
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective [2 + 2] cycloaddition of N-allenamides with cyclic N-sulfonylketimines: access to polysubstituted azetidines bearing quaternary stereocenters†
Ren-Rong
Liu
,
Jiang-Ping
Hu
,
Jian-Jun
Hong
,
Chuan-Jun
Lu
,
Jian-Rong
Gao
and
Yi-Xia
Jia
*
College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: yxjia@zjut.edu.cn
First published on 19th January 2017
Abstract
A Ni(ClO4)2-catalyzed enantioselective [2 + 2] cycloaddition of N-allenamides with cyclic N-sulfonylketimines was developed, which regioselectively occurred at the proximal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of the N-allenamides. Broad substrate scope of N-allenamides and cyclic N-sulfonylketimines was observed. A range of fused polysubstituted azetidines bearing quaternary stereocenters were afforded in good yields and with excellent enantioselectivities (up to 99%).
C bonds of the N-allenamides. Broad substrate scope of N-allenamides and cyclic N-sulfonylketimines was observed. A range of fused polysubstituted azetidines bearing quaternary stereocenters were afforded in good yields and with excellent enantioselectivities (up to 99%).
N-Allenamides are readily available allenic scaffolds and valuable building blocks in organic synthesis, and therefore have received considerable attention in the past decades.1,2 Significant effort has been devoted to the cycloaddition reactions of N-allenamides,3 and several enantioselective [n + 2] reactions have been explored as reliable approaches to synthesise a diverse array of carbo- or heterocyclic substructures (Scheme 1).4–6 Mascareñas et al. developed an elegant enantioselective intermolecular [4 + 2] cycloaddition reaction between N-allenamides and dienes using a gold/chiral carbene catalyst (Scheme 1a).4 González's and Zhang's groups disclosed [2 + 2] cycloadditions of N-allenamides with olefins, leading to optically active cyclobutanes with excellent enantioselectivities (Scheme 1b).5a,c Interestingly, in the latter case a dearomative [4 + 2] reaction of 3-styrylindoles was observed by varying indolic N-substituents.5c A dearomative [2 + 2] cycloaddition reaction of N-allenamides with indoles was realized by Bandini's group as a powerful route to access cyclobutane-fused indolines with excellent enantioselectivities.5b The [3 + 2] reaction was also developed by Chen and co-workers in an enantioselective cycloaddition of N-allenamides with nitrones (Scheme 1c).6a It should be noted that all of the above reactions mainly relied on N-allenamide activation to form an α,β-unsaturated imine intermediate and hence regioselectively occurred at its distal C
C bond. In sharp contrast, the reactivity of N-allenamide at the proximal CC bond has remained rarely studied.7 The only enantioselective example was reported by Zhang's group in an asymmetric [3 + 2] reaction involving in situ annulation of yne-enones (Scheme 1d).6b Meanwhile, all of the precedent examples exclusively relied on chiral noble gold catalysts. Moreover, despite these advances, no example of cycloaddition of N-allenamides with imines has appeared for the synthesis of nitrogen-bearing four-membered heterocycles even in their racemic version.1a Therefore, the development of new cycloaddition reactions of N-allenamides and a new catalyst system to synthesize important heterocyclic cores is highly attractive.
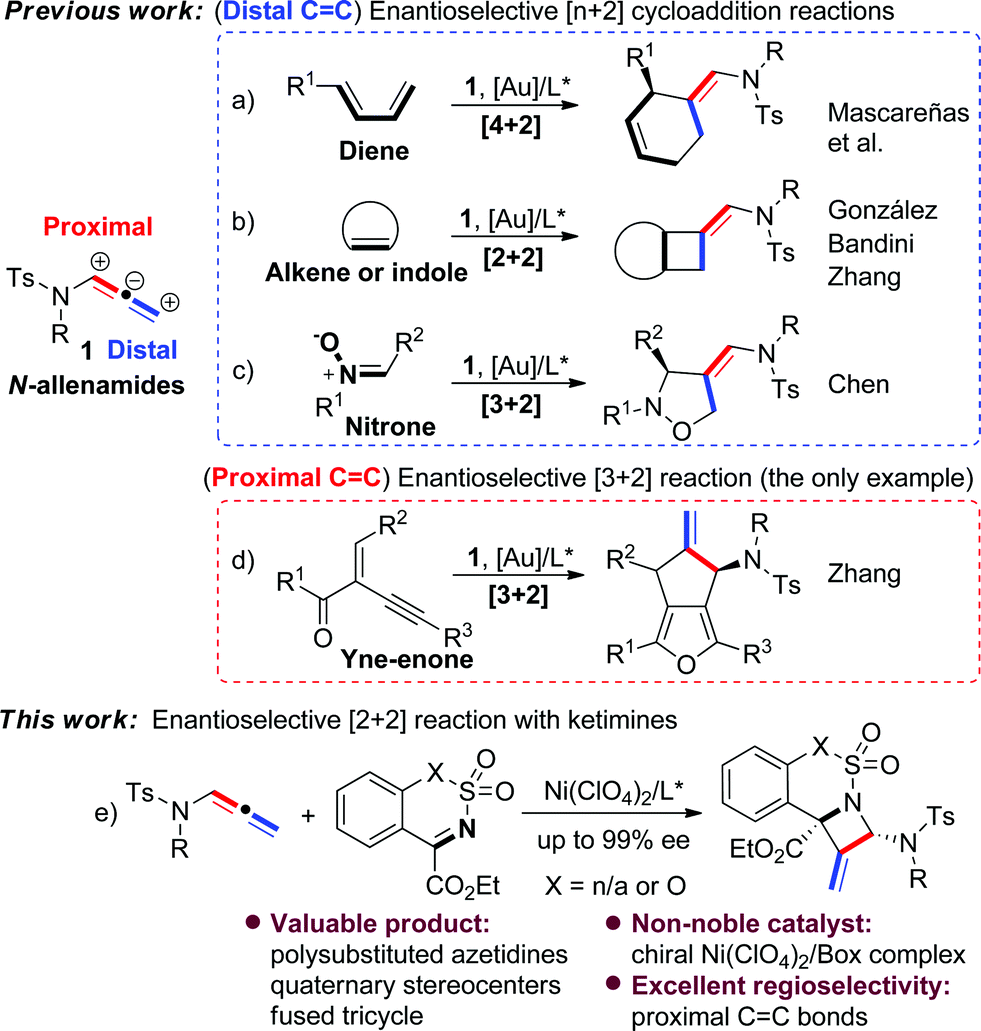 | ||
| Scheme 1 Reactivity of N-allenamides in enantioselective cycloaddition reactions. | ||
Azetidine derivatives are important entities in organic chemistry and are frequently occurring scaffolds in natural products and bioactive molecules. Enantioselective [2 + 2] reaction of imines provides a straightforward route to such strained four-membered heterocycles, including β-lactams.8,9 However, as a unique building block, N-allenamide has not been utilized in the [2 + 2] cycloaddition with imines. We envisioned that an enantioselective [2 + 2] cycloaddition of N-allenamides with imines might occur via an imine activation strategy using a chiral Lewis acid catalyst, where N-allenamide acts as a nucleophile (like enamide) to attack the imine at its more electron-rich proximal CC bond and hence delivers highly substituted azetidines through the cyclization that follows.10 Herein, we report the first enantioselective [2 + 2] cycloaddition reaction of N-allenamides with cyclic N-sulfonyl α-ketiminoesters11 to afford fused polysubstituted chiral azetidines bearing quaternary stereocenters with excellent enantioselectivities (Scheme 1e). A non-noble chiral nickel catalyst was applied for the first time in the cycloaddition reactions of N-allenamides.
Our study began with the model reaction between N-allenamide 1a and cyclic α-ketiminoester 2a. Pleasingly, an initial test revealed that the complex of Ni(ClO4)2·6H2O/chiral bisoxazoline L1 smoothly promoted the reaction in DCE and in the presence of 4 Å molecular sieves at room temperature, resulting in the desired product 3aa in 83% yield, albeit with a poor ee value (Table 1, entry 1). Subsequent ligand examination implied that chiral substituents of oxazoline were critical to enantioselectivity (entries 2–7). Ligands L2–L3 bearing isopropyl or tert-butyl substituents led to poor enantioselectivities (entries 2 and 3), while L4, containing phenyl substituents, was the best choice to give 3aa in 99% ee and 82% yield (entry 4).12 Inferior results were obtained when L5 and L6 bearing trans- or cis-diphenyl substituents were used (entries 5 and 6). Phosphine ligand L7 led to 3aa with a moderate yield and ee value (entry 7). Solvent investigation showed that excellent enantioselectivities with relatively lower yields were observed in CH2Cl2, THF, and toluene (entries 8–10). Either poor yield or poor enantioselectivity was obtained in Et2O or MeCN (entries 11 and 12). Changing the Lewis acid to Cu(OTf)2 or Mg(OTf)2 resulted in an almost racemic product (entries 13 and 14), while Zn(OTf)2 led to a relatively lower ee (entry 15). Other perchlorate metallic salts, such as Mg(ClO4)2, Zn(ClO4)2·6H2O, and Cu(ClO4)2·6H2O, were also tested and inferior results were obtained (entries 16–18). Finally, a poor yield was obtained in the absence of 4 Å MS due to the decomposition of N-allenamide 1a (entry 19). It is noteworthy that only one isomer was detected in these reactions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | L* | LA | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.2 mmol), LA (10 mol%), ligand (12 mol%), and 4 Å molecular sieves (100 mg) in solvent (1.0 M) at room temperature; L7: (S)-BINAP. b Isolated yield. c Determined by chiral HPLC. d Without 4 Å MS. | |||||
| 1 | L1 | Ni(ClO4)2·6H2O | DCE | 83 | 18 |
| 2 | L2 | Ni(ClO4)2·6H2O | DCE | 75 | 15 |
| 3 | L3 | Ni(ClO4)2·6H2O | DCE | 69 | 11 |
| 4 | L4 | Ni(ClO4)2·6H2O | DCE | 82 | 99 |
| 5 | L5 | Ni(ClO4)2·6H2O | DCE | 62 | 92 |
| 6 | L6 | Ni(ClO4)2·6H2O | DCE | 65 | 93 |
| 7 | L7 | Ni(ClO4)2·6H2O | DCE | 70 | 60 |
| 8 | L4 | Ni(ClO4)2·6H2O | CH2Cl2 | 57 | 96 |
| 9 | L4 | Ni(ClO4)2·6H2O | THF | 51 | 99 |
| 10 | L4 | Ni(ClO4)2·6H2O | Toluene | 60 | 95 |
| 11 | L4 | Ni(ClO4)2·6H2O | Et2O | 23 | 91 |
| 12 | L4 | Ni(ClO4)2·6H2O | CH3CN | 90 | 10 |
| 13 | L4 | Cu(OTf)2 | DCE | 65 | 7 |
| 14 | L4 | Mg(OTf)2 | DCE | 75 | <5 |
| 15 | L4 | Zn(OTf)2 | DCE | 70 | 85 |
| 16 | L4 | Mg(ClO4)2 | DCE | 70 | 28 |
| 17 | L4 | Zn(ClO4)2·6H2O | DCE | 55 | 46 |
| 18 | L4 | Cu(ClO4)2·6H2O | DCE | Trace | — |
| 19d | L4 | Ni(ClO4)2·6H2O | DCE | 45 | 99 |
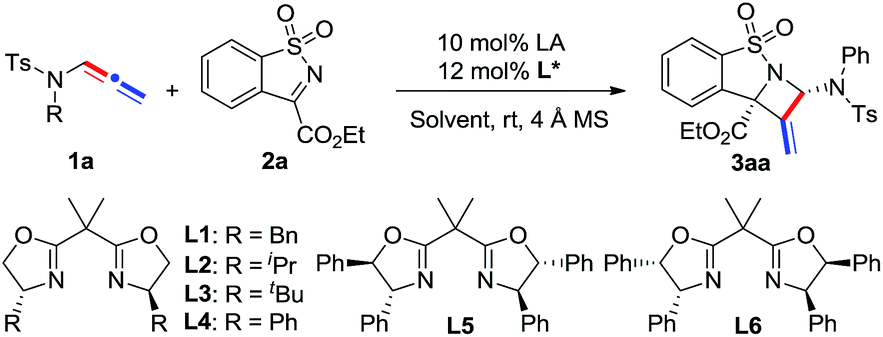
With the optimal reaction conditions found, we then investigated the scope of N-allenamides 1 and N-sulfonyl α-ketiminoesters 2. As shown in Table 2, α-ketiminoesters 2a–2d, bearing different ester groups, were first evaluated. Good yields and enantioselectivities were afforded in these reactions in spite of a slightly decreased ee value for the iso-propyl substrate 2b (entries 1–4). Substituents attached at the C5–C7 position of the α-ketiminoesters were then investigated (entries 5–14). The substituted groups, such as halide, –CF3, –OCF3, –OMe, and alkyl groups, were well tolerated to afford the corresponding products with excellent enantioselectivities and modest to good yields. The reaction yields of the α-ketiminoesters containing electron-donating substituents were slightly higher than those of the α-ketiminoesters bearing electron-deficient substituents (entries 5–7 vs. 8–11).
 | (1) |
 | (2) |
|
|
||||
|---|---|---|---|---|
| Entry | 1 (R2/R3) | 2 (R/R1) | Yieldb (%) | eec (%) |
| a Reaction conditions are identical to those in entry 4 in Table 1. b Isolated yield of the only isomer. c Determined by chiral HPLC. | ||||
| 1 | 1a (4-Me/H) | 2a (H/Et) | 82 (3aa) | 99 |
| 2 | 1a (4-Me/H) | 2b (H/iPr) | 75 (3ab) | 90 |
| 3 | 1a (4-Me/H) | 2c (H/Me) | 76 (3ac) | 99 |
| 4 | 1a (4-Me/H) | 2d (H/nBu) | 80 (3ad) | 98 |
| 5 | 1a (4-Me/H) | 2e (5-OMe/Et) | 77 (3ae) | 99 |
| 6 | 1a (4-Me/H) | 2f (5-Me/Et) | 73 (3af) | 97 |
| 7 | 1a (4-Me/H) | 2g (5-tBu/Et) | 70 (3ag) | 98 |
| 8 | 1a (4-Me/H) | 2h (5-Cl/Et) | 64 (3ah) | 99 |
| 9 | 1a (4-Me/H) | 2i (5-F/Et) | 70 (3ai) | 95 |
| 10 | 1a (4-Me/H) | 2j (5-CF3/Et) | 52 (3aj) | 99 |
| 11 | 1a (4-Me/H) | 2k (5-OCF3/Et) | 55 (3ak) | 98 |
| 12 | 1a (4-Me/H) | 2l (7-Cl/Et) | 64 (3al) | 94 |
| 13 | 1a (4-Me/H) | 2m (7-OCF3/Et) | 76 (3am) | 93 |
| 14 | 1a (4-Me/H) | 2n (6,7-(CH)4/Et) | 70 (3an) | 94 |
| 15 | 1b (4-Me/4-Me) | 2a (H/Et) | 75 (3ba) | 99 |
| 16 | 1c (4-Me/4-Br) | 2a (H/Et) | 67 (3ca) | 97 |
| 17 | 1d (4-Me/3-OMe) | 2a (H/Et) | 78 (3da) | 99 |
| 18 | 1f (H/H) | 2a (H/Et) | 70 (3fa) | 99 |
| 19 | 1g (4-tBu/H) | 2a (H/Et) | 80 (3ga) | 99 |
| 20 | 1h (4-OMe/H) | 2a (H/Et) | 70 (3ha) | 94 |
| 21 | 1i (4-CF3/H) | 2a (H/Et) | 66 (3ia) | 99 |
| 22 | 1j (3,4-(CH)4/H) | 2a (H/Et) | 80 (3ja) | 94 |
| 23 | 1k (4-Cl/H) | 2a (H/Et) | 72 (3ka) | 98 |
| 24 | 1k (4-Cl/H) | 2e (5-OMe/Et) | 75 (3ke) | 99 |
| 25 | 1k (4-Cl/H) | 2h (5-Cl/Et) | 90 (3kh) | 98 |
| 26 | 1b (4-Me/4-Me) | 2e (5-OMe/Et) | 85 (3be) | 98 |
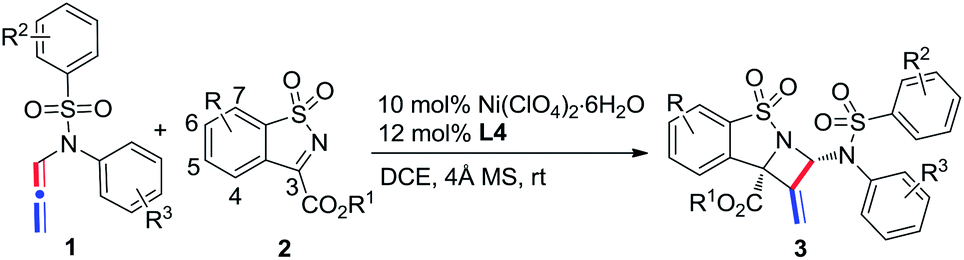
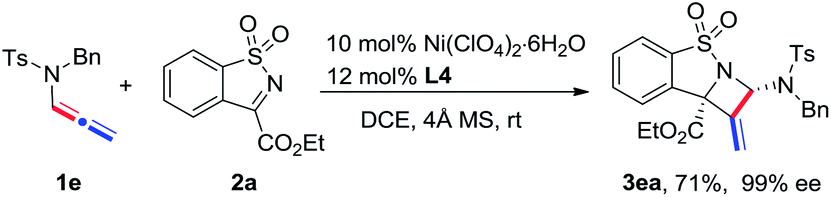
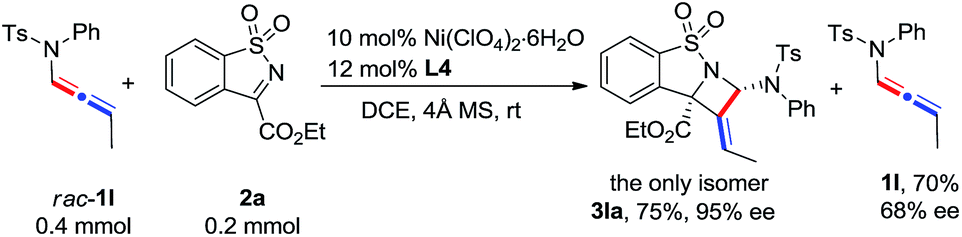
The scope of N-allenamide 4 was also evaluated. By varying the aryl substituents and the sulfonyl groups, a number of substituted N-allenamides were tested and afforded the expected products in good yields and with excellent ee values (entries 15–26). It is worth noting that the reaction of N-benzyl substrate 1e also produced the desired azetidine 3ea in 71% yield and 99% ee (eqn (1)). Moreover, a racemic 3-substituted N-allenamide 1l was also used in this reaction to give the desired product 3la in 75% yield and 95% ee (eqn (2)). Interestingly, a kinetic resolution process of N-allenamide 1l was observed and the remaining starting material was isolated in 70% yield and 68% ee, which represents a promising route to chiral N-allenamide.13 In addition, N-allenyl pyrrolidinone 4a and oxazolidinone 4b were also tested as substrates in this transformation, however, acrylaldehyde 5a was isolated as the major product instead of the desired cycloaddition adduct (Scheme 2). Although a poor yield of 5a was obtained for the reaction of 4a, the yield and ee were both remarkably improved by employing N-oxazolidinone 4b as the substrate. Several other α-ketiminoesters were then examined in the reactions with 4b to afford the corresponding acrylaldehydes 5b–5e in acceptable yields and with good to excellent enantioselectivities.
 | ||
| Scheme 2 Reaction of N-allenyl pyrrolidinone 4a and oxazolidinone 4b with α-ketiminoesters. | ||
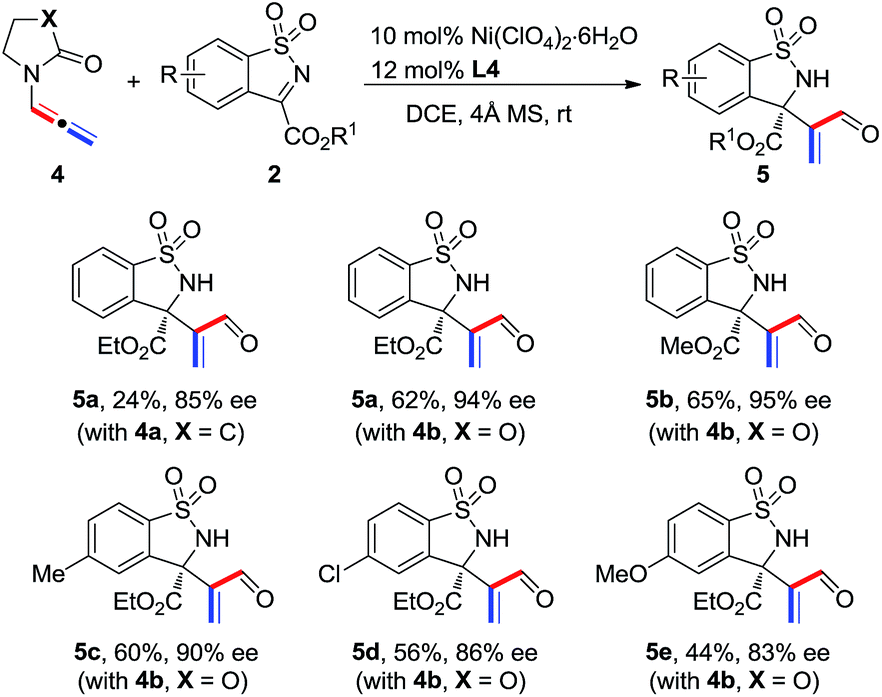
Cyclic six-membered N-sulfonyl α-ketimines were also examined as substrates in this reaction. The desired [2 + 2] adducts 7a and 7b were afforded with good yields and enantioselectivities (Scheme 3). In the case of a naphthyl-ketimine substrate, the pure adduct could not be obtained; however, acrylaldehyde 8c was isolated in 63% yield and with 82% ee by treating the mixture with 10 mol% TsOH after the reaction.
 | ||
| Scheme 3 Reaction of N-allenamide 1a with cyclic six-membered N-sulfonylketimines 6. | ||
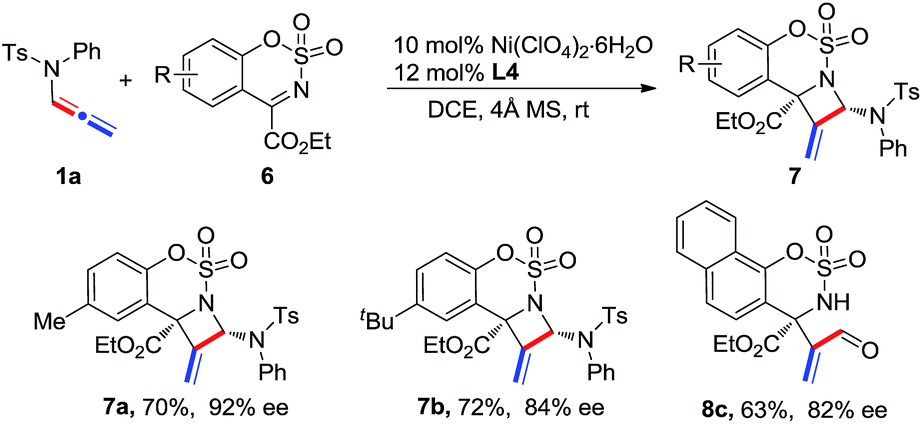
A gram-scale reaction of 1a with 2a was carried out to afford product 3aa in 75% yield and 99% ee, showing good reliability of the present process (Scheme 4). Synthetic transformations of 3aa were then performed. A Pd/C-catalyzed hydrogenation of 3aa afforded saturated azetidine 9 as a single isomer in 76% yield and 97% ee. Reduction of 3aa in the presence of LiAlH4 led to homoallylic alcohol 10 in 90% yield and 99% ee. TsOH-catalyzed hydrolysis of 3aa afforded acrylaldehyde 5a in 80% yield without any loss of enantioselectivity.
 | ||
| Scheme 4 Gram-scale reaction and synthetic transformations of product 3aa. a Conditions are identical to those in entry 4 in Table 1; b Pd/C (10 mol%), H2 (1 atm) in MeOH (0.1 M) at 25 °C for 1 h; c TsOH·H2O (10 mol%) in DCE (0.2 M) at 60 °C for 3 h; d LiAlH4 (2.0 equiv.) in THF (0.1 M) at 25 °C for 1 h. | ||
A tentative stepwise mechanism that accounts for the [2 + 2] reaction is proposed in Scheme 5. α-Ketiminoester 2 is firstly activated by the chiral nickel catalyst in a 1,4-binding coordination fashion to form intermediate A. Subsequent enantioselective nucleophilic addition of N-allenamide 1 or 4 to A generates intermediate B. The [2 + 2] product 3 was then regioselectively formed by the intramolecular cyclization of the amide with iminium (path a) and the catalyst was released simultaneously. The formation of acrylaldehyde 5 instead of the [2 + 2] adduct is probably owing to the hydrolysis of intermediate B when employing N-allenyl oxazolidinone 4b as the substrate (path b).
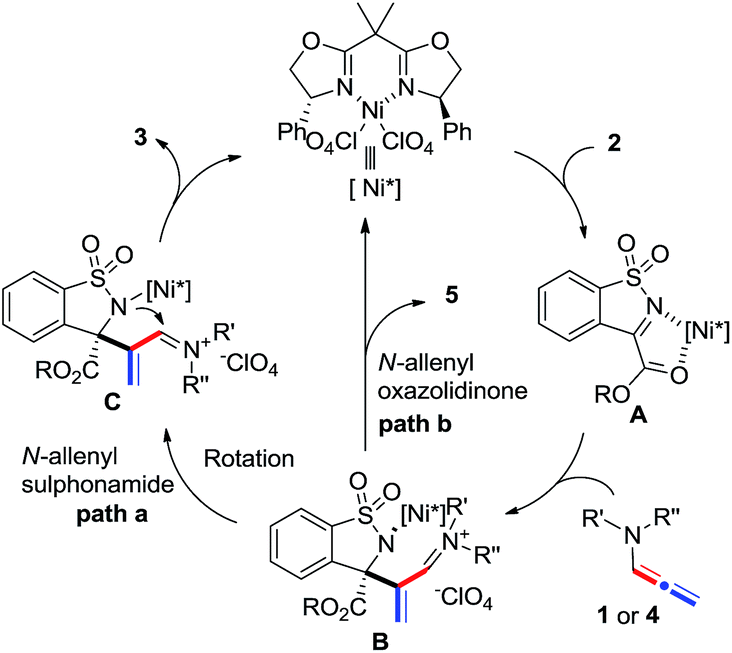 | ||
| Scheme 5 Proposed reaction pathway. | ||
Conclusions
In summary, we have developed a novel catalytic enantioselective [2 + 2] cycloaddition reaction of N-allenamides with cyclic N-sulfonyl α-ketiminoesters, which delivered a range of polysubstituted chiral azetidines bearing quaternary stereocenters as valuable bioactive compounds in good yields and with excellent enantioselectivities. The complex of Ni(ClO4)2 with commercially available chiral bisoxazoline ligands was utilized for the first time as a chiral catalyst in the cycloaddition reaction of N-allenamides. Broad substrate scope was observed for allenamides and cyclic N-sulfonylketimines. It is noteworthy that acrylaldehyde products were obtained when employing N-allenyl oxazolidinones as substrates.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21372202, 21402175, 21522207, and 21502169) and the Natural Science Foundation of Zhejiang Province (LR14B020001 and LQ15B020003). Y.-X. Jia dedicates this work to Professor Qi-Lin Zhou on the occasion of his 60th birthday.References
- For reviews: (a) T. Lu, Z. Lu, Z.-X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862 CrossRef CAS PubMed; (b) F. López and J. L. Mascareñas, Chem. Soc. Rev., 2014, 43, 2904 RSC; (c) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783 RSC; (d) L. Manoni and M. Bandini, Eur. J. Org. Chem., 2016, 19, 3135 CrossRef.
- For selected examples of the transformations of N-allenamides, see: (a) J. Huang and R. P. Hsung, J. Am. Chem. Soc., 2005, 127, 50 CrossRef CAS PubMed; (b) S. Montserrat, H. Faustino, A. Lledys, J. L. Mascareñas, F. Lypez and G. Ujaque, Chem.–Eur. J., 2013, 19, 15248 CrossRef CAS PubMed; (c) N. H. Slater, N. J. Brown, M. R. J. Elsegood and M. C. Kimber, Org. Lett., 2014, 16, 4606 CrossRef CAS PubMed; (d) M. Jia, G. Cera, D. Perrotta, M. Monari and M. Bandini, Chem.–Eur. J., 2014, 20, 9875 CrossRef CAS PubMed; (e) C. Romano, M. Jia, M. Monari, E. Manoni and M. Bandini, Angew. Chem., Int. Ed., 2014, 53, 13854 CrossRef CAS PubMed; (f) L. Rocchigiani, M. Jia, M. Bandini and A. Macchioni, ACS Catal., 2015, 5, 3911 CrossRef CAS; (g) J. Fernández-Casado, R. Nelson, J. L. Mascareñas and F. López, Chem. Commun., 2016, 52, 2909 RSC; (h) M. M. Mastandrea, N. Mellonie, P. Giacinto, A. Collado, S. P. Nolan, G. P. Miscione, A. Bottoni and M. Bandini, Angew. Chem., Int. Ed., 2015, 54, 14885 CrossRef CAS PubMed; (i) X. Yang and F. D. Toste, Chem. Sci., 2016, 7, 2653 RSC; (j) A. Ballesteros, P. Morán-Poladura and J. M. González, Chem. Commun., 2016, 52, 2905 RSC.
- For selected examples of racemic cycloaddition reactions of N-allenamides, see: (a) H. Faustino, F. López, L. Castedo and J. L. Mascareñas, Chem. Sci., 2011, 2, 633 RSC; (b) H. Faustino, P. Bernal, L. Castedo, F. López and J. L. Mascareñas, Adv. Synth. Catal., 2012, 354, 1658 CrossRef CAS; (c) P. Bernal-Albert, H. Faustino, A. Gimeno, G. A. Sensio, J. L. Mascareñas and F. López, Org. Lett., 2014, 16, 6196 CrossRef CAS PubMed; (d) H. Faustino, I. Varela, J. L. Mascareñas and F. López, Chem. Sci., 2015, 6, 2903 RSC.
- (a) J. Francos, F. Grande-Carmona, H. Faustino, J. Lglesias-Sigüenza, E. Díez, I. Alonso, R. Fernández, J. M. Lassaletta, F. López and J. L. Mascareñas, J. Am. Chem. Soc., 2012, 134, 14322 CrossRef CAS PubMed; (b) H. Faustino, I. Alonso, J. Mascareñas and F. López, Angew. Chem., Int. Ed., 2013, 52, 6526 CrossRef CAS PubMed.
- (a) S. Suárez-Pantiga, C. Hernández-Díaz, E. Rubio and J. M. González, Angew. Chem., Int. Ed., 2012, 51, 11552 CrossRef PubMed; (b) M. Jia, M. Monari, Q.-Q. Yang and M. Bandini, Chem. Commun., 2015, 51, 2320 RSC; (c) Y. Wang, P. Zhang, Y. Liu, F. Xia and J. Zhang, Chem. Sci., 2015, 6, 5564 RSC.
- (a) W. Zhou, X.-X. Li, G.-H. Li, Y. Wu and Z. Chen, Chem. Commun., 2013, 49, 3552 RSC; (b) Y. Wang, P. Zhang, D. Qian and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14849 CrossRef CAS PubMed.
- For a racemic reaction: E. López, J. González and L. A. López, Adv. Synth. Catal., 2016, 358, 1428 CrossRef.
- For reviews, see: (a) C. R. Pitts and T. Lectka, Chem. Rev., 2014, 114, 7930 CrossRef CAS PubMed; (b) C.-K. Pei and M. Shi, Chem.–Eur. J., 2012, 18, 6712 CrossRef CAS PubMed; (c) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437 CrossRef CAS PubMed; (d) B. Alcaide, P. Almendros and C. Aragoncillo, Curr. Opin. Drug Discovery Dev., 2010, 13, 685 CAS.
- For catalytic [2 + 2] reactions toward azetidines or β-lactams, see: (a) A. E. Taggi, A. M. Hafez, H. Wack, B. Young, D. Ferraris and T. Lectka, J. Am. Chem. Soc., 2002, 124, 6626 CrossRef CAS PubMed; (b) M. M.-C. Lo and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 4572 CrossRef CAS PubMed; (c) M.-C. Ye, J. Zhou and Y. Tang, J. Org. Chem., 2006, 71, 3576 CrossRef CAS PubMed; (d) Y.-R. Zhang, L. He, X. Wu, P.-L. Shao and S. Ye, Org. Lett., 2008, 10, 277 CrossRef CAS PubMed; (e) J.-B. Denis, G. Masson, P. Retailleau and J. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5356 CrossRef CAS PubMed; (f) S. Takizawa, F. A. Arteaga, Y. Yoshida, M. Suzuki and H. Sasai, Org. Lett., 2013, 15, 4142 CrossRef CAS PubMed; (g) E. Schaumann and H. Mrotzek, Tetrahedron, 1979, 35, 1965 CrossRef; (h) G.-L. Zhao, J.-W. Huang and M. Shi, Org. Lett., 2003, 5, 4737 CrossRef CAS PubMed; (i) T. Akiyama, K. Daidouji and K. Fuchibe, Org. Lett., 2003, 5, 3691 CrossRef CAS PubMed; (j) K. L. Jensen, D. U. Nielsen and T. F. Jamison, Chem.–Eur. J., 2015, 21, 7379 CrossRef CAS PubMed.
- For reviews and selected examples of enantioselective reactions of enamines with imines, see: (a) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599 CrossRef CAS PubMed; (b) R. Matsubara, Y. Nakamura and S. Kobayashi, Angew. Chem., Int. Ed., 2004, 43, 1679 CrossRef CAS PubMed; (c) N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 1080 CrossRef CAS PubMed; (d) M. Terada, K. Machioka and K. Sorimachi, J. Am. Chem. Soc., 2007, 129, 10336 CrossRef CAS PubMed; (e) H. Kiyohara, R. Matsubara and S. Kobayashi, Org. Lett., 2006, 8, 5333 CrossRef CAS PubMed; (f) F. Drouet, C. Lalli, H. Liu, G. Masson and J. Zhu, Org. Lett., 2011, 13, 94 CrossRef CAS PubMed.
- For selected examples of enantioselective transformations of cyclic N-sulfonylketimines, see: (a) T. Nishimura, A. Noishiki, G. C. Tsui and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 5056 CrossRef CAS PubMed; (b) Y. Luo, A. J. Carnell and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 6762 CrossRef CAS PubMed; (c) H. Wang, T. Jiang and M.-H. Xu, J. Am. Chem. Soc., 2013, 135, 971 CrossRef CAS PubMed; (d) C. Jiang, Y. Lu and T. Hayashi, Angew. Chem., Int. Ed., 2014, 53, 9936 CrossRef CAS PubMed; (e) H. B. Hepburn and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 11605 CrossRef CAS PubMed; (f) X. Yin, Y. Zheng, X. Feng, K. Jiang, X.-Z. Wei, N. Gao and Y.-C. Chen, Angew. Chem., Int. Ed., 2014, 53, 6245 CrossRef CAS PubMed; (g) G. Yang and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7540 CrossRef CAS PubMed; (h) R.-R. Liu, D.-J. Wang, L. Wu, B. Xiang, G.-Q. Zhang, J.-R. Gao and Y.-X. Jia, ACS Catal., 2015, 5, 6524 CrossRef CAS.
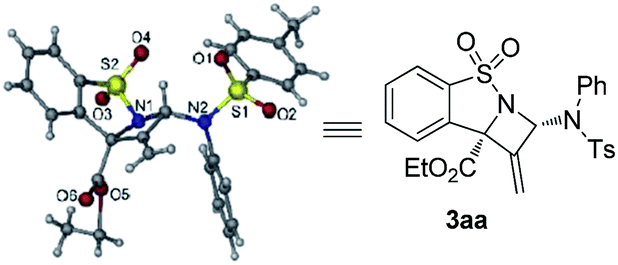
- The absolute configuration of 3aa was determined to be (2S,8S) based on single crystal X-ray analysis. CCDC 1495550 (3aa) contains the supplementary crystallographic data for this paper
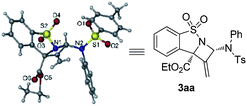 .
. - A. Armstrong and D. P. G. Emmerson, Org. Lett., 2009, 11, 1547 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data for all the new compounds, and chiral HPLC spectra for the products. CCDC 1495550. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05450a |
| This journal is © The Royal Society of Chemistry 2017 |