
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective uni- and bidirectional homologation of diborylmethane†
Daniel J.
Blair
 ,
Damiano
Tanini
,
Joseph M.
Bateman
,
Helen K.
Scott
,
Eddie L.
Myers
and
Varinder K.
Aggarwal
*
,
Damiano
Tanini
,
Joseph M.
Bateman
,
Helen K.
Scott
,
Eddie L.
Myers
and
Varinder K.
Aggarwal
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: V.Aggarwal@bristol.ac.uk
First published on 9th February 2017
Abstract
Diborylmethane can be homologated uni- and bidirectionally by using enantiomerically pure lithium-stabilized carbenoids to give 1,2- and 1,3-bis(boronic esters), respectively, in good yield and with excellent levels of enantio- and diastereoselectivity. The high sensitivity of the transformation to steric hindrance enables the exclusive operation of either manifold, effected through the judicious choice of the type of carbenoid, which can be a sparteine-ligated or a diamine-free lithiated benzoate/carbamate. The scope of the 1,2-bis(boronic esters) so generated is complementary to that encompassed by the asymmetric diboration of alkenes, in that primary–secondary and primary–tertiary 1,2-bis(boronic esters) can be prepared with equally high levels of selectivity and that functional groups, such as terminal alkynes and alkenes, are tolerated. Methods for forming C2-symmetric and non-symmetrical anti and syn 1,3-bis(boronic esters) are also described and represent a powerful route towards 1,3-functionalized synthetic intermediates.
Introduction
Investigations into the utility of organic molecules containing multiple C-sp3 boryl units continue to flourish, enabled in part by recent developments in the asymmetric diboration of alkenes.1 It is now clear that polyboronic esters are powerful intermediates in organic synthesis owing to the abundance of functional groups in which the C–B bonds can be transformed.2 Many of these methods are stereospecific and regioselective transformation of polyboronic esters is feasible through the judicious manipulation of steric effects3 and proximal functional groups.4 Despite these advances, the types of polyboronic esters available for further functionalization remain limited. For example, although primary–secondary and a very limited number of secondary–secondary 1,2-bis(boronic) esters are readily available with high levels of enantiopurity, obtained through the platinum-5 or rhodium-mediated6 diboration of the corresponding alkenes or the hydroxyl-directed diboration7 of enantioenriched alkenyl alcohols (Fig. 1A), 1,2-bis(boronic) esters with higher levels of substitution are not.8 Furthermore, metal-catalyzed diboration reactions are not tolerant of several functional groups, for example alkynes, thus further limiting the diversity of polyboronic esters available. Herein, we present an alternative method for preparing 1,2-bis(boronic esters)—the one-carbon homologation of diborylmethane 1. Specifically, when treated with enantioenriched lithiated benzoates or carbamates, diborylmethane 1 undergoes stereospecific homologation to give primary–secondary and primary–tertiary 1,2-bis(boronic esters). Furthermore, we show that diborylmethane can be homologated in both directions in a one-pot process to give secondary–secondary 1,3-bis(boronic esters), a class of polyboronic esters with similar potential for application to their 1,2-related counterparts.9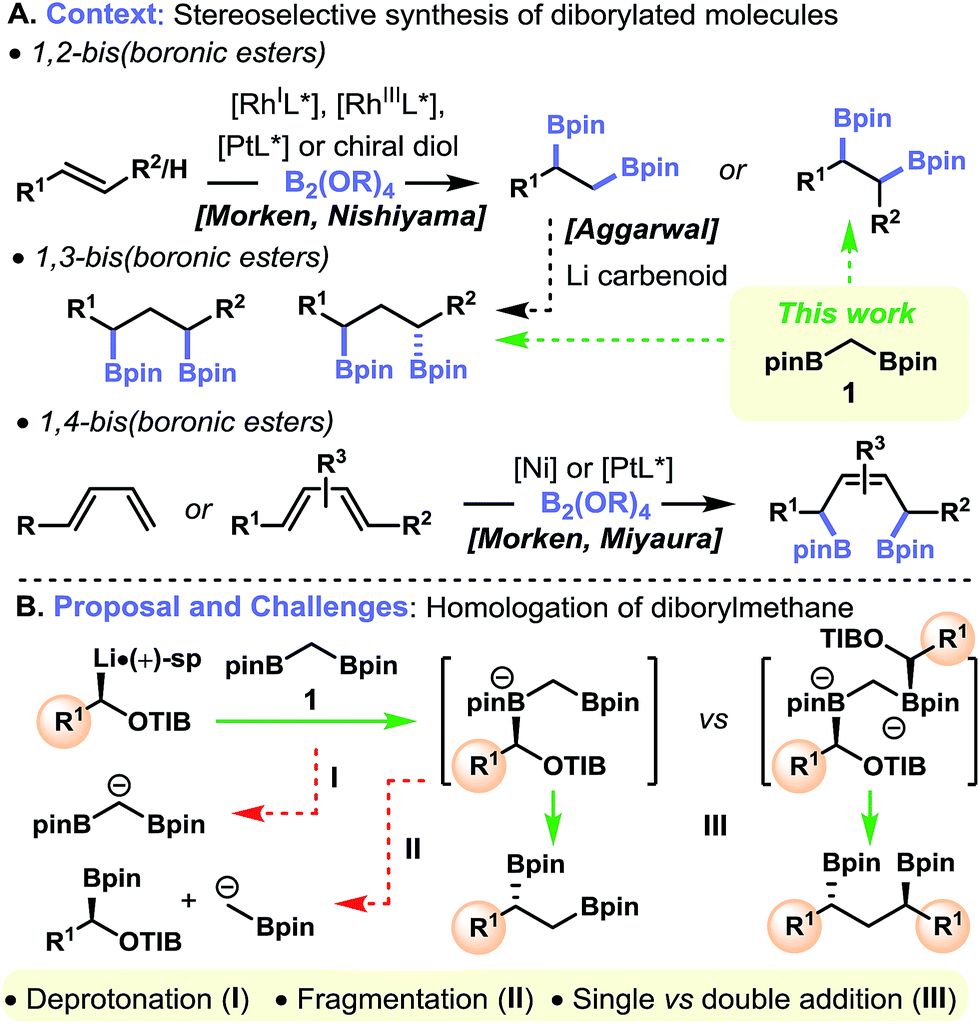 | ||
| Fig. 1 Context, proposal and challenges. | ||
We have recently developed efficient methodology for the homologation of boronic esters by using stereodefined lithiated carbamates10 and 2,4,6-triisopropylbenzoates11 (TIB esters). The transformation involves the stereoretentive complexation of the lithiated carbamate12/TIB ester13 with the boronic ester to form a boronate complex, which then undergoes a stereoinvertive 1,2-metallate rearrangement14 to generate a homologated boronic ester. Our attention was drawn to commercially available diborylmethane 1,15 which we envisioned serving as a valuable reagent for the elaboration of stereodefined carbenoids (Fig. 1B). Specifically, the complexation of one boryl moiety of diborylmethane 1 with a carbenoid followed by 1,2-metallate rearrangement would give a 1,2-bis(boronic ester). In a second manifold, the complexation of both boryl groups to the same or different carbenoids, thus forming a dianionic 1,1-bis(boronate), with subsequent rearrangement at both centers, would give a 1,3-bis(boronic ester), thus representing a three-component coupling with diborylmethane 1 acting as a linchpin. For these transformations to be successful, a number of key criteria would have to be met: (i) the acidic methylene group of diborylmethane 1 must not undergo deprotonation (I);16 (ii) fragmentation of the intermediate boronate complex to form stabilized α-boryl carbanions must not occur (II);15a (iii) single versus double homologation must be controlled (III) (Fig. 1B).
Results and discussion
(+)-Sparteine-ligated lithiated TIB ester 217 met all three of the criteria outlined above: by generating this carbenoid through the sparteine-mediated deprotonation of the corresponding benzoate and then treatment with diborylmethane 1, the single-homologation product 3 was isolated in high yield and with very high levels of enantiopurity (Fig. 2). Surprisingly, when using the corresponding diamine-free carbenoid (generated by tin–lithium exchange of the corresponding enantioenriched α-stannyl benzoate) the double-addition product 4 was obtained exclusively; none of the monoaddition product 3 was observed.18 In contrast, Shibata and co-workers have shown that the diamine-free Matteson-type carbenoid (LiCH2Cl), which was generated in situ, reacts with the more sterically hindered 1,1-diborylphenylethane to give the single-homologation product.19,20 Clearly the level of homologation of geminal diboryl compounds is very sensitive to the nature of the carbenoid employed.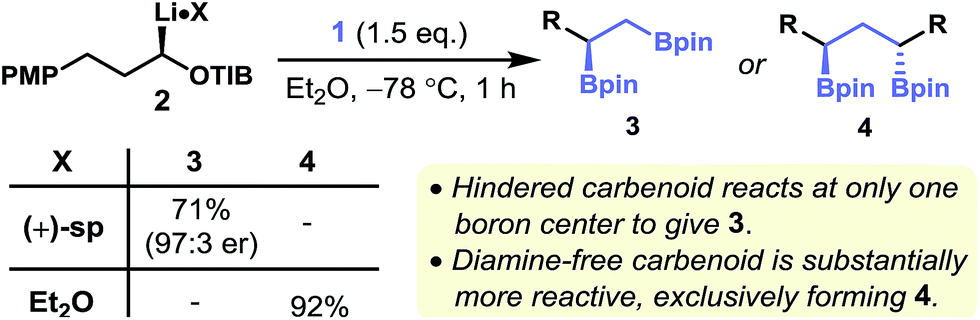 | ||
| Fig. 2 Key findings. | ||
Having established the optimal conditions for single and double homologation we set out to demonstrate the generality of our protocol for the synthesis of chiral non-racemic 1,2-bis(boronic esters). The homologation of diborylmethane 1 with lithiated primary benzoates containing a pyrrole-masked primary amine,21 a terminal olefin, and a steroidal group gave the corresponding 1,2-bis(boronic esters), 5–7, in high yield and enantiopurity (Fig. 3A). Additionally, the 1,2-bis(boronic ester) 5 was prepared on gram scale with comparable yields and selectivity. In contrast to metal-mediated diboration, the stereoselectivity of this homologative process appears to be insensitive to proximal stereogenic centers as epimeric 1,2-bis(boronic esters) 8 and 9 were prepared with similarly high levels of diastereoselectivity. The potential adverse effect of acidic functional groups on the carbenoid partner could be mitigated by their in situ protection as the corresponding conjugate bases by using excess s-BuLi. This protocol facilitated the diborylative transformation of a carbenoid bearing a terminal alkyne (to give 10). Again, this particular transformation is significant in that alkyne groups are incompatible with alkene diboration reactions.
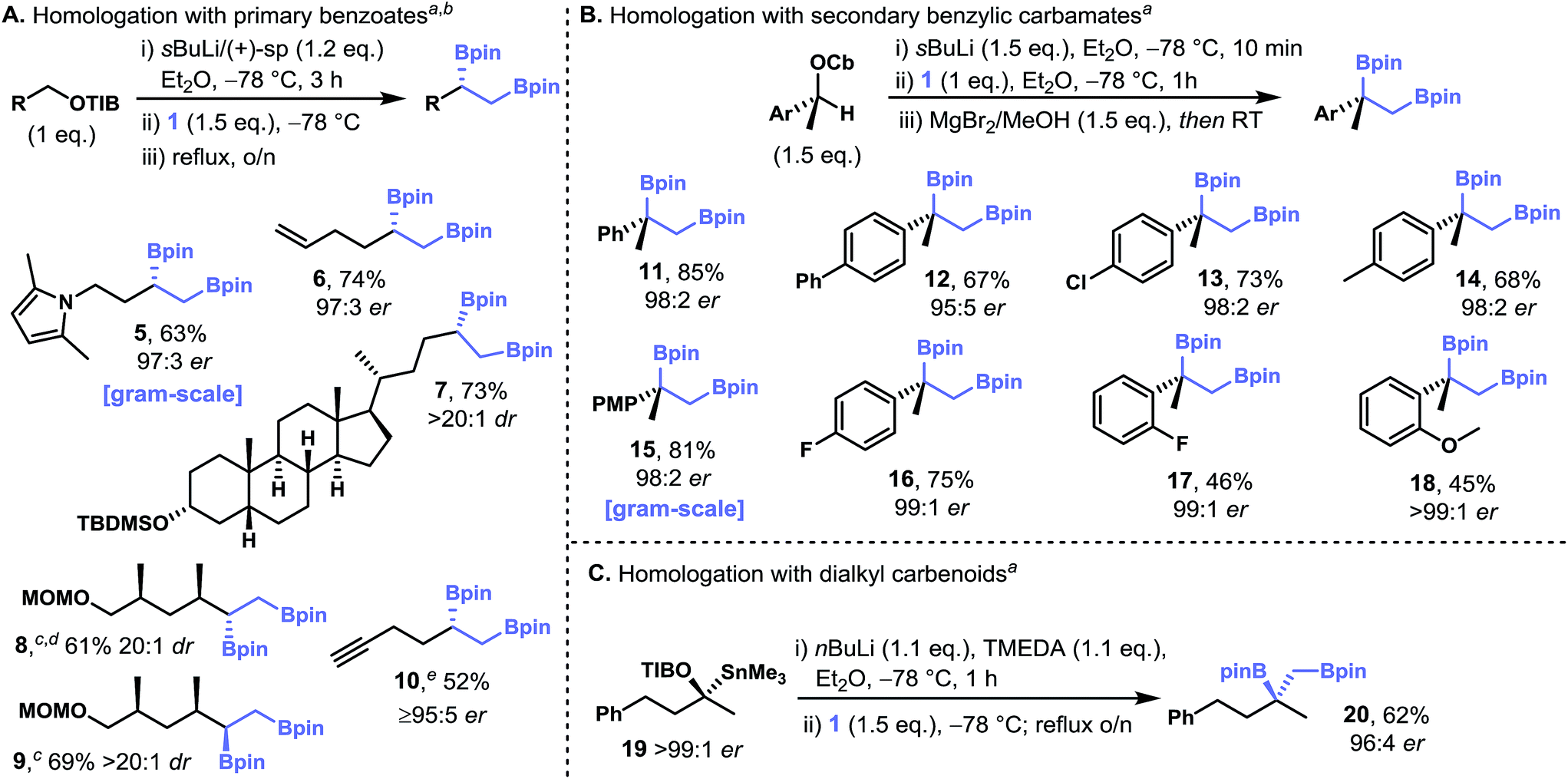 | ||
| Fig. 3 Stereoselective synthesis of 1,2-bis(boronic esters) from primary benzoates (A), secondary benzylic carbamates (B) and secondary dialkyl benzoates (C). Footnotes: (a) enantiomeric ratios determined by HPLC, GC or SFC analysis using chiral stationary phases. (b) Diastereomeric ratios determined by 1H NMR analysis. (c) Prepared from the corresponding carbamate (d) using (−)-sparteine in place of (+)-sparteine. (e) 2.2 equivalents of (+)-sparteine, sBuLi and 1. Abbreviations: Cb = N,N-diisopropyl carbamoyl, MOM = methoxymethyl, PMP = para-methoxyphenyl, TBS = tert-butyldimethylsilyl, TIB = 2,4,6-triisopropylbenzoyl, TMEDA = N,N,N′,N′-tetramethylethylenediamine, pin = pinacolato. | ||
Based on our observation that sterically hindered diamine-ligated primary carbenoids are highly selective for the single homologation of 1, we reasoned that secondary benzylic carbenoids, which can be generated through the stereospecific deprotonation of the corresponding carbamates in the absence of a diamine, should be similarly selective. We were also mindful that the products, primary–tertiary 1,2-bis(boronic esters), are not accessible in useful yields or levels of enantiopurity through the asymmetric diboration of 1,1-disubstituted alkenes, thus giving such a process extra significance. Pleasingly, the treatment of enantiopure (S)-1-phenylethanol-derived lithiated carbamate with diborylmethane 1 gave primary–tertiary 1,2-bis(boronic ester) 11 in high yield and with a high level of enantiopurity (98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 e.r.; Fig. 3B). The addition of a Lewis acid (MgBr2) and MeOH at −78 °C immediately after the formation of the boronate complex, yet prior to warming to room temperature (the 1,2-metallate rearrangement only occurs at elevated temperatures), was essential for achieving high yields and high levels of enantiopurity.10c
:2 e.r.; Fig. 3B). The addition of a Lewis acid (MgBr2) and MeOH at −78 °C immediately after the formation of the boronate complex, yet prior to warming to room temperature (the 1,2-metallate rearrangement only occurs at elevated temperatures), was essential for achieving high yields and high levels of enantiopurity.10c
These homologation reactions were insensitive to the electronic demands of the aromatic ring as p-Ph, p-Cl, p-Me, p-MeO and p-F substituted carbamates gave the desired 1,2-bis(boronic esters) 12–16 in high yields and high levels of enantioselectivity. These transformations were similarly effective on gram scale (15). Although the transformation of ortho-substituted benzylic carbamates (o-F and o-MeO: 17 and 18) gave more moderate yields of the products, the levels of enantiopurity remained very high. The transformation of secondary alkyl benzoates (i.e. non-benzylic)22 necessitated the generation of the chiral carbenoid from the corresponding stannane through tin–lithium exchange. The carbenoid so generated from 19 (>99:1 e.r.) reacted with diborylmethane 1 to give the corresponding 1,2-bis(boronic ester) 20 in 62% yield and with high levels of enantiospecificity (96:4 e.r.), the slight erosion of stereochemical information being most likely due to competing stereoinversion of the carbanion during the formation of the boronate complex.22,23
Our initial studies established that diamine-free primary lithiated benzoates react with diborylmethane 1 to give C2-symmetric secondary–secondary 1,3-bis(boronic esters), that is, both ends of the diborylmethane undergo homologation (Fig. 2). We explored the scope of this transformation by using a range of diamine-free lithiated benzoates, which were generated through tin–lithium exchange of the corresponding enantiopure α-stannyl benzoates (Fig. 4A). The double-homologation reactions of diborylmethane 1 with primary lithiated benzoates gave a range of C2-symmetric secondary–secondary 1,3-bis(boronic esters) (21–24) in high yield and near-perfect diastereoselectivity. In contrast, one-pot double homologation with the more hindered secondary benzylic lithiated carbamates proved more challenging: a low yield of C2-symmetric tertiary–tertiary 1,3-bis(boronic ester) 25 was obtained, even when using a large excess of the requisite lithiated carbamate (Fig. 4B). However, we overcame this limitation through the isolation of the mono-homologation product, 15, and resubjecting it to the homologation conditions, a protocol that gave tertiary–tertiary 1,3-bis(boronic ester) 25 in moderate yield and very high levels of diastereoselectivity. Presumably formation of the dianionic geminal bis(boronate), which would be required for the one-pot process, is disfavored due to steric hindrance. Attempts to homologate 15 with (+)-sparteine-ligated primary benzoates were unsuccessful, underscoring the sensitivity of primary–tertiary 1,2-bis(boronic esters) to steric hindrance. Pleasingly, both the selective homologation of the primary boronic ester moiety of 15 (to give secondary–tertiary 1,3-bis(boronic ester) 26) and the double homologation of 15 (to give secondary–secondary 1,4-bis(boronic ester) 27) could be carried out by using 0.9 and 2.1 equivalents, respectively, of diamine-free lithiated benzoate.
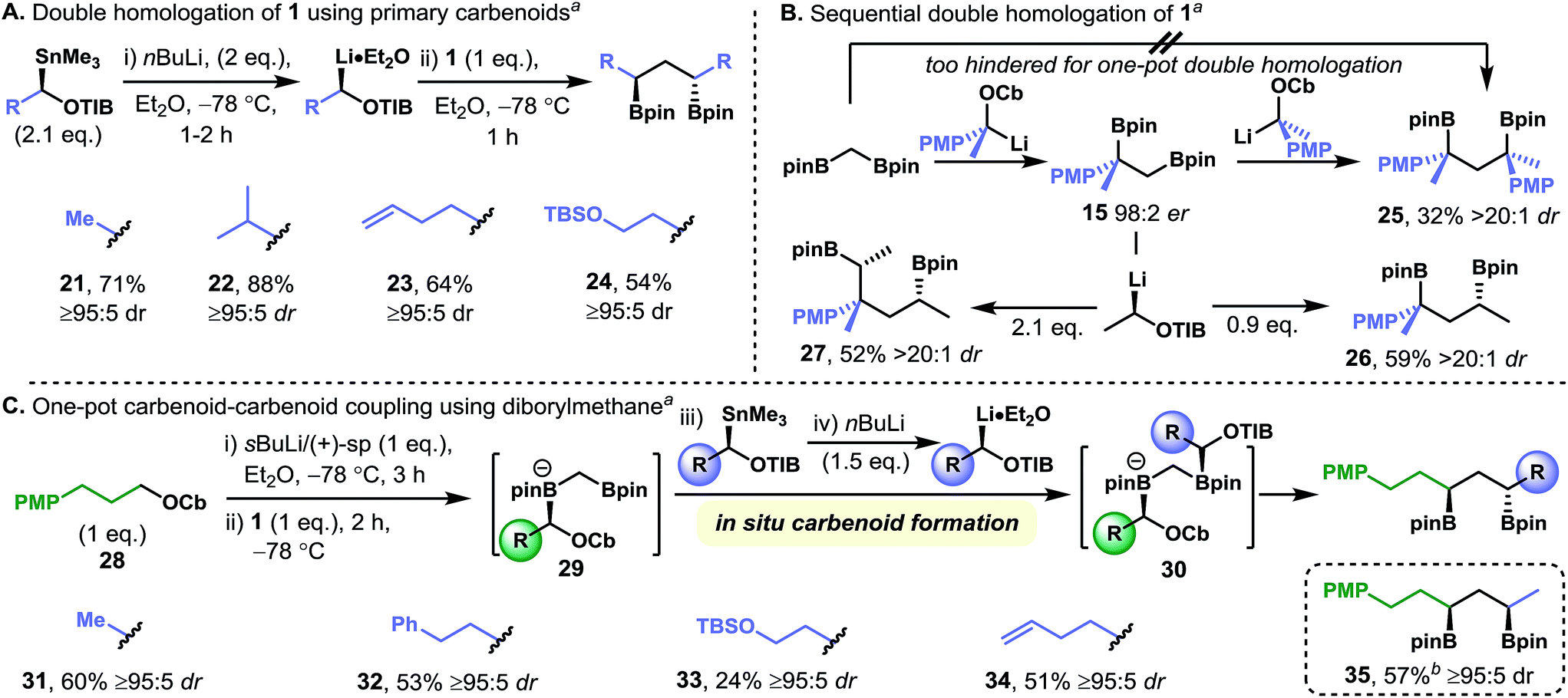 | ||
| Fig. 4 Synthesis of C2-symmetric-1,3-bis(boronic esters) from diborylmethane 1 by using diamine-free primary carbenoids (A) and stepwise double homologation with secondary benzylic carbamates (B). Also, the one-pot mixed homologation of diborylmethane to form non-symmetric 1,3-bis(boronic esters) (C). Footnotes: (a) diastereomeric ratios were determined by 1H NMR analysis. (b) Opposite enantiomer of stannane was used. Abbreviations: Cb = N,N-diisopropyl carbamoyl, PMP = para-methoxyphenyl, TBS = tert-butyldimethylsilyl, TIB = 2,4,6-triisopropylbenzoyl, pin = pinacolato. | ||
The regioselective single homologation of diborylmethane 1 with sparteine-ligated carbenoids, and the relative insensitivity of the diamine-free carbenoids to steric hindrance in their reactions with boronic esters, suggested that these types of carbenoids could be coupled in a one-pot three-component coupling reaction with diborylmethane 1 acting as a linchpin. Specifically, the generation of a (+)-sparteine-ligated carbenoid at −78 °C and the addition of diborylmethane 1 would give the mixed-valent diboryl species 29. Subsequent addition of an enantiomerically pure α-stannyl benzoate followed by an equivalent portion of n-BuLi would lead to rapid and selective tin–lithium exchange followed by the reaction of the resulting diamine-free carbenoid with the remaining boronic ester moiety to give the heteroleptic geminal 1,1-bis(boronate) 30. Finally, upon warming to room temperature 1,1-bis(boronate) 30 would undergo 1,2-metallate rearrangement at both boron centers to give the corresponding non-symmetrical secondary–secondary 1,3-bis(boronic esters) (Fig. 4C). This one-pot process was carried out for sparteine-ligated lithiated carbamate 2824 with a range of α-stannyl benzoates to give 1,3-anti-bis(boronic esters) 31–34 in moderate yield and with very high levels of diastereoselectivity. As is the nature of the transformation, 1,3-syn-bis(boronic esters), such as 35, can be formed with equal ease and selectivity simply by using the enantiomeric α-stannyl benzoate. This transformation is a rare example of a fragment coupling reaction in which 1,3-related stereo centers are formed directly with complete control of the relative and absolute configuration, suggesting that it could be used as a strategy for bringing together advanced fragments for the preparation of complex molecules.
We briefly explored a number C–B functionalisation reactions of primary–tertiary 1,2-bis(boronic ester) 15 (Fig. 5). Its subjection to standard Matteson homologation,25 Zweifel olefination26 and standard oxidation conditions gave the corresponding difunctionalisation products 36–38, respectively, in good yield. We were also particularly interested in whether selective functionalisation of either the primary or the tertiary boronic ester could be achieved. As expected, selective protodeboronation of the tertiary benzylic boronic ester of 15 (98:2 e.r.) was possible, but with only moderate enantiospecificity (primary boronic ester 39 was obtained in 88:12 e.r.).27 The group of Morken has shown that primary boronic esters can be transformed selectively in the presence of a vicinally-positioned secondary boronic ester under Suzuki cross-coupling conditions.3b However, although facile cross coupling of the primary boronic ester in 15 was evident, the conditions were also favourable for protodeboronation of the tertiary boronic ester, giving 1,2-diarylpropane 40 in 75% yield. Clearly, further exploration and optimisation of the conditions for the group-selective functionalisation of primary–tertiary bis(boronic esters) is warranted.
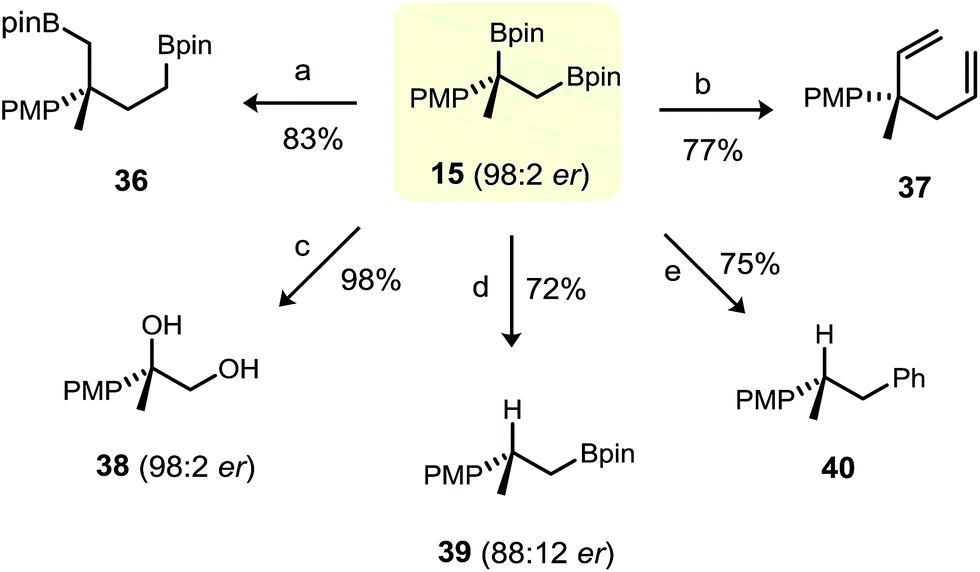 | ||
| Fig. 5 C–B functionalisation of primary–tertiary 1,2-bis(boronic ester) 15. Reaction conditions: (a) BrCH2Cl (6 eq.), n-BuLi (5 eq.), Et2O, −78 °C (20 min); then RT (1 h); (b) vinyllithium (4.0 eq.), Et2O, −78 °C, 45 min; then I2/THF (4.0 eq.), −78 °C, 30 min; then NaOMe/MeOH (8.0 eq.), −78 °C to RT. (c) aq. 2 M NaOH/aq. 30% H2O2 (2:1), THF, 0 °C to RT, 4 h. (d) TBAF·3H2O (3.0 eq.), toluene, 90 °C, 2 h. (e) Pd(OAc)2/RuPhos (1 mol%), bromobenzene (1.5 eq.), KOH (3 eq.), THF/H2O, 70 °C, 12 h. | ||
Conclusions
In summary, we have developed a regiodivergent strategy for the stereoselective synthesis of 1,2- and 1,3-bis(boronic esters) from commercially available diborylmethane 1. Using hindered chiral carbenoids we have been able to effect the selective single homologation of diborylmethane 1 to prepare a wide range of 1,2-bis(boronic esters). Generating primary chiral carbenoids in the absence of diamines enabled the exclusive double homologation of diborylmethane 1 to afford C2-symmetric 1,3-bis(boronic esters). The strategic combination of these carbenoids (sparteine-ligated and diamine-free) with diborylmethane 1, representing a formal carbenoid–carbenoid coupling reaction, allowed non-symmetrical 1,3-bis(boronic esters) to be prepared directly.Acknowledgements
We thank the European Research Council (FP7/2007-2013, ERC grant no. 246785; H2020/2015-2020, ERC grant no. 670668), and EPSRC (EP/I038071/1) for financial support. We thank Dominik Zetschok for technical assistance.References
- For reviews on the synthesis of polyborylated and dimetallated molecules, see: (a) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed; (b) H. E. Burks and J. P. Morken, Chem. Commun., 2007, 4717 RSC; (c) C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyás and E. Fernández, Org. Biomol. Chem., 2010, 8, 2667 RSC; (d) J. Takaya and N. Iwasawa, ACS Catal., 2012, 2, 1993 CrossRef CAS; (e) L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848 RSC; (f) J. R. Coombs and J. P. Morken, Angew. Chem., Int. Ed., 2016, 55, 2636 CrossRef CAS PubMed; (g) H. Yoshida, Chem. Rec., 2016, 16, 419 CrossRef CAS PubMed.
- Transformations of the carbon–boron bond: amination (a) V. Bagutski, T. G. Elford and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 1080 CrossRef CAS PubMed; (b) S. N. Mlynarski, A. S. Karns and J. P. Morken, J. Am. Chem. Soc., 2012, 134, 16449 CrossRef CAS PubMed; arylation (c) D. Imao, B. W. Glasspoole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024 CrossRef CAS PubMed; (d) T. Ohmura, T. Awano and M. Suginome, J. Am. Chem. Soc., 2010, 132, 13191 CrossRef CAS PubMed; (e) D. L. Sandrock, L. Jean-Gérard, C. –Y. Chen, S. D. Dreher and G. A. Molander, J. Am. Chem. Soc., 2010, 132, 17108 CrossRef CAS PubMed; (f) J. C. H. Lee, R. McDonald and D. G. Hall, Nat. Chem., 2011, 3, 894 CrossRef CAS PubMed; (g) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584 CrossRef CAS PubMed; for a review, see: (h) D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2015, 54, 1082 CrossRef CAS PubMed; fluorination (i) Z. Li, Z. Wang, L. Zhu, X. Tan and C. Li, J. Am. Chem. Soc., 2014, 136, 16439 CrossRef CAS PubMed; (j) C. Sandford, R. Rasappan and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10100 CrossRef CAS PubMed; heteroarylation (k) J. Llaveria, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10958 CrossRef CAS PubMed; (l) W. Jo, J. Kim, S. Choi and S. H. Cho, Angew. Chem., Int. Ed., 2016, 55, 9690 CrossRef CAS PubMed; alkylation (m) A. Ros and V. K. Aggarwal, Angew. Chem., Int. Ed., 2009, 48, 6289 CrossRef CAS PubMed; (n) G. Berthon and T. Hayashi, in Catalytic Asymmetric Conjugate Reactions, ed. A. Córdova, Wiley-VCH, Weinheim, Germany, 2010, ch. 1, pp. 1–70 Search PubMed; (o) J. Wang, T. Qin, T. G. Chen, L. Wimmer, J. T. Edwards, J. Cornella, B. Vokits, S. A. Shaw and P. S. Baran, Angew. Chem., Int. Ed., 2016, 55, 9676 CrossRef CAS PubMed; alkynylation (p) Y. Wang, A. Noble, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 4270 CrossRef CAS PubMed; see also: (q) Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed; (r) Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer International Publishing, Cham, Switzerland, 2015 Search PubMed.
- (a) S. P. Miller, J. B. Morgan, F. J. Nepveux and J. P. Morken, Org. Lett., 2004, 6, 131 CrossRef CAS PubMed; (b) S. N. Mlynarski, C. H. Schuster and J. P. Morken, Nature, 2013, 505, 386 CrossRef PubMed; (c) C. M. Crudden, C. Ziebenhaus, J. P. G. Rygus, K. Ghozati, P. J. Unsworth, M. Nambo, S. Voth, M. Hutchinson, V. S. Laberge, Y. Maekawa and D. Imao, Nat. Commun., 2016, 7, 11065 CrossRef PubMed.
- (a) E. Ballard and J. P. Morken, Synthesis, 2004, 9, 1321 Search PubMed; (b) N. F. Pelz, A. R. Woodward, H. E. Burks, J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2004, 126, 16328 CrossRef CAS PubMed; (c) A. R. Woodward, H. E. Burks, L. M. Chan and J. P. Morken, Org. Lett., 2005, 7, 5505 CrossRef CAS PubMed; (d) J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2006, 128, 74 CrossRef CAS PubMed; (e) K. Endo, T. Ohkubo, M. Hirokami and T. Shibata, J. Am. Chem. Soc., 2010, 132, 11033 CrossRef CAS PubMed; (f) L. T. Kliman, S. N. Mlynarski, G. E. Ferris and J. P. Morken, Angew. Chem., Int. Ed., 2012, 51, 521 CrossRef CAS PubMed; (g) H. Le, R. E. Kyne, L. A. Brozek and J. P. Morken, Org. Lett., 2013, 15, 1432 CrossRef CAS PubMed; (h) J. R. Coombs, L. Zhang and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 16140 CrossRef CAS PubMed; (i) B. Potter, A. A. Szymaniak, E. K. Edelstein and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 17918 CrossRef CAS PubMed; (j) T. P. Blaisdell and J. P. Morken, J. Am. Chem. Soc., 2015, 137, 8712 CrossRef CAS PubMed.
- (a) L. T. Kilman, S. N. Mlynarski and J. P. Morken, J. Am. Chem. Soc., 2009, 131, 13210 CrossRef PubMed; (b) J. R. Coombs, F. Haeffner, L. T. Kilman and J. P. Morken, J. Am. Chem. Soc., 2013, 135, 11222 CrossRef CAS PubMed.
- (a) J. B. Morgan, S. P. Miller and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 8702 CrossRef CAS PubMed; (b) S. Trudeau, J. B. Morgan, M. Shrestha and J. P. Morken, J. Org. Chem., 2005, 70, 9538 CrossRef CAS PubMed; (c) K. Toribatake and H. Nishiyama, Angew. Chem., Int. Ed., 2013, 52, 11011 CrossRef CAS PubMed; (d) K. Toribatake, S. Miyata, Y. Naganawa and H. Nishiyama, Tetrahedron, 2015, 71, 3203 CrossRef CAS.
- (a) T. P. Blaisdell, T. C. Caya, L. Zhang, A. Sanz-Marco and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 9264 CrossRef CAS PubMed; for other transition metal free diboration reactions, see: (b) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158 CrossRef CAS PubMed; (c) L. Fang, L. Yan, F. Haeffner and J. P. Morken, J. Am. Chem. Soc., 2016, 138, 2508 CrossRef CAS PubMed.
- To date primary–tertiary 1,2-bis(boronic esters) have been formed in at most 76% ee from α-methyl styrene (see ref. 6c).
- We have recently shown that primary–secondary 1,2-bis(boronic esters) can be homologated selectively at the primary boron moiety to give stereocontrolled 1,3-bis(boronic esters): (a) A. Fawcett, D. Nitsch, M. Ali, J. M. Bateman, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 14663 CrossRef CAS PubMed; 1,4-bis(boronic esters) are known; see: (b) H. E. Burks, L. T. Kilman and J. P. Morken, J. Am. Chem. Soc., 2009, 131, 9134 CrossRef CAS PubMed; (c) C. H. Schuster, B. Li and J. P. Morken, Angew. Chem., Int. Ed., 2011, 50, 7906 CrossRef CAS PubMed; (d) T. Ishiyama, M. Yamamoto and N. Miyaura, Chem. Commun., 1997, 689 RSC.
- (a) J. L. Stymiest, G. Dutheuil, A. Mahmood and V. K. Aggarwal, Angew. Chem., Int. Ed., 2007, 46, 7491 CrossRef CAS PubMed; (b) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS PubMed; (c) V. Bagutski, R. M. French and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 5142 CrossRef CAS PubMed; (d) M. P. Webster, B. M. Partridge and V. K. Aggarwal, Org. Synth., 2011, 88, 247 CrossRef CAS; for reviews on our work, see (e) H. K. Scott and V. K. Aggarwal, Chem.–Eur. J., 2011, 17, 13124 CrossRef CAS PubMed; (f) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed; see also (g) G. Besong, K. Jarowicki, P. J. Kocienski, E. Sliwinski and F. T. Boyle, Org. Biomol. Chem., 2006, 4, 2193 RSC.
- (a) R. Larouche-Gauthier, C. J. Fletcher, I. Couto and V. K. Aggarwal, Chem. Commun., 2011, 47, 12592 RSC; (b) M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183 CrossRef CAS PubMed.
- (a) D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282 CrossRef CAS; (b) A. Basu and S. Thayumanavan, Angew. Chem., Int. Ed., 2002, 41, 716 CrossRef CAS; (c) D. Hoppe, F. Marr and M. Bruggemann, in Organolithiums in Enantioselective Synthesis, ed. D. M. Hodgson, Springer, Heidelberg, 2003, p. 61 Search PubMed.
- (a) P. Beak and B. G. McKinnie, J. Am. Chem. Soc., 1977, 99, 5213 CrossRef CAS; (b) P. Beak, M. Baillargeon and L. G. Carter, J. Org. Chem., 1978, 43, 4255 CrossRef CAS; (c) P. Beak and L. G. Carter, J. Org. Chem., 1981, 46, 2363 CrossRef CAS.
- (a) S. P. Thomas, R. M. French, V. Jheengut and V. K. Aggarwal, Chem. Rec., 2009, 9, 24 CrossRef CAS PubMed; (b) D. S. Matteson, J. Org. Chem., 2013, 78, 10009 CrossRef CAS PubMed.
- Diborylmethane can be purchased from Sigma Aldrich (Catalogue No. 794287) or prepared on decagram scale from dibromomethane; see (a) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581 CrossRef CAS PubMed; for other selected recent applications of diborylmethane, see: (b) S. A. Murray, J. C. Green, S. B. Tailor and S. J. Meek, Angew. Chem., Int. Ed., 2016, 55, 9065 CrossRef CAS PubMed; (c) Y. Shi and A. H. Hoveyda, Angew. Chem., Int. Ed., 2016, 55, 3455 CrossRef CAS PubMed; (d) J. Park, J. Kim and S. H. Cho, Org. Lett., 2016, 18, 1210 CrossRef CAS PubMed; (e) K. Endo, T. Ishioka, T. Ohkubo and T. Shibata, J. Org. Chem., 2012, 77, 7223 CrossRef CAS PubMed.
- D. S. Matteson and R. J. Moody, Organometallics, 1982, 1, 20 CrossRef CAS.
- (+)-Sparteine was obtained from Ark Pharm (AK142759) and Manchester Organics (G50239), and (−)-sparteine was obtained from the University of Vienna, E-mail: Nuno.Maulide@univie.ac.at.
- Reversing the order of addition, that is, adding 1.0 equivalents of diamine-free lithiated benzoate to 1.5 equivalents of diborylmethane, gave a 3:1 mixture of single and double addition products. As there is no significant bias toward double homologation, selectivity for its formation under normal addition is due to rapid electrophilic trapping of both boron centres during the dropwise addition of diborylmethane and not cooperativity between the nascent mono-boronate complex and the lithiated benzoate.
- K. Endo, M. Hirokami and T. Shibata, Synlett, 2009, 1331 CrossRef CAS.
- Substituted chiral α-chloro carbenoids are known to react slowly with more hindered boronic esters, leading to reduced levels of stereoselectivity; see: P. R. Blakemore and M. S. Burge, J. Am. Chem. Soc., 2007, 129, 3068 CrossRef CAS PubMed.
- Dibenzyl amine containing carbamates can be lithiated in high e.r.; see: (a) P. Sommerfeld and D. Hoppe, Synlett, 1992, 764 CrossRef CAS; however, neither the use of the lithiated 3-dibenzylamino carbamate nor that of the benzoate were successful in homologating diborylmethane 1; the relatively basic nitrogen atom may interfere with the 1,2-metallate rearrangement through coordinating to lithium in the corresponding boronate complex; see also: (b) S. Roesner, J. M. Casatejada, T. G. Elford, R. P. Sonawane and V. K. Aggarwal, Org. Lett., 2011, 13, 5740 CrossRef CAS PubMed.
- A. P. Pulis, D. J. Blair, E. Torres and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 16054 CrossRef CAS PubMed.
- A. P. Pulis, Ph D thesis, University of Bristol, 2012.
- Benzoates may be used in place of carbamates but give reduced levels of diastereoselectivity owing to the corresponding lithiated benzoates being ca. 5–7% less enantioenriched.
- (a) K. M. Sadhu and D. S. Matteson, Organometallics, 1985, 4, 1687 CrossRef CAS; (b) H. C. Brown, S. M. Singh and M. V. Rangaishenvi, J. Org. Chem., 1986, 51, 3150 CrossRef CAS.
- (a) G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652 CrossRef CAS; (b) G. Zweifel, N. L. Polston and C. C. Whitney, J. Am. Chem. Soc., 1968, 90, 6243 CrossRef CAS; (c) D. A. Evans, T. C. Crawford, R. C. Thomas and J. A. Walker, J. Org. Chem., 1976, 41, 3947 CrossRef CAS PubMed.
- S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, synthetic details and NMR spectra. See DOI: 10.1039/c6sc05338f |
| This journal is © The Royal Society of Chemistry 2017 |