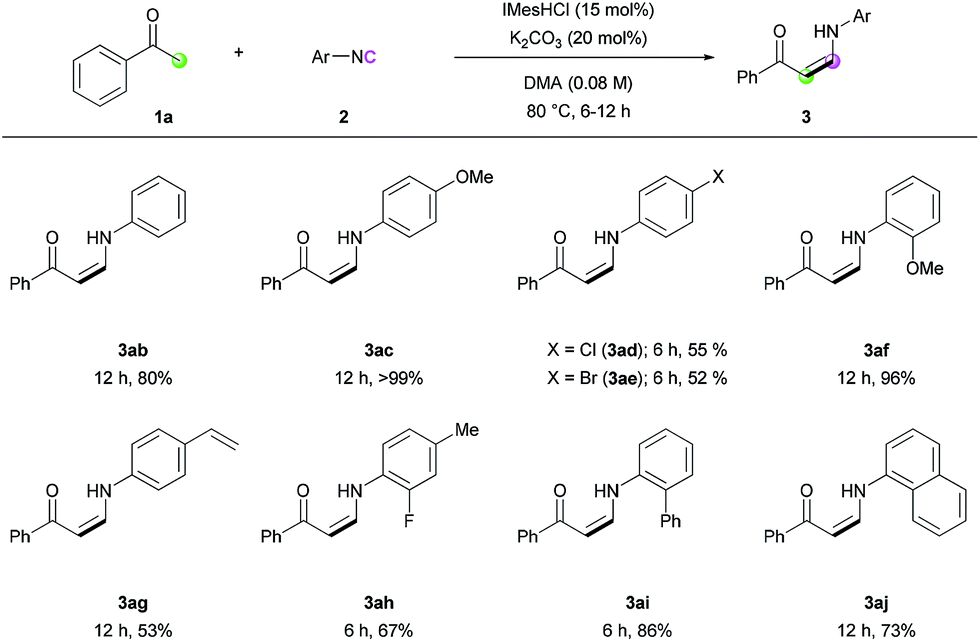
With the optimized conditions in hand, the scope of the reaction was investigated. Firstly, the reactions of various methyl ketones were performed (Table 2). para-Substituted acetophenones produced the corresponding enaminones in good to excellent yields (3aa–3ea). In particular, halide-containing substrates were tolerated under the reaction conditions demonstrating the advantage of this organocatalytic approach (3ca and 3da). ortho-Substituted acetophenones also gave excellent yields within 6 h (3fa and 3ga), and bis-substituted acetophenone 1h produced the corresponding (Z)-enaminone 3ha in 75% yield. In addition, heteroaromatic methyl ketones exhibited excellent reactivity (3ia–3ma). Non-aromatic methyl ketones could also be used in the reaction, albeit lower yields were obtained (3na and 3oa).
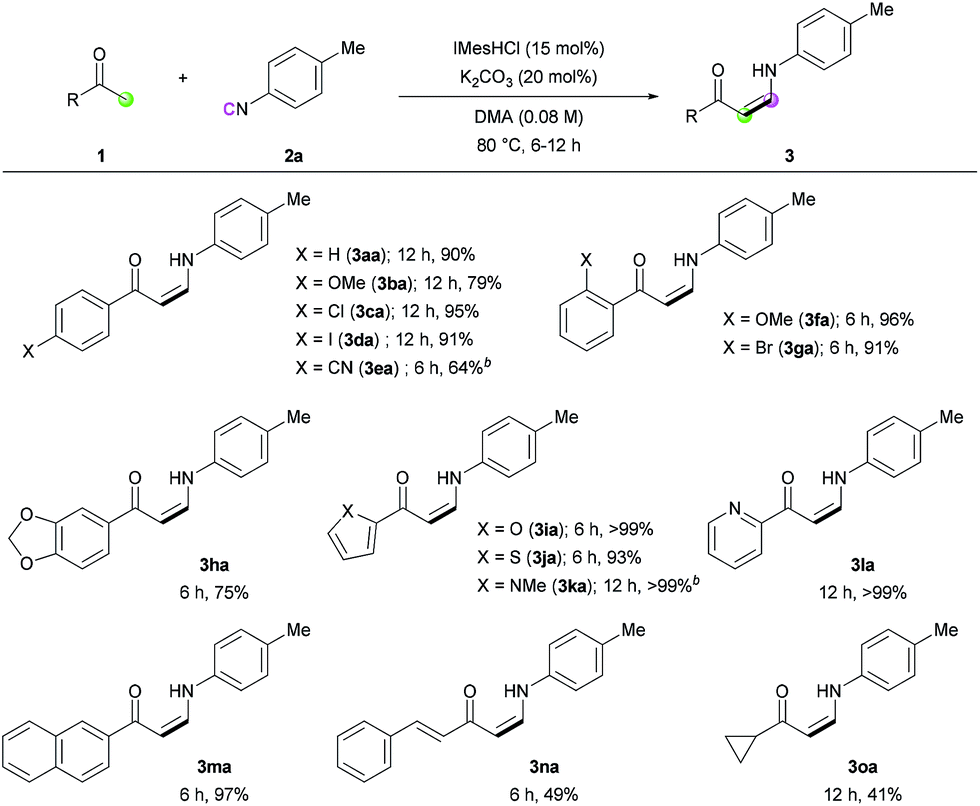
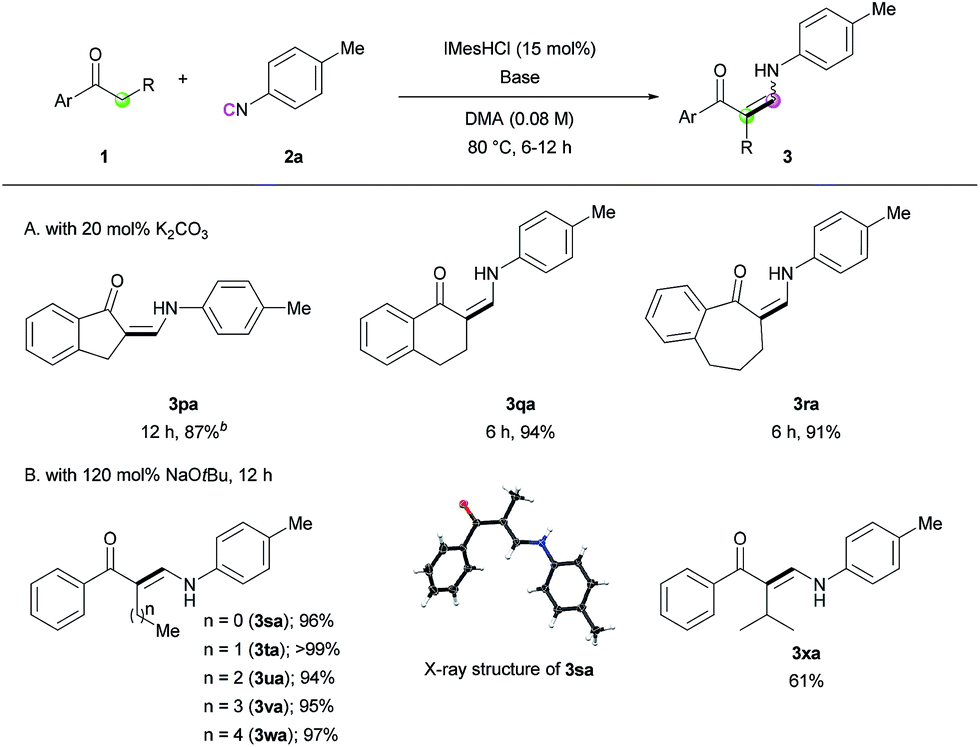
Next, we turned our attention toward the synthesis of trisubstituted enaminones. The synthesis of α-alkyl enaminones has been sparsely reported using specifically prepared starting materials,11 such as triazolyl alkanols,11a,11b β-dicarbonyl compounds,11c,11d and α,β-unsaturated carbonyl compounds.11e To our delight, our strategy could provide an alternative approach starting from readily available ketones (Table 3). Ring-fused acetophenone derivatives efficiently produced the corresponding trisubstituted (Z)-enaminones under the optimized conditions (3pa–3ra). In the case of propiophenone and other α-alkyl-substituted acetophenones, stoichiometric amounts of NaOtBu were used to facilitate enolate formation and protonation of the imidoyl intermediate by in situ generated tBuOH. Under these modified reaction conditions, α-substituted ketones with a linear alkyl chain or a branched alkyl group were all reactive, affording α-alkyl (E)-enaminones in good to excellent yields (3sa–3xa). The exclusive (E)-selectivity can be rationalized by the steric repulsion between the phenyl group and the α-alkyl substituent, as previously reported,12 and the (E)-configuration of 3sa was further determined by X-ray crystallography (see ESI†).
The reaction of various isocyanides was evaluated (Table 4). Phenyl isocyanide (2b) and p-methoxyphenyl isocyanide (2c) exhibited good to excellent reactivity. However, halogen-substituted isocyanides gave only moderate yields under several reaction conditions (3ad and 3ae). ortho-Substituted isocyanides (3af and 3ai), 4-styryl isocyanide (3ag), and disubstituted isocyanide (3ah) were all tolerated under the reaction conditions, with moderate to good reactivity. Biphenyl isocyanide (2i) and naphthyl isocyanide (2j) afforded the corresponding (Z)-enaminones in good yields (3ai and 3aj). Unfortunately, alkyl isocyanides were not reactive, presumably because the relatively low electrophilicity of the terminal carbon atom of the alkyl isocyanides prevents the nucleophilic attack of the NHC catalyst.
| a Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), IMesHCl (0.03 mmol), K2CO3 (0.04 mmol), and DMA (2.4 mL) at 80 °C for the indicated time. Isolated yields. |
|---|
|
Encouraged by the high efficiency of the developed reaction, we further evaluated the scope of the carbon nucleophiles (Scheme 2). When bis-acetyl compounds (1y and 1z) were treated with excess amounts of 2a, formation of bis-(Z)-enaminones was observed (3ya and 3za). It is noteworthy that such bis-enaminones have been synthesized using highly activated formamide acetal under harsh conditions, limiting the applicability.13 These examples clearly demonstrate the utility of our methodology for the synthesis of various complex enaminones. In addition, diethyl malonate (1A) and benzyl cyanide (1B) efficiently participated in the reaction (3Aa and 3Ba). To check the utility of the reaction further, a gram-scale synthesis was conducted with ketone 1f, and enaminone 3fa was produced in 75% yield (Scheme 2).
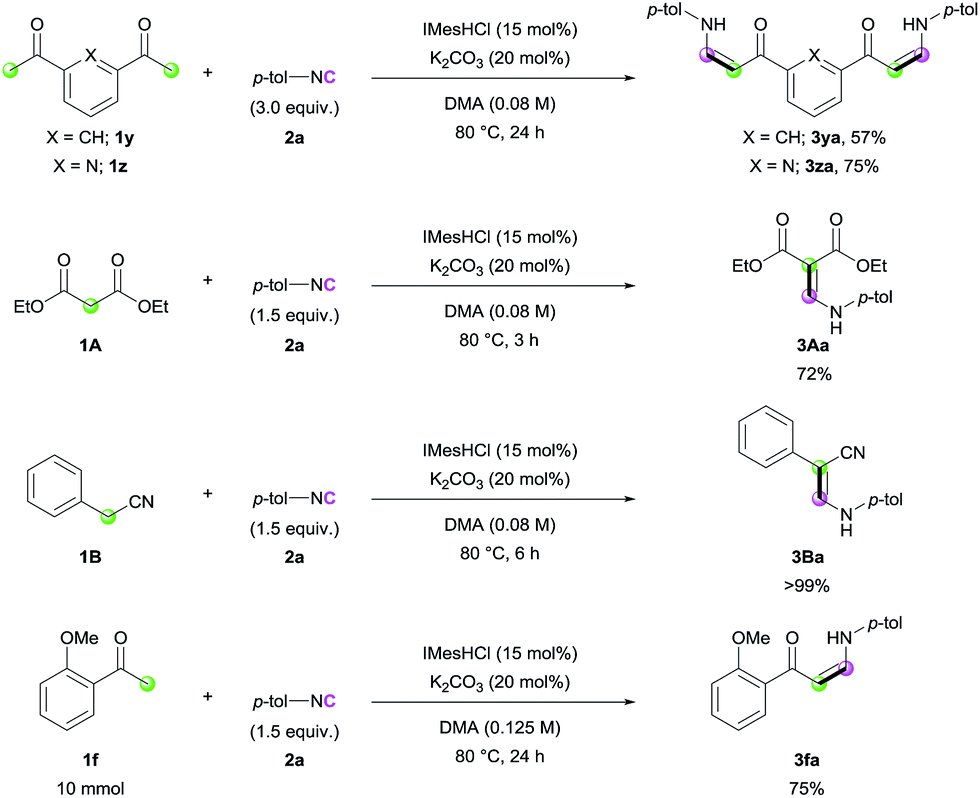 | ||
| Scheme 2 Expansion of the scope of carbon nucleophiles and gram-scale synthesis. | ||
In order to gain mechanistic insight into this transformation, control experiments were conducted (Scheme 3). When the standard reaction was performed with free carbene (IMes), a comparable yield of the desired product was obtained, suggesting that free carbene is involved in the catalytic cycle (Scheme 3A). To further reveal the role of the NHC catalyst, ethyl N-phenylformimidate (2b′), which can be considered as an analogue of the imine–azolium intermediate, was treated with 1a (Scheme 3B). Even in the presence of stoichiometric amounts of IMes, the reaction yield was not improved, suggesting that the major role of NHC is not ketone deprotonation. In contrast, the use of stoichiometric amounts of a strong base led to full conversion of the ketone to enaminone. This was further supported by the direct alkylation reaction via a stepwise approach with acetophenone and benzyl bromide (Scheme 3C). When a stoichiometric amount of KOtBu was used in the deprotonation step, enolate 1a′ was observed and α-benzylated product 4 was produced, whereas the use of IMes as a base was not effective for the same transformation. Even at an elevated temperature of 60 °C in THF, we could not observe 1a′ with IMes. From these observations,14 together with the literature data for the pKa values of acetophenone and related NHC salt,15 we concluded that the major role of the NHC catalyst is to activate the terminal carbon atom of the isocyanide, generating a more basic imidoyl intermediate, which deprotonates the ketone. Besides, we performed a stoichiometric reaction between an independently generated enolate 1a′ and isocyanide 2a to check the possibility of the direct addition of 1a′ to isocyanide. 3aa was obtained in a decreased isolated yield than that of the original catalytic conditions (55% vs. 86%), suggesting that a minor pathway of direction addition of enolate to isocyanide could exist (Scheme 3D). Considering the use of a stoichiometric amount of enolate anions for the reaction under an excess amount of isocyanide as the sole coupling partner, such direct addition process between isocyanide and enolate would not be the major pathway in our reaction.
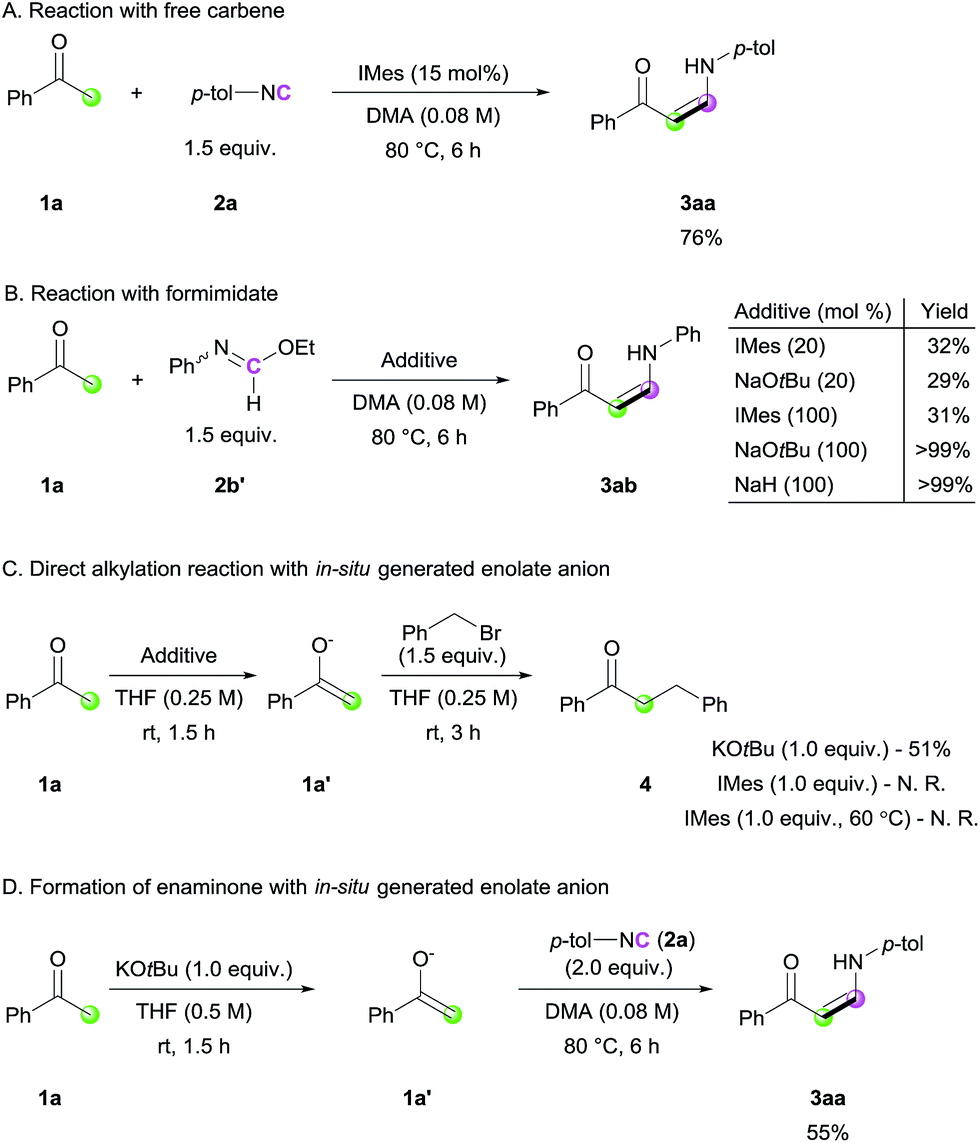 | ||
| Scheme 3 Control experiments. | ||
Based on the control experiments and earlier literature, a catalytic cycle is proposed in Scheme 4. The reaction of IMes with isocyanide generates an imidoyl intermediate (Int A), which can perform deprotonation to afford an imine–azolium intermediate (Int B). During this step, the proton source can be either the ketone, which directly produces the corresponding enolate (Path A), or unreacted imidazolium salt/protonated base (Path B). Trials for the observation of Int A or Int B through stepwise nucleophilic attack and protonation via1H NMR spectroscopy were conducted, but promising species were not detectable.16 We reasoned that those intermediates derived from IMes are transient species in catalytic conditions, which structures are relatively different from that of the stable ketenimine adduct. However, the instability of the intermediates would be a driving force to achieve overall turnover of the catalytic cycle in the reaction mixture. Deprotonation of the ketone by the regenerated base, i.e., Path B, is dominant when Path A is disfavored by steric repulsion interactions such as in the case of bulky ketones. These steps lead to the formation of activated nucleophiles (enolate) and electrophiles (Int B). Subsequent coupling and isomerization reactions produce the desired enaminone, with regeneration of IMes. Direct coupling between the generated enolate and isocyanide could be also possible, although it would be a minor pathway in the catalytic cycle.
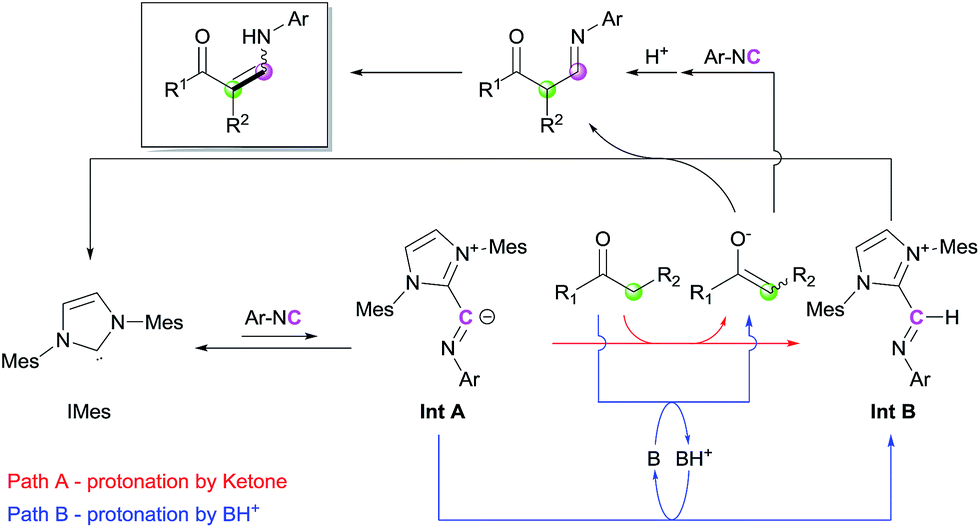 | ||
| Scheme 4 Proposed catalytic cycle. | ||
Conclusion
In conclusion, we demonstrated the first NHC-catalyzed C–C bond formation reaction between ketones and isocyanides. Using an imidazolium-based NHC catalyst, diverse enaminones were synthesized in excellent yields. We believe that the major role of the NHC in the catalytic cycle is to transform the isocyanide moiety into an imidoyl intermediate, followed by the formation of a highly electrophilic imine–azolium intermediate via a proton transfer process. The in situ generated enolate attacks the terminal carbon atom of the isocyanide to complete the catalytic cycle. In this strategy, isocyanides are used as new substrates for NHC organocatalysis. Detailed mechanistic studies and further applications of the developed strategy are in progress.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF-2014R1A2A1A11050028; NRF-2014R1A5A1011165, Center for New Directions in Organic Synthesis; NRF-2015M3D3A1A01065480) funded by the Korean Government. J. Kim was supported by the Global Ph.D Fellowship Program funded by the National Research Foundation of Korea (NRF-2014H1A2A1015749).Notes and references
-
(a) A. V. Lygin and A. de Meijere, Angew. Chem., Int. Ed., 2010, 49, 9094 CrossRef CAS PubMed
; (b) S. Sadjadi and M. M. Heravi, Tetrahedron, 2011, 67, 2707 CrossRef CAS
-
(a) T. Vlaar, E. Ruijter, B. U. Maes and R. V. Orru, Angew. Chem., Int. Ed., 2013, 52, 7084 CrossRef CAS PubMed
-
(a) Y. Ito, Y. Inubushi and T. Saegusa, Tetrahedron Lett., 1974, 15, 1283 CrossRef
-
(a) B. Pooi, J. Lee, K. Choi, H. Hirao and S. H. Hong, J. Org. Chem., 2014, 79, 9231 CrossRef CAS PubMed
-
(a) H. M. Walborsky and G. E. Niznik, J. Am. Chem. Soc., 1969, 91, 7778 CrossRef CAS
-
(a) K. Kobayashi, K. Yoneda, M. Mano, O. Morikawa and H. Konishi, Chem. Lett., 2003, 32, 76 CrossRef CAS
-
(a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed
-
(a) A. Halleux, Angew. Chem., Int. Ed. Engl., 1964, 3, 752 CrossRef
-
(a) A.-Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463 CrossRef CAS
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405 CrossRef CAS
-
(a) T. Miura, Y. Funakoshi, M. Morimoto, T. Biyajima and M. Murakami, J. Am. Chem. Soc., 2012, 134, 17440 CrossRef CAS PubMed
-
(a) J.-C. Zhuo and K. Schenk, Helv. Chim. Acta, 1997, 80, 2137 CrossRef CAS
-
(a) R. F. Abdulla and R. S. Brinkmeyer, Tetrahedron, 1979, 35, 1675 CrossRef CAS
- We tried to observe any interaction or reaction between IMes and 1a in C6D6via1H NMR at various temperatures, but no change was observed. Although deuterium incorporation on the α-position of 3-pentanone by IMes was reported in the previous GC-MS analysis, it was hard to observe the generation of significant amount of enolate anion directly from IMes and acetophenone in our experiments. See: E. M. Phillips, M. Riedrich and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 13179 CrossRef CAS PubMed
- The pKa value of acetophenone in DMSO is 24.7, whereas the calculated pKa value of 1,3-bis(2,6-dimethylphenyl)imidazolium
cation is ∼17. See:
(a) W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 7006 CrossRef CAS
- Reactions between stoichiometric amount of IMes and 2a were monitored by 1H NMR in benzene-d6 and THF-d8 respectively, but unidentificable broadened peaks were observed in both cases. We think that generated imidoyl intermediate could initiate oligomerization process, because fast conversion of 2a was observed. In the developed reaction, an appropriate pronucleophile, ketone in our reaction, could intercept reactive imidoyl intermediate and prevent the unwanted side reactions. For anion mediated oligomerization of isocyanide, see:
(a) C. Grundmann, Chem. Ber., 1958, 91, 1380 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full data for reaction conditions optimizations, detailed experimental procedures, and full characterization of substrates and products. Crystallographic data for compound 3sa. CCDC 1503347. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05266e |
| This journal is © The Royal Society of Chemistry 2017 |