
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of hydride bridges in a high-spin [Fe3(μ-H)3]3+ cluster: reversible H2/CO exchange and Fe–H/B–F bond metathesis†
Kevin J.
Anderton
 a,
Brian J.
Knight
a,
Arnold L.
Rheingold
c,
Khalil A.
Abboud
b,
Ricardo
García-Serres
d and
Leslie J.
Murray
*a
a,
Brian J.
Knight
a,
Arnold L.
Rheingold
c,
Khalil A.
Abboud
b,
Ricardo
García-Serres
d and
Leslie J.
Murray
*a
aCenter for Catalysis, University of Florida, 214 Leigh Hall P.O. Box 117200, Gainesville, FL 32611, USA. E-mail: murray@chem.ufl.edu
bDepartment of Chemistry, University of Florida, 214 Leigh Hall P.O. Box 117200, Gainesville, FL 32611, USA
cDepartment of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, MC 0358, La Jolla, CA 92093-0358, USA
dLaboratoire de Chimie de Biologie des Métaux, UMR 5249, Université Joseph Fourier, Grenoble-1, CNRS-CEA 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France
First published on 11th April 2017
Abstract
The triiron trihydride complex Fe3H3L (1) [where L3− is a tris(β-diketiminate)cyclophanate] reacts with CO and with BF3·OEt2 to afford (FeICO)2FeII(μ3-H)L (2) and Fe3F3L (3), respectively. Variable-temperature and applied-field Mössbauer spectroscopy support the assignment of two high-spin (HS) iron(I) centers and one HS iron(II) ion in 2. Preliminary studies support a CO-induced reductive elimination of H2 from 1, rather than CO trapping a species from an equilibrium mixture. This complex reacts with H2 to regenerate 1 under a dihydrogen atmosphere, which represents a rare example of reversible CO/H2 exchange and the first to occur at high-spin metal centers, as well as the first example of a reversible multielectron redox reaction at a designed high-spin metal cluster. The formation of 3 proceeds through a previously unreported net fluoride-for-hydride substitution, and 3 is surprisingly chemically inert to Si–H bonds and points to an unexpectedly large difference between the Fe–F and Fe–H bonds in this high-spin system.
Introduction
Beginning with Hieber's discovery of H2Fe(CO)4 and HCo(CO)4,1 the synthesis and reactivity studies of transition metal hydrides have remained an active area of chemical research.2 Mononuclear metal hydrides have applications in industry (e.g., hydroformylation), electrocatalysis, and chemical synthesis.3 In addition, polynuclear transition metal hydrides have garnered substantial interest as these species have either been identified as or are proposed to be key intermediates in reactions in biological and chemical systems. For example, hydrides bound to iron centers are known or proposed in the Haber–Bosch4 and Fischer–Tropsch5 processes, dihydrogen oxidation/proton reduction by the nickel–iron and iron–iron hydrogenases,6 and the catalytic cycles for nitrogenases7 and nickel–iron carbon monoxide dehydrogenase.8 Molecular systems designed to model the multimetallic activation of H2 to generate and probe the reactivity of polynuclear metal hydrides generally use strong field donors (e.g., CO or phosphines).9 It remains unclear how parameters such as the metal coordination number and spin state tune reactivity of metal hydrides. This consideration is important in the context of heterogeneous and biological metal hydride clusters in which high- or intermediate-spin metal centers are typically present. The NiFe and FeFe hydrogenases are exceptions to the biological trend as the metal centers are low spin.Among the most important reactions of transition metal hydrides are reductive elimination (re) and oxidative addition (oa) of H2, which are ubiquitous in organometallic chemistry, including such processes as catalytic asymmetric hydrogenation.10 The study of these reactions in polynuclear high-spin iron clusters is of particular interest because of mechanistic questions related to the reduction of N2 to NH3 by the iron molybdenum cofactor in nitrogenase from Azotobacter vinlandii, which has recently been proposed to store reducing equivalents as hydrides that unmask low-valent iron centers upon elimination of H2 to facilitate binding and reduction of N2.7 Relatedly, the chemistry of metal hydrides is also germane to chemical hydrogen storage materials for fuel cell applications, in which repetitive cycling between H2 re and oa are important reactions for discharge and recharging, respectively.11 Ideally, the former would be stimuli-dependent (e.g., reduced pressure, light, exogenous agent) to allow for controlled and on-demand H2 release. A fundamental understanding of this type of reactivity is thus important to ongoing research efforts, even if transition metal hydrides lack sufficient energy density for use in such applications. Exposure to N2, reduced pressure, or visible light irradiation are known to instigate reductive elimination of two hydride donors from transition metal complexes.3c,12 In those cases where N2 is involved, the interplay between N2 binding and H2 loss is unclear. Ideally then, compounds that can cycle between H2 re and oa as a function of a chemical stimulus provide important insight towards tuning the energy demand for discharge and recharge of transition metal hydrides.
We previously reported the synthesis and reactivity of a series of trimetallic trihydride complexes, M3(μ-H)3L (M = FeII, CoII, ZnII), supported by the tris(β-diketiminate)cyclophanate, L3−.13 In those reports, we highlighted the surprisingly specific reactivity of these compounds for CO2. These metal-hydride compounds were unreactive towards other potential substrates, including nitriles, aldehydes, and ketones. Notably, the ZnII analog demonstrated stability even towards methanol and water. In an effort to expand the substrate scope, we report here the reactivity of Fe3H3L (1) with carbon monoxide, which resulted in H2 elimination to generate a formal triiron(I/I/II) compound, (FeICO)2FeII(μ3-H)L, and with BF3·OEt2, which afforded Fe3F3L through a net fluoride-for-hydride substitution. To our knowledge, the H2/CO reactivity demonstrated here is the first example of a reversible multielectron redox reaction occurring at a designed high-spin multimetallic compound. Similar reactivity under a minimal driving force is typically invoked as a hallmark of biological catalysis.
Results and discussion
Reaction of 1 with carbon monoxide (∼1 atm) in THF results in a rapid color change from red to dark yellow-green (broad absorption with λmax = 320 nm, Fig. S14, ESI†) and formation of (FeICO)2FeII(μ3-H)L (2) in quantitative yield by 1H NMR spectroscopy and in good crystalline yield (Scheme 1, 69%). A new resonance corresponding with H2 is also observed in in situ NMR spectra of reaction mixtures, implying H2 re upon reaction of 1 with CO. This formulation of 2 is supported by single-crystal X-ray diffraction (vide infra) and combustion analysis. We also observe a strong IR absorption at 1846 cm−1 in spectra of 2 synthesized from 12CO, which shifts to 1804 cm−1 for the 13CO labelled 2 (2-13CO) (theoretical/observed Δν(12CO–13CO) = 41/42 cm−1).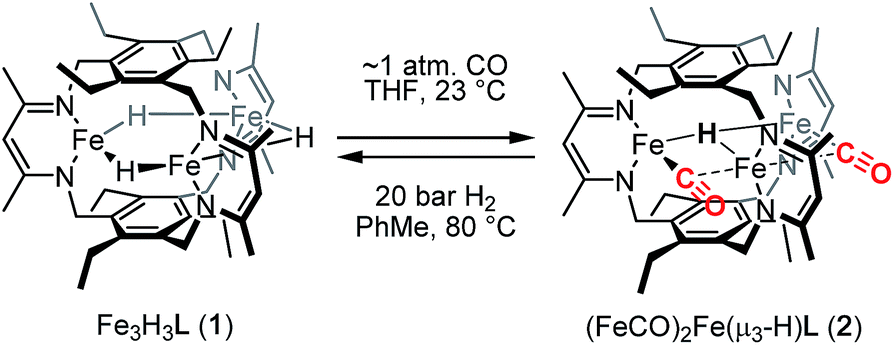 | ||
| Scheme 1 Preparation of (FeCO)2Fe(μ3-H)L (2). | ||
In the solid-state structure of 2, two iron centers (Fe2 and Fe3) are in a distorted trigonal pyramidal geometry (τ4 = 0.75) with each ligated by one semi-bridging CO, one ligand β-diketiminate (or nacnac) arm, and a μ3-hydride (Fig. 1, left). The calculated values for α (0.35 and 0.39) and the Fe2/Fe3–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond angles (163.7(2)° and 162.2(2)°) agree with Curtis' correlation14 for bent semi-bridging CO ligands. However, bent FeI–CO bonds are known for terminal carbonyls (e.g., ∼173° for a (nacnac)FeI(CO)2 complex),15a and the geometric constraints imposed by L3− and μ3-H− likely limit the distance and angular relationships as compared with pseudo-octahedral metal centers. The Fe2/Fe3–NL bond distances for 2 are significantly longer than for monometallic nacnac Fe(I) carbonyls (>2.05 Å vs. 1.97–1.98 Å),15 suggesting differing iron spin states or steric effects. The Fe–C bond distances (1.829(2) and 1.830(2) Å) are within the range of those reported for the (β-diketiminato)iron(I) di- and tri(carbonyl) complexes (1.79–1.87 Å)15 and for [PhTptBu]FeI(CO) (1.789(3) Å)16 but substantially longer than those reported for Peters' trigonal bipyramidal Fe(I)–CO series (1.679–1.769 Å).17 The energy of ν(CO) for 2 is also lower than those reported for terminal FeI–CO species consistent with an interaction with Fe1.
O bond angles (163.7(2)° and 162.2(2)°) agree with Curtis' correlation14 for bent semi-bridging CO ligands. However, bent FeI–CO bonds are known for terminal carbonyls (e.g., ∼173° for a (nacnac)FeI(CO)2 complex),15a and the geometric constraints imposed by L3− and μ3-H− likely limit the distance and angular relationships as compared with pseudo-octahedral metal centers. The Fe2/Fe3–NL bond distances for 2 are significantly longer than for monometallic nacnac Fe(I) carbonyls (>2.05 Å vs. 1.97–1.98 Å),15 suggesting differing iron spin states or steric effects. The Fe–C bond distances (1.829(2) and 1.830(2) Å) are within the range of those reported for the (β-diketiminato)iron(I) di- and tri(carbonyl) complexes (1.79–1.87 Å)15 and for [PhTptBu]FeI(CO) (1.789(3) Å)16 but substantially longer than those reported for Peters' trigonal bipyramidal Fe(I)–CO series (1.679–1.769 Å).17 The energy of ν(CO) for 2 is also lower than those reported for terminal FeI–CO species consistent with an interaction with Fe1.
 | ||
| Fig. 1 Solid-state structure of 2 viewed from the side (left) and top (right). C, N, O, and Fe atoms are depicted as gray, blue, red, and orange 75% thermal ellipsoids, and the μ3-hydride as a black sphere. All other H-atoms as well as solvent molecules are omitted for clarity. | ||
Comparison of the Fe–C and C–O bond distances in 2 with those reported for other complexes featuring the monocarbonyl iron motif demonstrates that 2 is atypical with unusually long C–O bonds given the Fe–C bond lengths (Fig. S16–S17†). We cannot discount libration effects as one reason for the unexpected differences in the bond lengths, although these values are reasonable given the IR data. The (nacnac)Fe(H)(CO) units in 2 represent the second example of a four coordinate FeI bearing only one CO donor, the first being [PhTptBu]FeI(CO).16 The Fe(H)(CO) fragment is known for P- and N-donor compounds,18 hydrogenase model compounds,19 and in metal carbonyl cluster chemistry,9 but is unknown for lower than 5-coordinate iron centers. Fe1 is held within a distorted trigonal bipyramidal coordination environment comprising the μ3-hydride and two N-atoms from the nacnac arm, and two long axial interactions of 2.481(2) Å and 2.545(2) Å to the semi-bridging CO donors.20 The bond metrics suggest minimal direct overlap between the iron centers in 2, which is evident from the formal shortness ratios for the Fe1 contacts (FSR = 1.24 and 1.31).21 Specifically, the Fe2–Fe3 (3.8612(4) Å) and Fe2/Fe3–Fe1 (2.9038(5), 3.0428(5) Å) distances are longer than typically associated with iron–iron bonds. The cyclophanate is distorted based on the bond metrics and angles relative to those of the M3X3L series (M = Mn, Fe, Co, Zn, Cu; X = Br−, Cl−, and H−), which highlights the surprising flexibility of L3−.22 The (μn-H)Fen fragment (n > 2) was previously unknown in the high-spin state (vide infra), and 2 is the first example of an internal μ3-hydride in polyiron chemistry; face-capping μ3-hydrides are well-known in iron carbonyl clusters.23
Reductive elimination of H2 from 1 should afford a triiron complex with 20 d electrons with formal oxidation states of FeI2FeII. This assignment is supported by the zero-field Mössbauer spectrum (Fig. 2), which comprises two different quadrupole doublets with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 area ratio. From the Mössbauer parameters, we assign the doublet with double integration as FeI (δ = 0.66 mm s−1; ΔEQ = 2.60 mm s−1) and corresponding to Fe2 and Fe3, whereas the parameters of the other doublet are characteristic of high-spin FeII (δ = 0.98 mm s−1; ΔEQ = 2.15 mm s−1) and attributed to Fe1. The δ values assigned to Fe2 and Fe3 are comparable to those for the high-spin (β-diketiminate)-supported iron(I) complexes with two tert-butylisocyanides24 or one dinitrogen bound25 (c.f. 0.64 or 0.62, respectively), and distinct from the analogous low-spin iron(I) di- and tri(carbonyl)26 and tris(tert-butylisocyanide) compounds (c.f. 0.12–0.25).24 This comparison suggests that 2 likely contains high-spin FeI centers, which is rare for a CO ligated iron center with tris(pyrazolyl)borate FeI(CO) compounds as the only characterized examples.16,27 Applying a magnetic field of 2 T at 4.8 K resulted in a slight line broadening for the doublet assigned to Fe(II), revealing an internal field of less than 4 T (Fig. S19†). This is indicative of an integer-spin system with a relatively well-isolated MS = 0 ground state within the spin manifold. On the contrary, the doublet assigned to Fe(I) is magnetically split between −3 mm s−1 and +4 mm s−1, revealing a much larger internal field. Such behavior excludes the possibility of strongly coupled Fe centers. Consistently, we observe a broad signal near g = 16 in X-band EPR spectra collected in parallel mode on 2 (Fig. S13†). Similar spectra are reported for biological and other synthetic polyiron systems and are attributed to weak coupling between integer-spin multiplets.28 As a first approximation, we have simulated the high-field Mössbauer spectra assuming three uncoupled iron centers, two of which account for the two Fe(I) centers, with identical parameters. The overall splittings were well reproduced assuming an S = 3/2 ground state for the Fe(I). Additionally, the solution magnetic susceptibility of 2 (μeff = 5.6 μB) is lower than that of all triiron(II) complexes supported by L3− (6.2–7.5 μB) and is therefore consistent with the FeI2FeII oxidation state assignment.13b,35
:1 area ratio. From the Mössbauer parameters, we assign the doublet with double integration as FeI (δ = 0.66 mm s−1; ΔEQ = 2.60 mm s−1) and corresponding to Fe2 and Fe3, whereas the parameters of the other doublet are characteristic of high-spin FeII (δ = 0.98 mm s−1; ΔEQ = 2.15 mm s−1) and attributed to Fe1. The δ values assigned to Fe2 and Fe3 are comparable to those for the high-spin (β-diketiminate)-supported iron(I) complexes with two tert-butylisocyanides24 or one dinitrogen bound25 (c.f. 0.64 or 0.62, respectively), and distinct from the analogous low-spin iron(I) di- and tri(carbonyl)26 and tris(tert-butylisocyanide) compounds (c.f. 0.12–0.25).24 This comparison suggests that 2 likely contains high-spin FeI centers, which is rare for a CO ligated iron center with tris(pyrazolyl)borate FeI(CO) compounds as the only characterized examples.16,27 Applying a magnetic field of 2 T at 4.8 K resulted in a slight line broadening for the doublet assigned to Fe(II), revealing an internal field of less than 4 T (Fig. S19†). This is indicative of an integer-spin system with a relatively well-isolated MS = 0 ground state within the spin manifold. On the contrary, the doublet assigned to Fe(I) is magnetically split between −3 mm s−1 and +4 mm s−1, revealing a much larger internal field. Such behavior excludes the possibility of strongly coupled Fe centers. Consistently, we observe a broad signal near g = 16 in X-band EPR spectra collected in parallel mode on 2 (Fig. S13†). Similar spectra are reported for biological and other synthetic polyiron systems and are attributed to weak coupling between integer-spin multiplets.28 As a first approximation, we have simulated the high-field Mössbauer spectra assuming three uncoupled iron centers, two of which account for the two Fe(I) centers, with identical parameters. The overall splittings were well reproduced assuming an S = 3/2 ground state for the Fe(I). Additionally, the solution magnetic susceptibility of 2 (μeff = 5.6 μB) is lower than that of all triiron(II) complexes supported by L3− (6.2–7.5 μB) and is therefore consistent with the FeI2FeII oxidation state assignment.13b,35
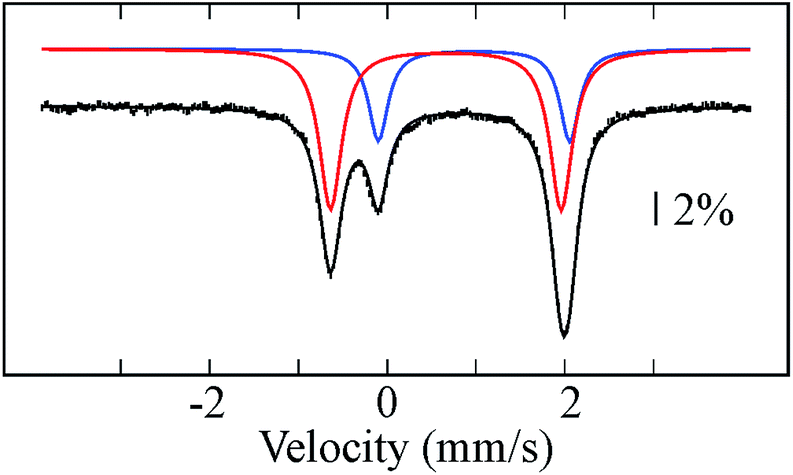 | ||
| Fig. 2 Mössbauer spectrum of 2 recorded at 80 K and zero-applied field. Black bars and colored lines represent the experimental data points and simulated quadrupole doublets, respectively (Table S1†). Solid black line is a composite spectrum obtained by combining individual doublets. | ||
The formal metal oxidation state assignments and the overall cofactor charge in the resting state (E0) of FeMoco remain unclear. Early formulations considered Mo(IV) and various ratios of Fe(II) and Fe(III); some of the Fe(II) and Fe(III) centers could be treated as mixed-valent pairs (i.e., 2Fe2.5+).29a–e Currently, the Mo is proposed to be a non-Hund's S = ½ Mo(III) center based on high-energy resolution fluorescence detected Mo K-edge XAS.29f Spatially resolved anomalous scattering (SpReAD) experiments support the Mo(III) assignment and suggest an [MoIIIFeII3FeIII4S9C]1− cluster.29g Recent DFT studies by Bjornsson, et al. benchmarked to the Mössbauer parameters reported by Yoo, et al. are consistent with the SpReAD result or the related isoelectronic mixed-valent formalism (i.e., [MoIIIFeIIFe2.5+4FeIII2S9C]1−).29h Similar ambiguities arise for the one-electron reduced (MR) and the CO-bound forms of FeMoco, although differences in the experimental conditions (i.e., extracted FeMoco vs. holo-MoFe protein) may account for these discrepancies.7,29a–c,30 Relatedly, the extent of charge delocalization may be critical to dinitrogen activation. Local iron(I/0) character would be required should N2 binding and activation by FeMoco parallel synthetic systems.42 Our results and the possible coordination number changes for iron center(s) in FeMoco during catalytic turnover31,32 hint that these uncertainties should be expected in the reduced states of FeMoco. For example, the isomer shift and quadrupole splittings for Fe2 and Fe3 could lead to the mistaken assignment of these centers as di-rather than mono-valent, since isomer shifts of 0.66 mm s−1 are well within the range of many iron(II) species.
We previously suggested that loss of H2 from 1 may be sterically controlled based on the large H⋯H distances (2.93–3.05 Å).13b H2 re from 1 is not observed either thermally or under broad wavelength irradiation. For an H2 re step to precede CO binding, an H2 re/oa equilibrium would likely have to exist in which any liberated H2 reacts rapidly to regenerate 1, but affords 2 in the presence of high concentrations of CO relative to H2 (Scheme 2). H2/D2 exchange should occur in this mechanism, thereby scrambling the respective isotopic labels. No changes were observed in 1H NMR spectra recorded on reaction mixtures of Fe3D3L (1-D3) with H2 (∼1 atm) at temperatures up to 80 °C for 20 h (Fig. S5†). The lack of observable H/D exchange supports CO binding preceding H2 loss and contrasts the exchange reported for dimeric β-diketiminate complexes.33 As noted in Scheme 2, we cannot exclude a tightly associated Kubas-type complex between the eliminated H2 and the triiron species, which does not exchange with dissolved H2 or D2. CO can then coordinate to the Kubas-type species, liberating H2 and generating 2. However, such tightly bound M–H2 complexes on a high-spin metal center are rare,11a and this pathway assumed unlikely. A similar substrate-binding prior to H2 elimination was recently reported for reaction of a di(μ-hydrido)diiron(II) complex with diazo compounds.34 In the simplest mechanism, retention of one hydride in 2 would arise from an intramolecular step for H2 loss upon reaction of 1 with CO (Scheme 2). Previously, we provided crystallographic evidence demonstrating that the β-diketiminate and benzene substituents on the ligand for two triiron complexes could interdigitate in the presence of K+ cations.35 A similar bis(triiron) intermediate could form during H2 re; however, the predicted long H⋯H distance and the absence of K+ cations suggests that such an intermediate is also unlikely. Consistent with an intracluster H2 re mechanism, we observe only H2 and not HD in the 1H-NMR spectra of reactions containing 1 and 1-D3 under a CO atmosphere (Fig. S7†).
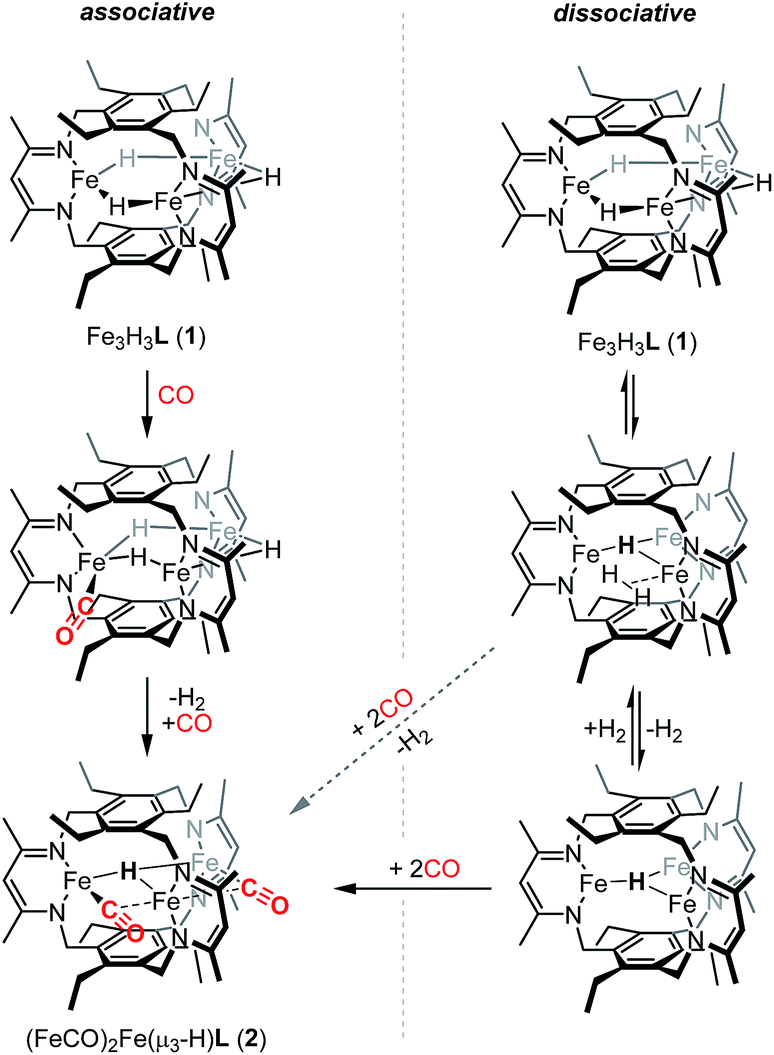 | ||
| Scheme 2 Possible associative (left) and dissociative (right) pathways for formation of 2, in which the two pathways can intersect depending on whether H2 loss succeeds (diagonal dashed arrow) or precedes (horizontal arrow) CO coordination. | ||
Given that we have hitherto been unable to isolate FeI-containing complexes for L3−,35 we were intrigued if reactivity characteristic of FeI centers could be accessed starting from 2. To that end, we examined the conversion of 2 to 1 upon addition of H2. Notably, we observed complete reaction of a toluene solution of 2 with H2 at 20 bar and 80 °C to yield 1 in 75(7)% spectroscopic yield as a 6.3:1 mixture of 1 and an as-yet undetermined decomposition product. Although reversible H2 re/oa both with and without coordination/dissociation of N2 are common reactions,3c,11a the reversible H2 re/oa with CO coordination/dissociation that we observe here is a very rare transformation. Despite the ubiquity of organometallic carbonyls and hydrides, this reaction has been previously reported only for one series of low-spin Group VIII HM3(CO)9X compounds.36 Because the reactions of mononuclear or self-assembled dinuclear β-diketiminate iron hydrides with CO lead to low-spin polycarbonyl products whose formation is presumably irreversible,40b we hypothesize that our use of a relatively rigid polynucleating ligand is the key factor that enables the unusual ability to convert between the two species that we observe. Additionally, the interchange between 1 and 2 represents a reversible two-electron redox reaction occurring at a high-spin polyiron cluster, and the starting species 1 demonstrates unusually high substrate specificity.13b As such, the reactivity demonstrated here bears resemblance to the biological reactivity of multimetallic cofactors; the proposed dihydride species in the E4 state of FeMoco is stable to protonation but readily undergoes reversible re/oa of H2. These observations suggest that the bio-inspired design of polynucleating ligands to stabilize high-spin clusters of base metals is a promising strategy for developing base-metal promoted multi-electron chemistry similar to that performed by multimetallic enzymes in biology.
If the dihydrogen reductive elimination is intramolecular, the distance between two hydride donors must necessarily decrease from the solid-state structure of 1 to form H2. We posit that CO binds to 1 and stabilizes a structure in which two H− donors are sufficiently close to liberate H2. Fluxional coordination of hydrides37 was proposed in 1 based on our previous reaction study, and a shift of a μ-hydride in 1 towards the internal cavity or to a μ3 position would generate an open coordination site for CO to bind. With this in mind, we hypothesized that small Lewis acids could coordinate to one or more hydrides in 1 and stabilize such a distorted conformation of [Fe3H3]3+. We probed the reaction of 1 with BF3·OEt2 as others have isolated species with R3B⋯H–M interactions.38 To our knowledge, the M–H⋯BF3 adduct is rare with the only reported example for a bis(cyclopentadienyl)niobium dihydride.39 We considered, however, the size of the R group and the need for an electron deficient borane in our choice of BF3.13b
Reaction of 1 with BF3·OEt2 resulted in formation of a bright yellow solution of Fe3(μ-F)3L, 3, (λmax = 321 nm, 392 nm, 431 nm, Fig. S15†) in near quantitative spectroscopic yield and moderate crystalline yield (Scheme 3, 57%). Our assignment of 3 is supported by combustion analysis, HR-ESI/MS(+), and 1H-NMR; the latter is consistent with a D3h symmetric species in solution and on the method timescale. The solution magnetic susceptibility of 3 is 7.4 μB; consistent with that of similar triiron(II) complexes.13b,35 The solid-state structure of 3 evidences three bridging fluoride ligands and a planar [Fe3F3]3+ core in 3 (Fig. 3) with comparable bond metrics to related mono- and di-metallic complexes (Table S2†) as well as those in tri-iron(III) and -iron(II) complexes of L3−.13b,35,40 Given the quantitative conversion of 1 to 3, the reaction is a net metathesis of Fe–H for B–F bonds. Consistently, treatment of the reaction mixture with NEt3 allowed observation of BH3 as the amine–borane adduct, Et3NBH3, in 11B-NMR spectra. To our knowledge, this reaction represents the first example of a controlled B–F/Fe–H for B–H/Fe–F bond metathesis. This transformation is enthalpically disfavored (∼21 kcal mol−1 per B–F/Fe–H bond metathesis) based on the average empirical values for the respective bonds.41 The fact that this reaction proceeds at ambient temperature, however, implies that the bond dissociation energies for the Fe–H and Fe–F bonds in 1 and 3 differ significantly from the reported average empirical values. This deviation from the expected bond enthalpies is likely due to the ligand's unique geometric constraints and the differences in bridging ligand properties of fluoride versus hydride. The B–F/Fe–H bond metathesis further illustrates the unexpected reactivity profile of 1 and related trihydride compounds.13 Previously, monometallic (nacnac)iron fluoride complexes were reported to react rapidly with trialkylsilanes to generate the corresponding iron hydride complexes.40a However, 3 is unreactive towards the silanes tested based on IR and 1H-NMR spectroscopy. This reaction should be only slightly enthalpically disfavored relative to the reaction of 1 with BF3, but our result is consistent with an unusually large difference between the Fe–F and Fe–H bond strengths in 3vs.1. Other factors such as approach of the silyl reagent, access to the transition state geometry, or poor access to the fluoride ligands may also modulate the reaction rate.
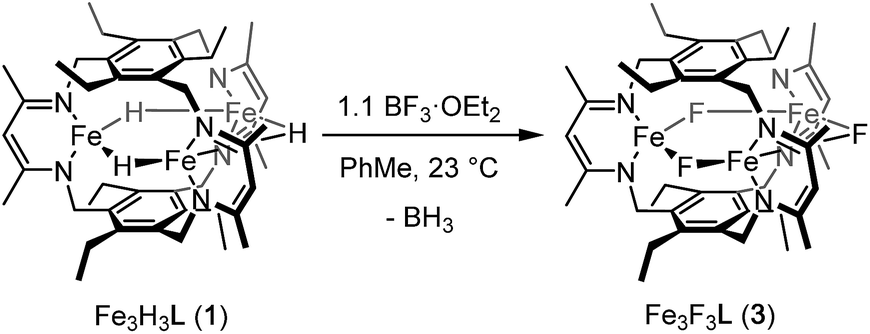 | ||
| Scheme 3 Preparation of Fe3F3L (3). | ||
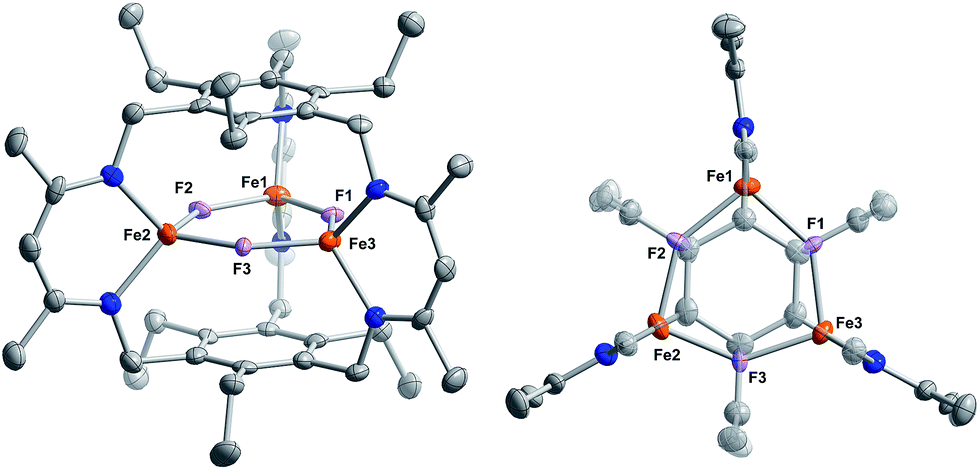 | ||
| Fig. 3 Solid-state structure of 3 viewed from the side (left) and top (right). C, N, O, F, and Fe atoms are depicted as gray, blue, red, fuchsia, and orange 75% thermal ellipsoids. All H-atoms as well as solvent molecules are omitted for clarity. | ||
Conclusions
We have expanded on the prior reactivity studies reported for 1, demonstrating that CO triggers reductive elimination of H2 from 1 to generate an unusual high-spin mixed-valent triiron(I/I/II) complex, 2, and that reaction with BF3·OEt2 affords the tri(μ-fluoride) species 3. These results add to the growing evidence that hydrides act as effective masks or protecting groups for low-valent high-spin metal centers. In addition, 2 reacts with H2 to regenerate 1, which is a rare example of reversible H2 re/oa involving CO and the first to occur at high-spin metal centers. Ongoing work focuses on continuing to develop the multielectron reactivity profiles of 1 as well as that of 2.Acknowledgements
LJM: University of Florida (UF), ACS Petroleum Research Fund (ACS-PRF 52704-DNI3), National Science Foundation (CHE-1464876, CHE-1048604), J. Goodsell and Prof. A. Angerhofer for help collecting EPR data. RGS: Labex ARCANE (ANR-11-LABX-0003-01). KAA: National Science Foundation (CHE-0821346) and UF for funding of the purchase of the X-ray equipment. KJA: UF University Scholars Program.Notes and references
- (a) W. Hieber and F. Leutert, Naturwissenschaften, 1931, 19, 360 CrossRef CAS; (b) W. Hieber and H. Schulten, Z. Anorg. Allg. Chem., 1937, 232, 17 CrossRef CAS.
- (a) Recent Advances in Hydride Chemistry, ed. M. Peruzzini and R. Poli, Elsevier, New York, 2001 Search PubMed; (b) G. S. McGrady and G. Guilera, Chem. Soc. Rev., 2003, 32, 383 RSC.
- (a) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed; (b) M. R. Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974 CrossRef PubMed; (c) J. Ballmann, R. F. Munhá and M. D. Fryzuk, Chem. Commun., 2010, 46, 1013 RSC; (d) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916 CrossRef CAS PubMed.
- (a) Ammonia: Catalysis and Manufacture, ed. A. Nielsen, Springer-Verlag, Berlin, 1995 Search PubMed; (b) H.-P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547 RSC.
- (a) C. K. Rofer-DePoorter, Chem. Rev., 1981, 81, 447 CrossRef CAS; (b) M. J. Overett, R. O. Hill and J. R. Moss, Coord. Chem. Rev., 2000, 206, 581 CrossRef.
- (a) W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef CAS PubMed; (b) H. Ogata, K. Nishikawa and W. Lubitz, Nature, 2015, 520, 571 CrossRef PubMed.
- (a) B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041 CrossRef CAS PubMed; (b) D. Lukoyanov, N. Khadka, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2016, 138, 1320 CrossRef CAS PubMed; (c) D. Lukoyanov, N. Khadka, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2016, 138, 10674 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149 CrossRef CAS PubMed.
- (a) E. L. Muetterties, R. R. Burch and A. M. Stolzenberg, Annu. Rev. Phys. Chem., 1982, 33, 89 CrossRef CAS; (b) R. D. Adams and B. Captain, Angew. Chem., Int. Ed., 2007, 47, 252 CrossRef PubMed.
- J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130 CrossRef CAS PubMed.
- (a) G. J. Kubas, Chem. Rev., 2007, 107, 4152 CrossRef CAS PubMed; (b) B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121 CrossRef CAS.
- (a) A. S. Weller and J. S. McIndoe, Eur. J. Inorg. Chem., 2007, 4411 CrossRef CAS; (b) R. N. Perutz and B. Procacci, Chem. Rev., 2016, 116, 8506 CrossRef CAS PubMed.
- (a) D. M. Ermert, I. Ghiviriga, V. J. Catalano, J. Shearer and L. J. Murray, Angew. Chem., Int. Ed., 2015, 54, 7047 CrossRef CAS PubMed; (b) Y. Lee, K. J. Anderton, F. T. Sloane, D. M. Ermert, K. A. Abboud, R. García-Serres and L. J. Murray, J. Am. Chem. Soc., 2015, 137, 10610 CAS.
- R. J. Klingler, W. M. Butler and M. D. Curtis, J. Am. Chem. Soc., 1978, 100, 5034 CrossRef CAS.
- (a) A. R. Sadique, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2008, 47, 784 CrossRef CAS PubMed; (b) J. M. Smith, A. R. Sadique, T. R. Cundari, K. R. Rodgers, G. Lukat-Rodgers, R. J. Lachicotte, C. J. Flaschenriem, J. Vela and P. L. Holland, J. Am. Chem. Soc., 2006, 128, 756 CrossRef CAS PubMed.
- J. L. Kisko, T. Hascall and G. Parkin, J. Am. Chem. Soc., 1998, 120, 10561 CrossRef CAS.
- (a) S. D. Brown, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 322 CrossRef CAS PubMed; (b) C. E. MacBeth, S. B. Harkins and J. C. Peters, Can. J. Chem., 2005, 83, 332 CrossRef CAS; (c) Y. Lee and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 4438 CrossRef CAS PubMed; (d) J. Rittle and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15898 CrossRef CAS PubMed.
- (a) P. Bhattacharya, J. A. Krause and H. Guan, Organometallics, 2011, 30, 4720 CrossRef CAS; (b) N. Gorgas, B. Stöger, L. F. Veiros, E. Pittenauer, G. Allmaier and K. Kirchner, Organometallics, 2014, 33, 6905 CrossRef CAS PubMed; (c) E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234 CrossRef CAS PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245 CrossRef CAS PubMed.
- Fe1 is disordered in the structure solution: two positions are modelled with minor occupancies of 3.5% and 2.5% (Fig. S18, ESI†). Electron density corresponding to the μ3-hydride required partial occupancy of a light atom to model satisfactorily (96% H, 4% O); the contaminant could not be detected by combustion analysis performed on multiple samples of 2.
- S. J. Tereniak and C. C. Lu, Group 8 Metal-Metal Bonds, in Molecular Metal-Metal Bonds, ed. S. T. Liddle, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed.
- (a) G. L. Guillet, F. T. Sloane, D. M. Ermert, M. W. Calkins, M. K. Peprah, E. S. Knowles, E. Čižmár, K. A. Abboud, M. W. Meisel and L. J. Murray, Chem. Commun., 2013, 49, 6635 RSC; (b) Y. Lee, K. A. Abboud, R. García-Serres and L. J. Murray, Chem. Commun., 2016, 52, 9295 RSC; (c) D. M. Ermert, J. B. Gordon, K. A. Abboud and L. J. Murray, Inorg. Chem., 2015, 54, 9282 CrossRef CAS PubMed.
- (a) C. Femoni, M. C. Iapalucci, G. Longoni, S. Zacchini and S. Zarra, Inorg. Chem., 2009, 48, 1599 CrossRef CAS PubMed; (b) C. Femoni, M. C. Iapalucci, G. Longoni and S. Zacchini, Dalton Trans., 2011, 40, 8685 RSC.
- S. M. Bellows, W. W. Brennessel and P. L. Holland, Eur. J. Inorg. Chem., 2016, 3344 CrossRef.
- S. A. Stoian, J. Vela, J. M. Smith, A. R. Sadique, P. L. Holland, E. Münck and E. L. Bominaar, J. Am. Chem. Soc., 2006, 128, 10181 CrossRef CAS PubMed.
- K. C. MacLeod, D. J. Vinyard and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 10226 CrossRef CAS PubMed.
- W. H. Harman, Personal communication.
- (a) M. P. Hendrich, E. Munck, B. G. Fox and J. D. Lipscomb, J. Am. Chem. Soc., 1990, 112, 5861 CrossRef CAS; (b) H. G. Jang, M. P. Hendrich and L. Que, Inorg. Chem., 1993, 32, 911 CrossRef CAS; (c) D. P. Goldberg, J. Telser, C. M. Bastos and S. J. Lippard, Inorg. Chem., 1995, 34, 3011 CrossRef CAS.
- (a) S. J. Yoo, H. C. Angove, V. Papaefthymiou, B. K. Burgess and E. Münck, J. Am. Chem. Soc., 2000, 122, 4926 CrossRef CAS; (b) B. Hedman, P. Frank, S. F. Gheller, A. L. Roe, W. E. Newton and K. O. Hodgson, J. Am. Chem. Soc., 1988, 110, 3798 CrossRef CAS; (c) H.-I. Lee, B. J. Hales and B. M. Hoffman, J. Am. Chem. Soc., 1997, 119, 11395 CrossRef CAS; (d) R. A. Venters, M. J. Nelson, P. A. McLean, A. E. True, M. A. Levy, B. M. Hoffman and W. H. Orme-Johnson, J. Am. Chem. Soc., 1986, 108, 3487 CrossRef CAS; (e) T. V. Harris and R. K. Szilagyi, Inorg. Chem., 2011, 50, 4811 CrossRef CAS PubMed; (f) R. Bjornsson, F. A. Lima, T. Spatzal, T. Weyhermüller, P. Glatzel, E. Bill, O. Einsle, F. Neese and S. DeBeer, Chem. Sci., 2014, 5, 3096 RSC; (g) T. Spatzal, J. Schlesier, E.-M. Burger, D. Sippel, L. Zhang, S. L. A. Andrade, D. C. Rees and O. Einsle, Nat. Commun., 2016, 7, 10902 CrossRef CAS PubMed; (h) R. Bjornsson, F. Neese and S. DeBeer, Inorg. Chem., 2017, 56, 1470 CrossRef CAS PubMed.
- (a) C. J. Pickett, K. A. Vincent, S. K. Ibrahim, C. A. Gormal, B. E. Smith and S. P. Best, Chem.–Eur. J., 2003, 9, 76 CrossRef CAS PubMed; (b) D. Lukoyanov, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2010, 132, 2526 CrossRef CAS PubMed; (c) P. E. Doan, J. Telser, B. M. Barney, R. Y. Igarashi, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2011, 133, 17329 CrossRef CAS PubMed.
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Science, 2014, 345, 1620 CrossRef CAS PubMed.
- J. B. Varley, Y. Wang, K. Chan, F. Studt and J. K. Nørskov, Phys. Chem. Chem. Phys., 2015, 17, 29541 RSC.
- T. R. Dugan, E. Bill, K. C. MacLeod, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2014, 53, 2370 CrossRef CAS PubMed.
- S. M. Bellows, N. A. Arnet, P. M. Gurubasavaraj, W. W. Brennessel, E. Bill, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2016, 138, 12112 CrossRef CAS PubMed.
- Y. Lee, F. T. Sloane, G. Blondin, K. A. Abboud, R. García-Serres and L. J. Murray, Angew. Chem., Int. Ed., 2015, 54, 1499 CrossRef CAS PubMed.
- (a) J. B. Keister, M. W. Payne and M. J. Muscatella, Organometallics, 1983, 2, 219 CrossRef CAS; (b) L. M. Bavaro, P. Montangero and J. B. Keister, J. Am. Chem. Soc., 1983, 105, 4977 CrossRef CAS; (c) L. M. Bavaro and J. B. Keister, J. Organomet. Chem., 1985, 287, 357 CrossRef CAS; (d) T. K. Dutta, X. Meng, J. C. Vites and T. P. Fehlner, Organometallics, 1987, 6, 2191 CrossRef CAS.
- (a) H. D. Kaesz and S. A. R. Knox, J. Am. Chem. Soc., 1971, 93, 4594 CrossRef CAS; (b) J. Powell, M. R. Gregg and J. F. Sawyer, J. Chem. Soc., Chem. Commun., 1987, 1029 RSC.
- A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873 CrossRef CAS PubMed.
- A. Antiñolo, F. Carrillo-Hermosilla, J. Fernández-Baeza, S. García-Yuste, A. Otero, J. Sánchez-Prada and E. Villaseñor, J. Organomet. Chem., 2000, 609, 123 CrossRef.
- (a) J. Vela, J. M. Smith, Y. Yu, N. A. Ketterer, C. J. Flaschenriem, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2005, 127, 7857 CrossRef CAS PubMed; (b) Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6624 CrossRef CAS PubMed; (c) Y. Lee, I.-R. Jeon, K. A. Abboud, R. García-Serres, J. Shearer and L. J. Murray, Chem. Commun., 2016, 52, 1174 RSC.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC, Boca Raton, 2007 Search PubMed.
- Selected examples: (a) M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780 CrossRef CAS PubMed; (b) J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef CAS PubMed; (c) T. J. Del Castillo, N. B. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 5341 CrossRef CAS PubMed; (d) S. Kuriyama, K. Arashiba, K. Nakajima, Y. Matsuo, H. Tanaka, K. Ishii, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2016, 7, 12181 CrossRef CAS PubMed; (e) P. J. Hill, L. R. Doyle, A. D. Crawford, W. K. Myers and A. E. Ashley, J. Am. Chem. Soc., 2016, 138, 13521 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and theoretical procedures and figures, crystallographic details, and theoretical structures. CCDC 1523783 and 1523784. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05583d |
| This journal is © The Royal Society of Chemistry 2017 |