
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent unprotected peptide macrocyclisation by palladium-mediated cysteine arylation†
Anthony J.
Rojas
 a,
Chi
Zhang
a,
Ekaterina V.
Vinogradova‡
a,
Nathan H.
Buchwald
a,
John
Reilly
b,
Bradley L.
Pentelute
*a and
Stephen L.
Buchwald
*a
a,
Chi
Zhang
a,
Ekaterina V.
Vinogradova‡
a,
Nathan H.
Buchwald
a,
John
Reilly
b,
Bradley L.
Pentelute
*a and
Stephen L.
Buchwald
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: blp@mit.edu; sbuchwal@mit.edu
bNovartis, Novartis Institutes for Biomedical Research Inc., Cambridge, Massachusetts 02139, USA
First published on 24th March 2017
Abstract
Macrocyclic peptides are important therapeutic candidates due to their improved physicochemical properties in comparison to their linear counterparts. Here we detail a method for a divergent macrocyclisation of unprotected peptides by crosslinking two cysteine residues with bis-palladium organometallic reagents. These synthetic intermediates are prepared in a single step from commercially available aryl bis-halides. Two bioactive linear peptides with cysteine residues at i, i + 4 and i, i + 7 positions, respectively, were cyclised to introduce a diverse array of aryl and bi-aryl linkers. These two series of macrocyclic peptides displayed similar linker-dependent lipophilicity, phospholipid affinity, and unique volume of distributions. Additionally, one of the bioactive peptides showed target binding affinity that was predominantly affected by the length of the linker. Collectively, this divergent strategy allowed rapid and convenient access to various aryl linkers, enabling the systematic evaluation of the effect of appending unit on the medicinal properties of macrocyclic peptides.
Introduction
The selective inhibition of protein–protein interactions (PPIs) is a general tactic that may lead to the development of therapeutics for targets once thought “undruggable.”1 One approach to achieve this is the use of peptide inhibitors, which feature high potency, selectivity, and often have low toxicity.2–4 Linear peptides, however, are readily degraded by proteases, exhibit low cell-permeability, and often adopt disordered conformations that affect their target binding affinity.5 Peptide macrocyclisation has emerged as a compelling strategy to overcome these issues.6–9 Compared to their linear counterparts, macrocyclic peptides often display enhanced binding affinity, resistance to protease degradation, and in certain cases, readily enter cells.4,10 However, few systematic studies of the pharmacokinetic and physicochemical properties (e.g. lipophilicity and phospholipid affinity) have been reported.Numerous synthetic tools for peptide macrocyclisation have been developed in recent years. One notable strategy is the crosslinking of two amino acid side chains. Classic techniques utilise lysine and glutamic acid/aspartic acid residues for lactam formation.11 More recently, these techniques have been augmented by protocols involving ring-closing metathesis,12 cycloadditions,13,14 oxime formation,15 alkylation,16 thiol–ene reaction,17 arylation chemistry by nucleophilic aromatic substitution (SNAr),18,19 and metal-mediated reactions.20 Many of these macrocyclisation protocols rely on unnatural amino acids requiring multistep synthesis to diversify their structures. Few methods allow for modular tuning of the linker structure without altering the amino acid sequences.21–24 Previous studies have indicated that the nature of the linker strongly influence the stability, structure, binding affinity, and cell permeability of macrocyclic peptides.18,19,21 However, it is unclear how linkers influence the pharmacokinetic properties such as lipophilicity, phospholipid affinity, volume of distribution, and Human Serum Albumin (HSA) binding profiles. It remains uncertain if these physicochemical properties can be modulated, generally, based on linker design or if their impact is peptide sequence dependent. Understanding the effect of linkers on these parameters will aid in assessing the “drug likeness” of macrocyclic peptides and facilitate the development of future therapeutics.
Recently, we have described a palladium-mediated method for cysteine arylation (Fig. 1A), using biaryldialkylphosphine-based complexes of type LPd(Ar)(X) (X = Cl, Br, OTf; L = ligand) as arylating reagents.25 Preliminary application to peptide macrocyclisation was demonstrated using a 4,4′-benzophenone-linked bis-palladium reagent along with a few others, resulting in effective crosslinking of two cysteine residues at the i, i + 4 positions of an unprotected peptide.25 Here we report expanding this method to synthesis of aryl linkers of varying length, rigidity, and electronic properties (Fig. 1B) and a systematic study of the effect of linkers on the lipophilicity, HSA binding, phospholipid affinity, and target binding affinity of these macrocyclic peptides. In particular, a series of bis-palladium reagents were readily prepared from commercially available aryl bis-halides. These air-stable palladium reagents were used to macrocyclise two bioactive peptides: a peptide (NYAD-1) that binds the HIV-1 C-terminal capsid protein (C-CA) with cysteine residues at the i, i + 4 positions and a p53/MDM2 inhibitor analogue based on the p53 protein with cysteine residues at the i, i + 7 positions.10,26,27 These two model systems are employed in peptide macrocyclisation studies and would let us determine if the resulting physicochemical trends were dependent on peptide sequences or crosslinking positions. The generality of this approach should allow for the further development of therapeutic reagents that can be finely tuned to meet the challenges during the development of peptide therapeutics.
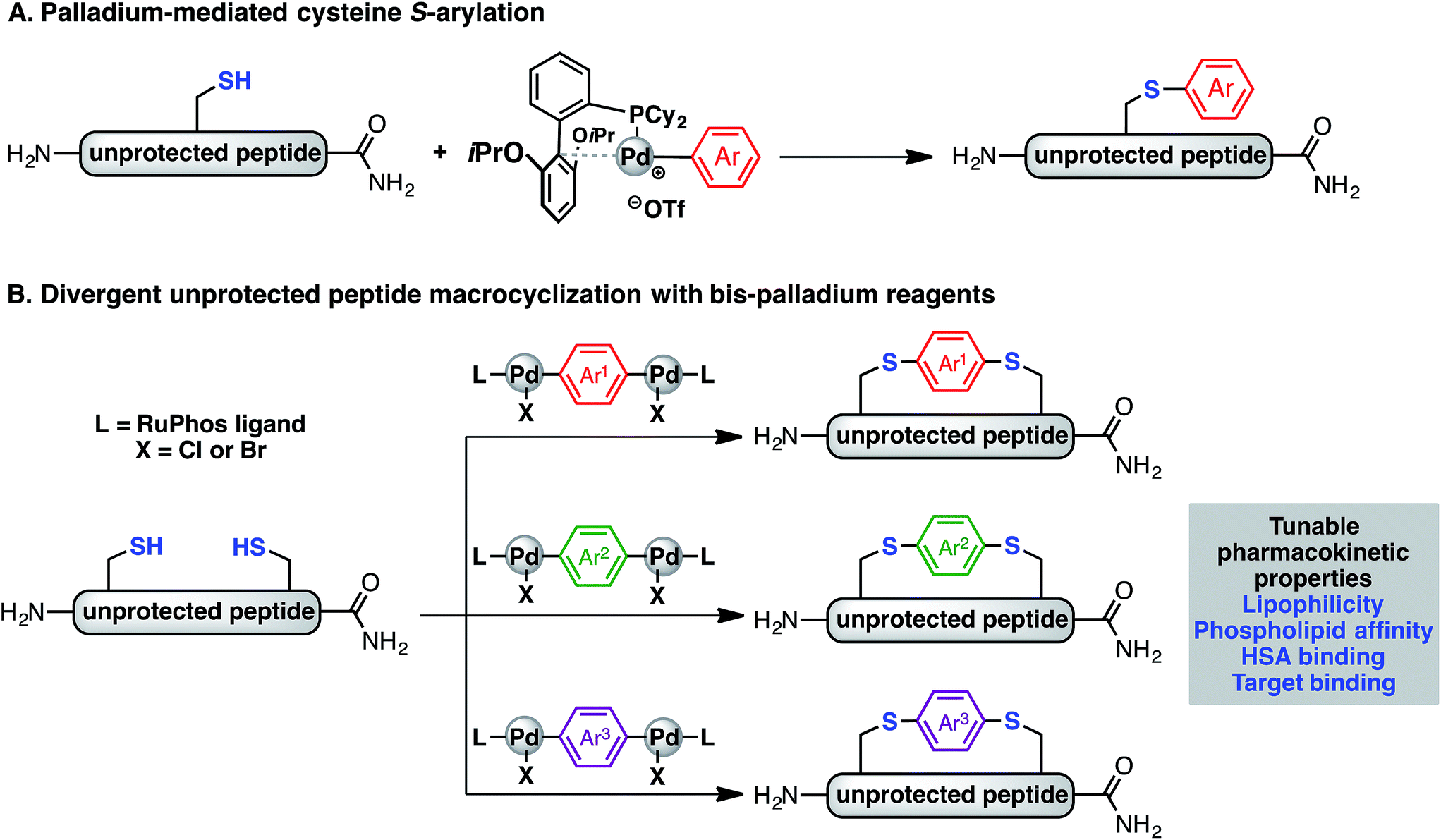 | ||
| Fig. 1 S-Arylation and macrocyclisation of unprotected peptides. This work expanded the palladium-mediated S-arylation chemistry (A) to peptide macrocyclization (B), providing a diverse array of aryl linkers with the capability to tune and manipulate the physiochemical properties of the resulting macrocyclic peptides. | ||
Reported isolated yields are an average of two runs after purification. Oxidative addition complexes are air-stable and readily characterized by solution 1H, 31P, and 19F NMR (where applicable), elemental analysis, FT-IR (see ESI† for characterization).
Results and discussion
Synthesis of bis-palladium oxidative addition complexes
To initiate our study, a series of bis-palladium complexes (OA-Y, Table 1) were synthesized as macrocyclisation reagents. 2-Dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl (RuPhos) was selected as the ligand for palladium, in combination with a series of aryl, biaryl, and biaryl ether dihalides, eight oxidative addition complexes were synthesized (OA-A through OA-I). These were chosen to probe the effects of linker length, rigidity, and degree of fluorination on the physicochemical properties of the macrocyclised peptide.
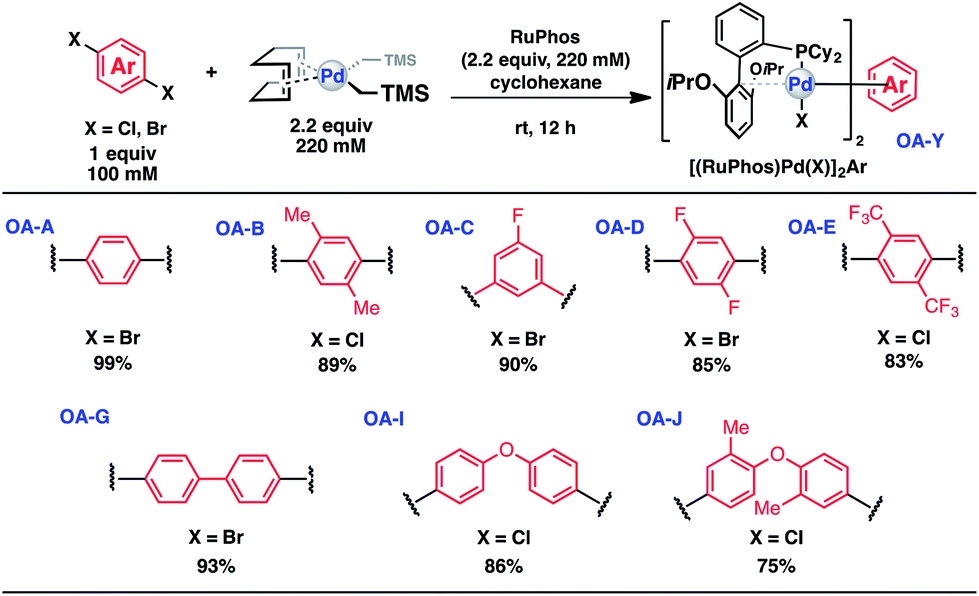
|
As shown in Table 1, the bis-palladium macrocyclisation reagents were prepared in good to excellent yields, regardless of the substitution pattern of the aryl dihalide. These oxidative addition complexes could be stored under air at 4 °C for extended periods without loss of reactivity, as demonstrated by similar reaction profiles of a freshly prepared sample and a sample stored in a freezer at 4 °C for more than two years (see ESI† for details).
Synthesis of macrocyclic peptides
Using a combination of batch and rapid flow solid phase peptide synthesis (SPPS),28 peptides P1 and P2 were synthesized on resin, cleaved, and later purified by reverse-phase HPLC to afford the starting material for the subsequent macrocyclisation reactions (see ESI† for details). Peptide P1 is a variant of a known inhibitor of the HIV-1 C-terminal capsid protein (C-CA)27 and peptide P2 is a mutant of a peptide that disrupts p53/MDM2 interaction based on the p53 protein.10 Cysteine residues were placed at i, i + 4 and i, i + 7 positions for P1 and P2, respectively. As such, these two systems will allow us to probe whether linkers will have the same effect when the peptide sequence and the position of the crosslinking is varied.Fine-tuning of the reaction conditions led to an optimised protocol for peptide macrocyclisation (Fig. 2, top). Exposure of the purified peptides P1 and P2 to these conditions afforded macrocyclisation products in good to excellent yields (70–99%, Fig. 2) as determined by LC-MS analysis of the crude reaction mixtures (see ESI† for details). As shown by a representative LC-MS trace, the crude reaction mixtures generally contained only desired macrocyclic product along with a few by-products derived from the macrocyclisation reagent (see ESI† for full characterization of each purified macrocyclic peptide). Upon completion of the reaction, the crude reaction mixture was treated with 3-thiopropionic acid,25,29 subjected to centrifugation, and the supernatant liquid was lyophilized and subsequently purified by reverse-phase HPLC. ICP-MS analysis of the purified peptides showed that more than 99% of palladium was removed using this purification protocol. Importantly, dimerization of two peptides was not observed. Side reactions whereby two peptides form dimers are common complications for two-component macrocyclisation reactions, but were not found here.6 The perfluorinated macrocyclic peptides P1-F, P2-F, P1-H, and P2-H (Table 3) were synthesized by previously reported SNAr chemistry.18
 | ||
| Fig. 2 Macrocyclisation of two unprotected peptides with cysteine residues at i, i + 4 (P1) and i, i + 7 (P2) positions proceeds with high efficiency. Peptide sequences are shown with one-letter code. Yields determined by integrating total ion current (TIC) chromatograms from HPLC-MS analysis of the crude reactions and represent an average of two runs (see ESI† for details). | ||
Physicochemical properties of macrocyclic peptides
After purifying the conjugates, we sought to investigate how the linking units affect the lipophilicity, human serum albumin (HSA) binding profiles, and if possible, the phospholipid affinity and volume of distribution for these macrocyclic peptides.Lipophilicity is a critical parameter for drug formulation and which determines whether a compound will have the tendency to dissolve into lipid or aqueous environments.18 This is an important consideration for pharmaceutical discovery and part of Lipinski's ‘Rule of 5’.30 For neutral small molecules, lipophilicity is often measured as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P where P is the partitioning coefficient between octanol and water. For compounds potentially exhibiting zwitterionic character, a more appropriate measure is logD where D is the distribution coefficient, which measures the overall distribution of both ionised and neutral forms of the compound when the aqueous phase is buffered to a particular pH.20 We sought to determine whether this parameter could be manipulated for macrocyclic peptides by varying just the aryl linkers. Being able to successfully modulate this parameter without disrupting other activity profiles could be a useful tactic for peptide drug development.
P where P is the partitioning coefficient between octanol and water. For compounds potentially exhibiting zwitterionic character, a more appropriate measure is logD where D is the distribution coefficient, which measures the overall distribution of both ionised and neutral forms of the compound when the aqueous phase is buffered to a particular pH.20 We sought to determine whether this parameter could be manipulated for macrocyclic peptides by varying just the aryl linkers. Being able to successfully modulate this parameter without disrupting other activity profiles could be a useful tactic for peptide drug development.
Using known methods,31 retention times of the HPLC profiles of cyclised and linear variants of peptides P1 and P2 were measured and correlated to octanol–water distribution in solutions buffered to pH 7.4. Retention times, and mass spectrometric data were correlated to literature values for the logD of other compounds and then used to assign numerical values of lipophilicity to these peptides.
Fig. 3A shows how these logD7.4 values can be manipulated between 1.8 for uncyclised P1-K to 2.7 for P1-C and P1-J. Interestingly, P1-G and P1-H exhibit the same logD7.4 value of 2.0, indicating that incorporation of fluorine substituents does not necessarily lead to increased lipophilicity. Peptides P1-B and P1-E (methyl substituted and p-trifluoromethyl phenyl linkers), however, do differ slightly with logD7.4 values of 2.3 and 2.4, respectively.
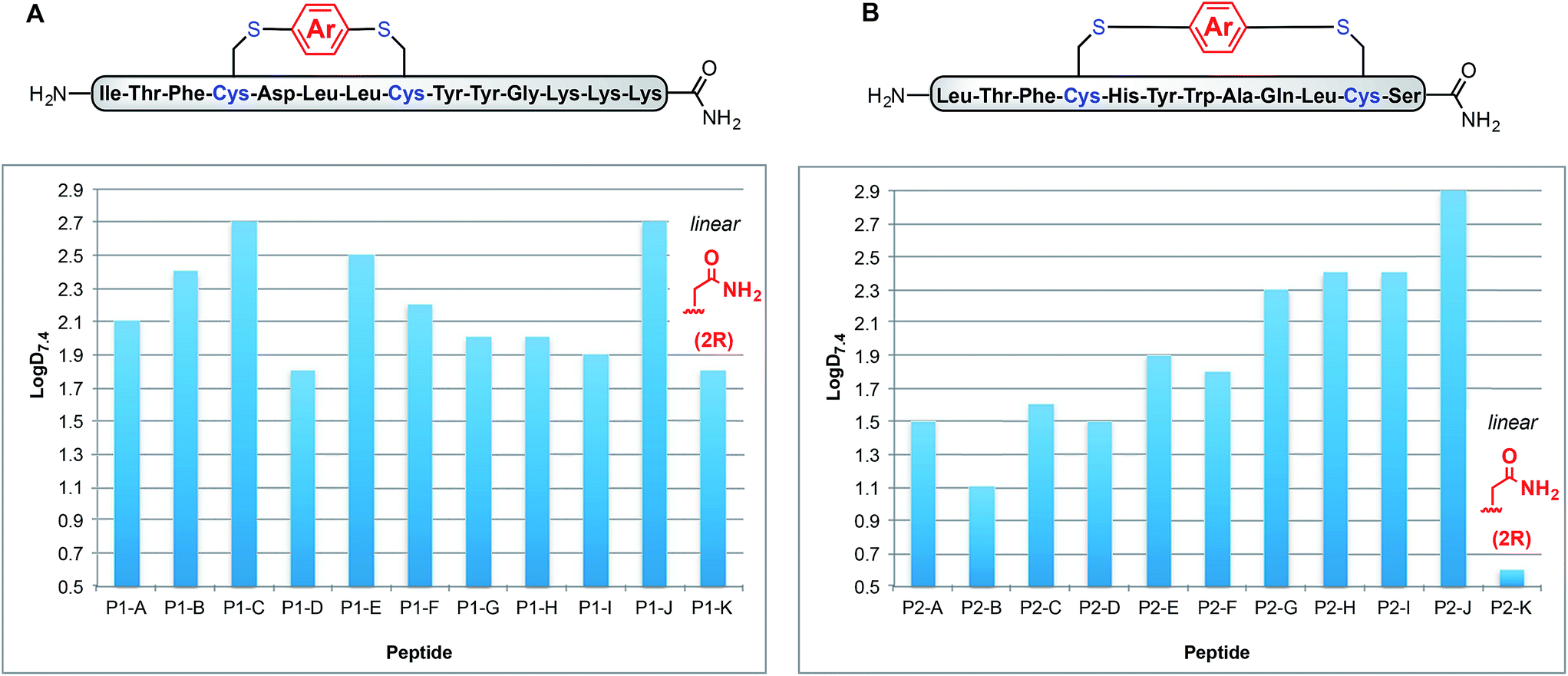 | ||
| Fig. 3 Lipophilicity measurements of two series of macrocyclic peptides show dependency on relationship between linker structure and amino acid sequence. LogD at pH 7.4, a measurement of lipophilicity for amphiphilic compounds, as measured by dissolving peptides in DMSO followed by rapid LC-UV-MS correlated to water/octanol partitioning for each macrocyclic peptide. Lipophilicity shows dependence on both crosslinker and amino acid composition. | ||
To determine whether the measured lipophilicity depends on peptide sequence and crosslinking positions, we also cyclised a variant of the P53/MDM2 inhibitor, P2, and measured lipophilicity values using the same methods (Fig. 3B).
Notably, the cyclisation of peptide P2 significantly increased the lipophilicity values from 0.6 for uncyclised P2-K to 2.9 for P2-J. The methylated diaryl ether cross linker again yielded the highest logD values for both peptides. However, the change in logD does not solely depend on the linker, rather a combination of linker and peptide. For instance, P1-C has the same logD as P1-J, but for P2-C the lipophilicity is intermediate. Another notable example is for linker B, peptides P1-B and P2-B display contrasting lipophilicities.
Another parameter governing therapeutic utility is human serum albumin (HSA) binding. HSA, a prominent protein in plasma, is capable of binding many different classes of ligands. This binding is partially responsible for the pharmacokinetic behaviour of many drugs and can strongly influence their efficacy and delivery.32 By using a stationary phase comprising immobilized HSA, the propensity of a molecule to bind to this protein can be determined by HPLC methods.33 Although minor deviations in HSA binding were observed, all macrocyclised P1 and P2 variants were found to exhibit high HSA binding percentage, regardless of the aryl crosslinker (see ESI† for details).
Measurement of gradient HPLC retention times on immobilized artificial membrane columns allows for phospholipid binding analysis (CHI IAM7.4). In conjunction with the lipophilicity results, modelling of volume of distribution (VD) can be performed on the modified peptides.34 VD relates administered doses of drugs to their distribution between tissues and plasma in vivo. Knowledge of this parameter, along with clearance times, allows for the duration of drug exposure to be estimated, making VD another key pharmacokinetic parameter in the development of new therapeutics.
The results presented in Table 2 are for peptides based on the p53/MDM2 inhibitor P2. Undesirable physicochemical properties can lead to a promiscuous profile and increased adverse effects. For a given lipophilicity, basic compounds tend to be less selective.35 Phospholipid affinity, a measure of this property, was therefore determined for the macrocyclised peptides. Four P2 peptides P2-G-J demonstrate high phospholipid affinity and considerably higher predicted volumes of distribution and would be expected to have a higher promiscuity index than other P2 peptides. This is an example of when the HPLC biomimetic methodologies can offer a perspective in structure activity relationships for pharmaceuticals. VD values could not be obtained for the P1 variants due to the high levels of binding to the immobilized artificial membrane column, which could be indicative of large tissue to plasma ratios in vivo.
VD values34
| Peptide | CHI IAM pH 7.4 | logVD |
|---|---|---|
| P2-A | 47.3 | 0.46 |
| P2-B | 36.7 | −0.26 |
| P2-C | 48.2 | 0.49 |
| P2-D | 46.1 | 0.35 |
| P2-E | 49.9 | 0.59 |
| P2-F | 49.2 | 0.59 |
| P2-G | 64.5 | 2.37 |
| P2-H | 59.2 | 1.54 |
| P2-I | 61.8 | 1.90 |
| P2-J | 70.0 | 3.42 |
| P2-K | 32.4 | 0.13 |
Binding of P1 variants to HIV-1 C-terminal capsid protein (C-CA)
Tuning the physiochemical properties without diminishing the activity of a peptide is valuable for developing peptide therapeutics. We chose P1 variants to study how the linkers affect the binding of these macrocyclic peptides to C-CA. Biotinylated P1 variants were prepared for immobilization on the streptavidin biosensors and were sampled against serially diluted concentrations of C-CA using BioLayer Interferometer (see ESI† for details). Two control peptides with scrambled sequences, P4-A and P4-K, were also measured to ensure that the observed binding events were not due to nonspecific binding of the aryl linkers (Table 3).| Peptide | Cross-linker | K D (μM) |
|---|---|---|
| P1-A |
|
9.2 ± 0.4 |
| P1-B |
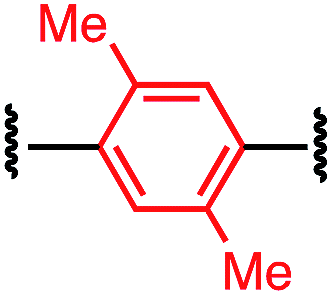
|
9.7 ± 0.4 |
| P1-C |
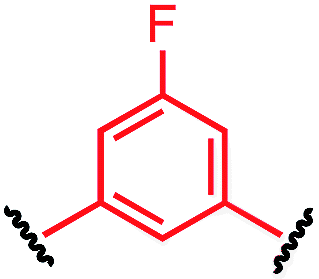
|
21.0 ± 2.4 |
| P1-D |
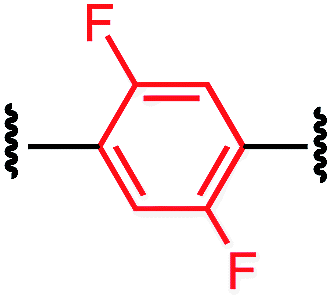
|
9.5 ± 0.5 |
| P1-E |
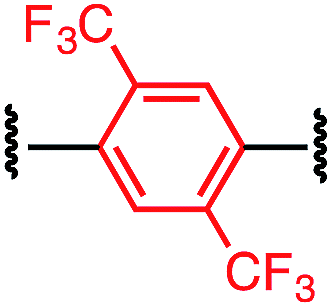
|
12.0 ± 0.8 |
| P1-F |
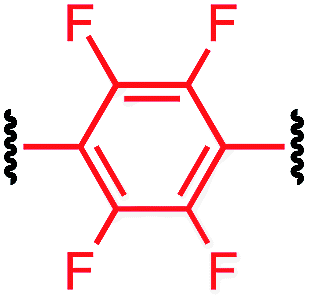
|
12.0 ± 0.6 |
| P1-G |
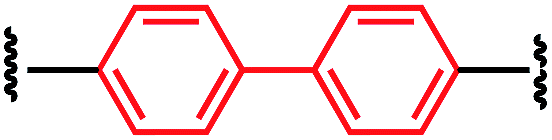
|
61 ± 35 |
| P1-H |

|
>100 |
| P1-I |
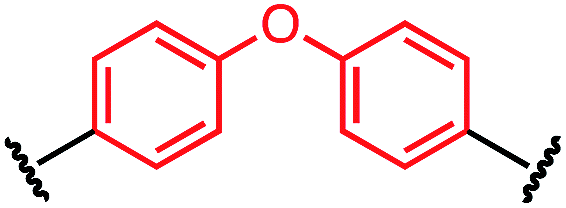
|
56 ± 21 |
| P1-J |
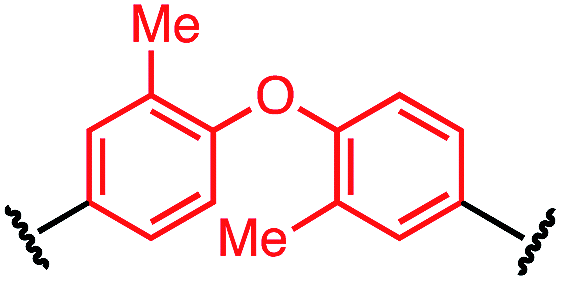
|
>100 |
| P1-K |
|
5.5 ± 0.6 |
| P4-A |
|
>100 |
| P4-K |

|
>100 |
The linear control peptide (P1-K) showed an equilibrium dissociation constant (KD) of 5.5 ± 0.6 μM in line with previous literature reports.27,36 All of the mono-aryl linkers with 1,4-substitutions (P1-B, P1-D, P1-E, and P1-F) had KD comparable to the linear control. However, variant P1-C with 1,3-substitution showed slightly lower binding affinity (KD = 21.0 ± 2.4 μM). Moreover, macrocyclic peptides with the biphenyl and biaryl ether crosslinkers all showed little to no binding affinity to C-CA. Taken together, these results indicated that the length of the crosslinker impact binding to the target. It is worth noting that, although differing greatly in their lipophilicity (Fig. 3), the 1,4-substituted mono-aryl linkers (P1-B, P1-D, P1-E, and P1-F) did not exhibit significant differences in binding affinity C-CA.
Conclusions
We have shown that palladium-mediated S-arylation chemistry is a “shovel ready” technology for rapid and straightforward divergent macrocyclisation of peptides; linkers, sequences, and crosslinking positions can be varied. Preparation of bis-palladium crosslinking reagents is straightforward and one-step from commercially available aryl dihalides. The linker had a significant affect on the physicochemical properties and in some cases the target binding affinity was maintained.The design and eventual use of macrocyclic peptides in the clinic is uncharted, especially when compared to small molecules. Here we show by combining powerful chemistry, and simple chromatographic techniques to measure the physicochemical properties of each variant we could rank order peptides on basis of protein binding, lipophilicity, phospholipid affinity, and predicted volume of distribution. These measurements may find application in medicinal chemistry by providing a means to filter out peptides with undesirable physicochemical characteristics such as high phospholipid affinity at an early stage within the drug discovery process.
Acknowledgements
Financial support for this work was provided by the National Institutes of Health (GM-46059), the MIT start-up fund (B. L. P.), a Damon Runyon Cancer Research Foundation Award (B. L. P.) and the Sontag Foundation Distinguished Scientist Award (B. L. P.). A. J. R. gratefully acknowledges support from the National Science Foundation Graduate Fellowship. C. Z. is the recipient of the George Büchi Research Fellowship, the Koch Graduate Fellowship in Cancer Research of MIT, and the Bristol-Myers Squibb Graduate Fellowship in Synthetic Organic Chemistry. The authors acknowledge the Biological Instrument Facility of MIT for providing the Octet BioLayer Interferometry System (NIH S10OD016326) and the Varian 300 spectrometer used for portions of this work was purchased with funds from NSF (grant CHE-9808061). We thank Dr Yuri Tulchinsky and the Center for Environmental Health Sciences for assistance with ICP-MS and Drs Yiming Wang and Michael Pirnot for assistance on the preparation of this manuscript.Notes and references
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS PubMed.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305(80), 1466–1470 CrossRef CAS PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CAS.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- N. S. Robertson and A. G. Jamieson, Rep. Org. Chem., 2015, 5, 65–74 CAS.
- L. K. Henchey, A. L. Jochim and P. S. Arora, Curr. Opin. Chem. Biol., 2008, 12, 692–697 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K.-H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS PubMed.
- A. M. Felix, E. P. Heimer, C. T. Wang, T. J. Lambros, A. Fournier, T. F. Mowles, S. Maines, R. M. Campbell, B. B. Wegrzynski, V. Toome, D. Fry and V. S. Madison, Int. J. Pept. Protein Res., 1988, 32, 441–454 CrossRef CAS PubMed.
- (a) H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS; (b) S. J. Miller and R. H. Grubbs, J. Am. Chem. Soc., 1995, 117, 5855–5856 CrossRef CAS.
- S. A. Kawamoto, A. Coleska, X. Ran, H. Yi, C. Y. Yang and S. Wang, J. Med. Chem., 2012, 55, 1137–1146 CrossRef CAS PubMed.
- M. M. Madden, A. Muppidi, Z. Li, X. Li, J. Chen and Q. Lin, Bioorg. Med. Chem. Lett., 2011, 21, 1472–1475 CrossRef CAS PubMed.
- C. M. Haney, M. T. Loch and W. S. Horne, Chem. Commun., 2011, 47, 10915–10917 RSC.
- F. M. Brunel and P. E. Dawson, Chem. Commun., 2005, 2552–2554 RSC.
- Y. Wang and D. H. C. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- G. Lautrette, F. Touti, H. G. Lee, P. Dai, B. L. Pentelute, G. Lautrette, H. G. Lee, P. Dai and B. L. Pentelute, J. Am. Chem. Soc., 2016, 138, 8340–8343 CrossRef CAS PubMed.
- L. Mendive-Tapia, S. Preciado, J. García, R. Ramón, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Sköld, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvönen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. Degrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed.
- A. Muppidi, K. Doi, C. P. Ramil, H. G. Wang and Q. Lin, Tetrahedron, 2014, 70, 7740–7745 CrossRef CAS PubMed.
- Y. Zou, A. M. Spokoyny, C. Zhang, M. D. Simon, H. Yu, Y.-S. Lin and B. L. Pentelute, Org. Biomol. Chem., 2013, 12, 566–573 Search PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- J. Phan, Z. Li, A. Kasprzak, B. Li, S. Sebti, W. Guida, E. Schönbrunn and J. Chen, J. Biol. Chem., 2010, 285, 2174–2183 CrossRef CAS PubMed.
- H. Zhang, Q. Zhao, S. Bhattacharya, A. A. Waheed, X. Tong, A. Hong, S. Heck, F. Curreli, M. Goger, D. Cowburn, E. O. Freed and A. K. Debnath, J. Mol. Biol., 2008, 378, 565–580 CrossRef CAS PubMed.
- M. D. Simon, P. L. Heider, A. Adamo, A. A. Vinogradov, S. K. Mong, X. Li, T. Berger, R. L. Policarpo, C. Zhang, Y. Zou, X. Liao, A. M. Spokoyny, K. F. Jensen and B. L. Pentelute, ChemBioChem, 2014, 15, 713–720 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Chem. Commun., 2011, 47, 1698–1700 RSC.
- P. J. Lipinski, C. A. Lombardo, F. Dominy and B. W. Feeney, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- J. A. Arnott and S. L. Planey, Expert Opin. Drug Discovery, 2012, 7, 863–875 CrossRef CAS PubMed.
- M. Fasano, S. Curry, E. Terreno, M. Galliano, G. Fanali, P. Narciso, S. Notari and P. Ascenzi, IUBMB Life, 2005, 57, 787–796 CrossRef CAS PubMed.
- J. Reilly, D. Etheridge, B. Everatt, Z. Jiang, C. Aldcroft, P. Wright, I. Clemens, B. Cox, N. J. Press, S. Watson, D. Porter, C. Springer and R. A. Fairhurst, J. Liq. Chromatogr. Relat. Technol., 2011, 34, 317–327 CrossRef CAS.
- F. Hollosy, K. Valko, A. Hersey, S. Nunhuck, G. Keri, C. Bevan, F. Hollósy, K. Valkó, A. Hersey, S. Nunhuck, G. Kéri and C. Bevan, J. Med. Chem., 2006, 49, 6958–6971 CrossRef CAS PubMed.
- E. Lounkine, M. J. Keiser, S. Whitebread, D. Mikhailov, J. Hamon, J. L. Jenkins, P. Lavan, E. Weber, A. K. Doak, S. Côté, B. K. Shoichet and L. Urban, Nature, 2012, 486, 361–367 CAS.
- A. A. Vinogradov, Z. N. Choo, K. A. Totaro and B. L. Pentelute, Org. Lett., 2016, 18, 1226–1229 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc05454d |
| ‡ Present address: Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California, 92037, USA. |
| This journal is © The Royal Society of Chemistry 2017 |