
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regio- and stereospecific rhodium-catalyzed allylic alkylation with an acyl anion equivalent: an approach to acyclic α-ternary β,γ-unsaturated aryl ketones†
Ben W. H.
Turnbull
a,
Jungha
Chae
b,
Samuel
Oliver
b and
P. Andrew
Evans
 *a
*a
aDepartment of Chemistry, Queen's University, 90 Bader Lane, Kingston, K7L 3N6, Ontario, Canada. E-mail: Andrew.Evans@chem.queensu.ca
bDepartment of Chemistry, The University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
First published on 31st March 2017
Abstract
The regio- and stereospecific rhodium-catalyzed allylic alkylation of secondary allylic carbonates with cyanohydrin pronucleophiles facilitates the direct construction of acyclic α-ternary β,γ-unsaturated aryl ketones. Interestingly, this study illustrates the impact of deaggregating agents on regiocontrol and the electronic nature of the aryl component to suppress olefin isomerization. In addition, we demonstrate that the dimethylamino substituent, which modulates the pKa of the α-ternary β,γ-unsaturated aryl ketone, provides a useful synthetic handle for further functionalization via Kumada cross-coupling of the aryl trimethylammonium salt. Finally, the stereospecific alkylation of a chiral nonracemic secondary allylic carbonate affords the enantioenriched α-ternary aryl ketone, which was employed in a formal synthesis of trichostatic acid to illustrate that the allylic alkylation proceeds with net retention of configuration.
Introduction
The ability to reverse the natural polarity of a functional group using an umpolung synthon can often provide a strategic advantage by circumventing the underlying limitations associated with a more conventional approach.1 For example, the combination of acyl anion equivalents with the asymmetric transition metal-catalyzed allylic alkylation reaction (AAA)2–5 provides an attractive strategy for the construction of acyclic α-ternary β,γ-unsaturated carbonyl derivatives (Scheme 1A). Although this approach avoids some of the challenges associated with asymmetric enolate alkylation, namely, polyalkylation, epimerization and electrophile scope,6–8 the reaction requires the construction of an intermediate that can either suppress the isomerization of the olefin to the thermodynamically more stable α,β-unsaturated derivative or requires the removal of the olefin prior to the unveiling of the carbonyl motif.9 In addition, many of the acyl anion equivalents that have been successfully deployed in the asymmetric transition metal-catalyzed allylic alkylation reaction require additional functionalization steps to reveal the carbonyl moiety at the desired oxidation state, which detracts from the overall efficiency and utility of this approach (Scheme 1B). For example, Helmchen et al. reported malononitrile as a methoxycarbonyl anion surrogate in the asymmetric iridium-catalyzed allylic substitution reaction, which requires selective oxidation and functionalization to afford the corresponding methyl ester.5d In addition, Breitler and Carreira demonstrated the utility of N,N-dialkylhydrazones as a formyl anion equivalent, albeit the acidic conditions required to provide the α-ternary aldehyde necessitate prior hydrogenation of the alkene to avoid isomerization.5f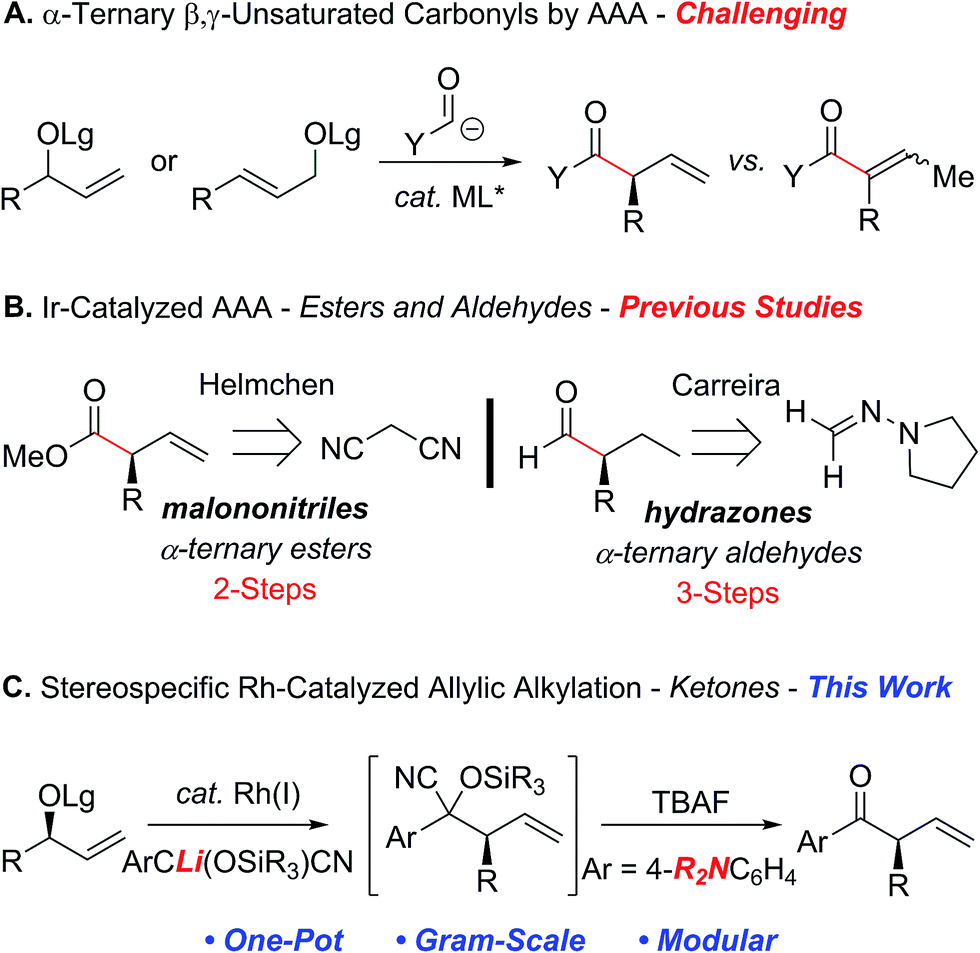 | ||
| Scheme 1 Factors affecting the development of the rhodium-catalyzed allylic alkylation with an acyl anion equivalent. | ||
In a program directed toward the development of rhodium-catalyzed allylic substitution reactions,10,11 we recently reported a highly regio- and stereospecific reaction of chiral nonracemic tertiary allylic alcohol derivatives with an acyl anion equivalent, which provides a convenient approach to quaternary stereogenic centers.12 For instance, tert-butyldimethylsilyl-protected cyanohydrins derived from the corresponding aryl and alkenyl aldehydes,13 function as acyl anion equivalents that can be unmasked in situ to afford the requisite ketone. Hence, we envisioned that the application of this strategy to acyclic chiral nonracemic secondary allylic carbonates should provide a direct one-pot approach to enantiomerically enriched α-ternary ketones, provided the isomerization of the olefin could be suppressed.14,15 To this end, we envisioned that the electronic nature of the aryl ketones could be tailored to modulate olefin isomerization and permit further functionalization. Herein, we now describe the first rhodium-catalyzed allylic alkylation of secondary allylic carbonates with cyanohydrin pronucleophiles to facilitate the construction of acyclic α-ternary β,γ-unsaturated aryl ketones (Scheme 1C).
Results and discussion
In accord with our supposition, preliminary studies demonstrated that the aryl ketones were indeed prone to facile isomerization. For example, treatment of the secondary allylic carbonate 1a′ (Lg = CO2Me) with the lithium anion of the aryl cyanohydrin 2a′ in the presence of [Rh(COD)Cl]2modified with triphenyl phosphite at −10 °C for ca. 16 hours, followed by the addition of tetra-n-butylammonium fluoride (TBAF) at −40 °C16 furnished the trisubstituted enone 5a′ in 41% yield with poor regioselectivity, albeit as a single geometrical isomer (E/Z ≥19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Table 1, entry 1).17 Hence, we reasoned that the isomerization could be suppressed by employing a more electron-rich aryl cyanohydrin to modulate the pKa of the α-proton in the ketone product. Interestingly, while the 4-methoxy-substituted aryl cyanohydrin 2aa′ afforded less of the trisubstituted α,β-unsaturated ketone (entry 2), the 4-dimethylamino derivative 2a reverses the outcome to afford the α-ternary β,γ-unsaturated ketone 3a (entry 3) in good yield and with modest regiocontrol. In an effort to further improve the efficiency and selectivity of this process, we elected to examine the effect of the leaving group, which can often have a dramatic impact on the outcome of metal-catalyzed allylic substitution reactions.10 To this end, the more bulky tert-butyl carbonate 1a (Lg = CO2tBu) improves the formation of 3a whilst maintaining similar regioselectivity (entry 4). Additional studies probed the effect of the phosphite ligand on regioselectivity, in which tris(tert-butyldimethylsilyl) phosphite provided further improvement in regiocontrol (entry 5). Finally, we envisioned the addition of an additive to coordinate the alkali metal would deaggregate the nucleophile and thereby increase the rate of allylic alkylation to provide improved selectivity. Gratifyingly, in accord with this hypothesis, the addition of DMPU provided significant improvement in regioselectivity to afford the α-ternary β,γ-unsaturated aryl ketone 3a in 89% yield and with ≥19:1 selectivity (entry 6).
:1) (Table 1, entry 1).17 Hence, we reasoned that the isomerization could be suppressed by employing a more electron-rich aryl cyanohydrin to modulate the pKa of the α-proton in the ketone product. Interestingly, while the 4-methoxy-substituted aryl cyanohydrin 2aa′ afforded less of the trisubstituted α,β-unsaturated ketone (entry 2), the 4-dimethylamino derivative 2a reverses the outcome to afford the α-ternary β,γ-unsaturated ketone 3a (entry 3) in good yield and with modest regiocontrol. In an effort to further improve the efficiency and selectivity of this process, we elected to examine the effect of the leaving group, which can often have a dramatic impact on the outcome of metal-catalyzed allylic substitution reactions.10 To this end, the more bulky tert-butyl carbonate 1a (Lg = CO2tBu) improves the formation of 3a whilst maintaining similar regioselectivity (entry 4). Additional studies probed the effect of the phosphite ligand on regioselectivity, in which tris(tert-butyldimethylsilyl) phosphite provided further improvement in regiocontrol (entry 5). Finally, we envisioned the addition of an additive to coordinate the alkali metal would deaggregate the nucleophile and thereby increase the rate of allylic alkylation to provide improved selectivity. Gratifyingly, in accord with this hypothesis, the addition of DMPU provided significant improvement in regioselectivity to afford the α-ternary β,γ-unsaturated aryl ketone 3a in 89% yield and with ≥19:1 selectivity (entry 6).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 1 where Lg = | 2 where X = | Phosphite L |
b/lb (3 + 5):(4 + 6) |
3:5c,d |
Yield of 3/5e (%) | ||
|
a All reactions were performed on a 0.25 mmol scale using 2.5 mol% [Rh(COD)Cl]2, 10 mol% L, 1.3 equiv. 2 and 1.8 equiv. LiHMDS in THF (2.5 mL) at −10 °C for ca. 16 hours, followed by the addition of 4.0 equiv. TBAF at −40 °C.
b Regioselectivity was determined by 500 MHz 1H NMR analysis of the reaction mixtures before deprotection of the cyanohydrin adducts.
c The ratio of 3:5 was determined by 500 MHz 1H NMR analysis of the crude ketones.
d Geometrical selectivity (E/Z ≥ 19:1) was determined by 500 MHz 1H NMR analysis of the crude enones.
e Isolated yields of the branched regioisomer.
f Reaction was conducted in THF (2.25 mL) and DMPU (0.25 mL).
|
||||||||
| 1 | CO2Me | a′ | H | a′ | P(OPh)3 | 2:1 |
≤1:19 |
41 |
| 2 | ′′ | ′′ | OMe | aa′ | ′′ | 3:1 |
1:3 |
62 |
| 3 | ′′ | ′′ | NMe2 | a | ′′ | 5:1 |
8:1 |
59 |
| 4 | CO2tBu | a | ′′ | ′′ | ′′ | 5:1 |
13:1 |
72 |
| 5 | ′′ | ′′ | ′′ | ′′ | P(OTBS)3 | 10:1 |
≥19:1 |
80 |
| 6 | CO 2 t Bu | ′′ | NMe 2 | a | P(OTBS) 3 |
≥19:1 |
≥19:1 |
89 |
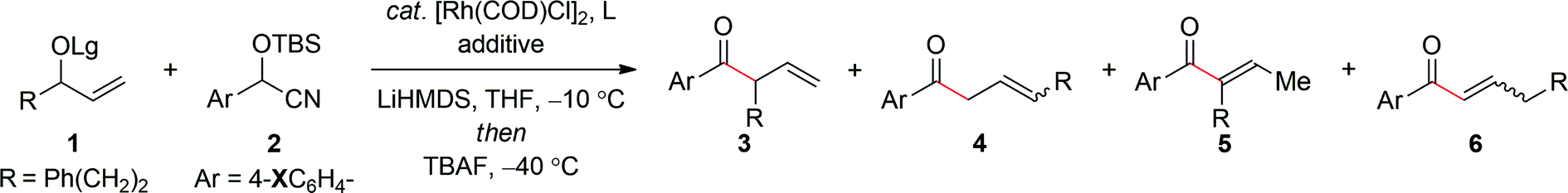
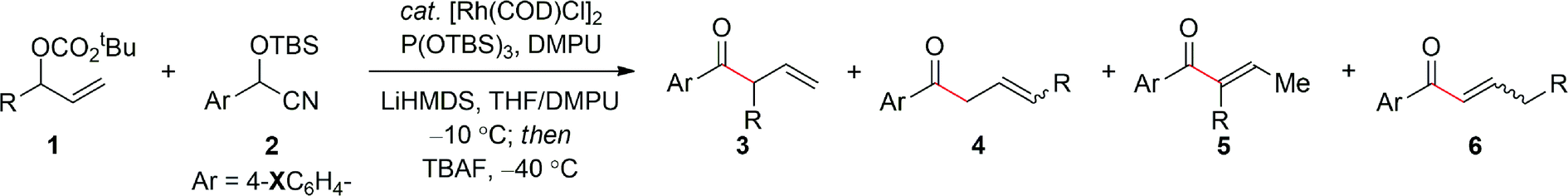
Table 2 summarizes the application of the optimized reaction conditions (Table 1, entry 6) to the synthesis of a range of α-ternary β,γ-unsaturated aryl ketones 3 using electron-rich aryl cyanohydrins. The reaction is tolerant of a number of important substituents within the allylic carbonate moiety. For example, in addition to the phenethyl derivative, a benzyl group (entries 1–2), long and short alkyl chains (entries 3–4), including branched alkyl chains (entries 5–6) afford the corresponding aryl ketones 3a–f in excellent yield and selectivity. Additionally, the ability to employ a range of functionalized groups (entries 7–9) is also particularly noteworthy given the problems often associated with these types of electrophiles in conventional enolate alkylation reactions. Finally, a number of related electron-rich aryl cyanohydrins were examined as pronucleophiles for this process, namely, those containing pyrrolidine, piperidine and morpholine substituents, which also facilitate the allylic alkylation without isomerization to the trisubstituted α,β-unsaturated aryl ketone (entries 10–12). Hence, this process affords a great deal of versatility, despite the necessity to utilize a 4-dialkylamino-substituted cyanohydrin to suppress olefin isomerization.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 R = | 2 X = |
b/lb (3 + 5):(4 + 6) |
3:5c |
Yield of 3d (%) | |
|
a All reactions were performed on a 0.5 mmol reaction scale using 2.5 mol% [Rh(COD)Cl]2, 10 mol% P(OTBS)3, 1.3 equiv. 2 and 1.8 equiv. LiHMDS in THF (4.5 mL) and DMPU (0.5 mL) at −10 °C for ca. 16 hours, followed by the addition of 4.0 equiv. TBAF at −40 °C.
b Regioselectivity was determined by 500 MHz 1H NMR analysis of the isolated ketones.
c The ratio of 3:5 was determined by 500 MHz 1H NMR analysis of the isolated ketones.
d Isolated yields.
|
||||||
| 1 | Ph(CH2)2 | NMe2 | a | ≥19:1 |
≥19:1 |
89 |
| 2 | PhCH2 | ′′ | b | ≥19:1 |
≥19:1 |
83 |
| 3 | Pr | ′′ | c | ≥19:1 |
≥19:1 |
86 |
| 4 | Me | ′′ | d | ≥19:1 |
≥19:1 |
81 |
| 5 | i Bu | ′′ | e | ≥19:1 |
≥19:1 |
73 |
| 6 | i Pen | ′′ | f | ≥19:1 |
≥19:1 |
74 |
| 7 | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)2 CH(CH2)2 |
′′ | g | ≥19:1 |
≥19:1 |
71 |
| 8 | BnOCH2 | ′′ | h | ≥19:1 |
≥19:1 |
71 |
| 9 | BnO(CH2)2 | ′′ | i | ≥19:1 |
≥19:1 |
74 |
| 10 | Ph(CH2)2 | N(CH2)4 | j | ≥19:1 |
≥19:1 |
76 |
| 11 | Ph(CH2)2 | N(CH2)5 | k | ≥19:1 |
≥19:1 |
70 |
| 12 | Ph(CH2)2 | N[(CH2)]2O | l | ≥19:1 |
≥19:1 |
67 |
Scheme 2 outlines additional studies to highlight the synthetic utility of this protocol using the chiral nonracemic secondary allylic carbonate (R)-1d, which addresses the stereospecificity of this process. In accord with our previous studies using enantiomerically enriched tertiary carbonates,12 the electron-poor tris(2,2,2-trifluoroethyl)-phosphite ligand proved optimal, furnishing the acyclic ketone (R)-3d with excellent regiocontrol (b/l ≥19:1) and conservation of enantiomeric excess (95% cee) on gram scale (Scheme 2A).18 Stereoselective reduction of the resulting aryl ketone (R)-3d with the Corey–Bakshi–Shibata reagent19 afforded the secondary alcohol 7 with excellent diastereocontrol, which was subsequently protected as the benzyl ether 8a and methyl ether 8b under standard reaction conditions to provide the intermediates required to functionalize the dimethylamino group and establish the absolute configuration in the allylic alkylation. Scheme 2B illustrates the metal-catalyzed cross-coupling of the dimethylamino group in order to broaden the scope of this process in the context of the aryl ketone component.20,21 For instance, Reeves and coworkers recently reported an elegant one-pot palladium-catalyzed Kumada coupling with aryl trimethylammonium salts at room temperature.22 To this end, the quaternization of the amine 8a with methyl triflate followed by palladium-catalyzed Kumada coupling with phenylmagnesium bromide furnished the biaryl 9, which illustrates the synthetic utility of the dialkylamino motif. Additional studies focused on establishing the stereochemical course of the allylic alkylation through a formal synthesis of trichostatic acid, which is a hydrolysis product of the potent histone deacetylase inhibitor (+)-trichostatin A (Scheme 2C).23 Oxidative cleavage of the olefin 8b using Jin's modification of the Lemieux-Johnson oxidation,24 furnished the known aldehyde 1025 which confirmed the alkylation proceeds via net retention of configuration. Overall, this work represents an important advance for the metal-catalyzed allylic alkylation of secondary allylic alcohol derivatives with an acyl anion equivalent to prepare acyclic α-ternary β,γ-unsaturated aryl ketones in a highly regio- and stereospecific manner.
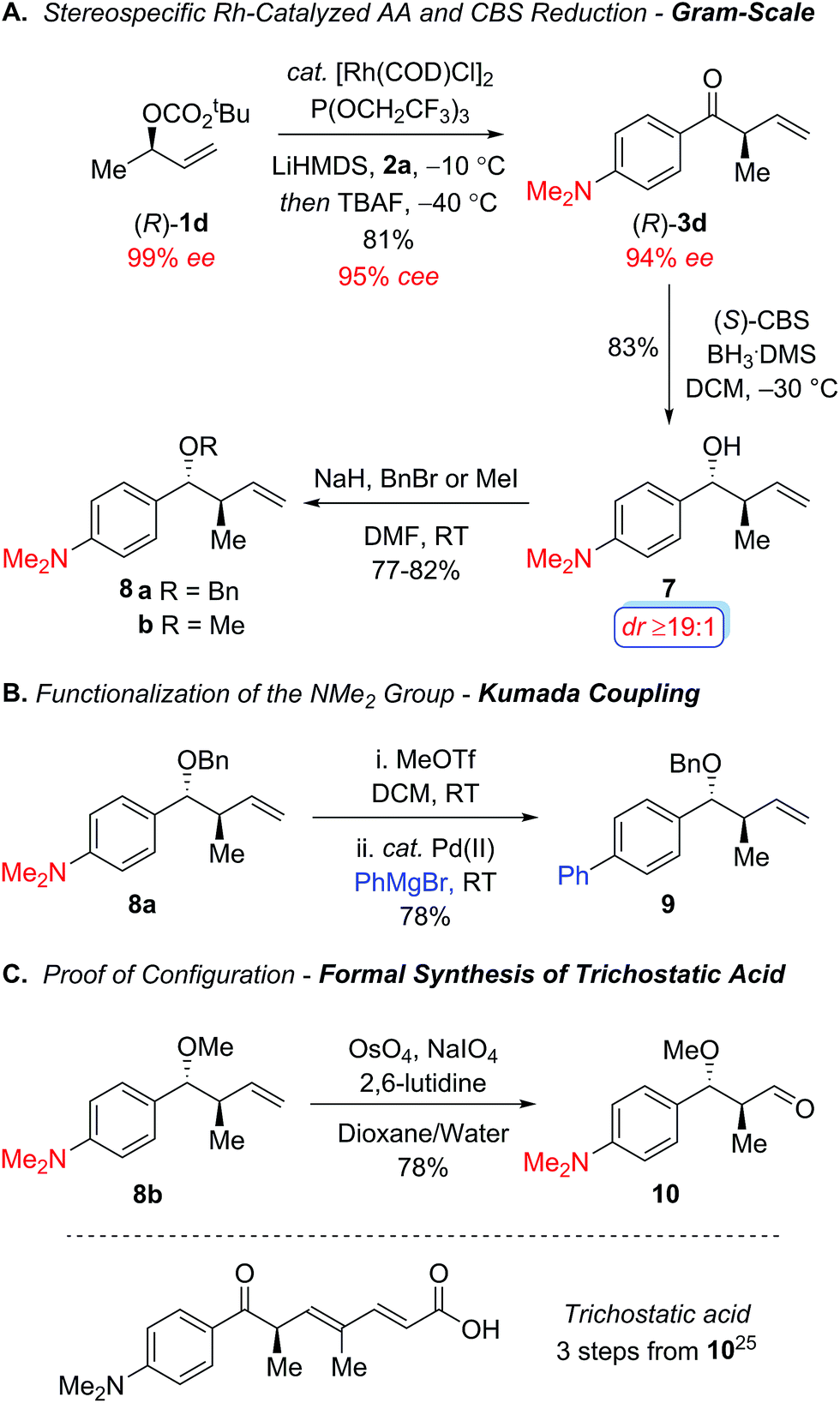 | ||
| Scheme 2 Stereospecific rhodium-catalyzed allylic alkylation to form α-ternary aryl ketone (R)-3d and further functionalization reactions. | ||
Conclusions
In conclusion, we have developed an approach to acyclic α-ternary β,γ-unsaturated aryl ketones via the rhodium-catalyzed allylic alkylation of secondary allylic carbonates with an acyl anion equivalent. The ability to control the degree of isomerization using a dimethylamino-substituted aryl cyanohydrin to modulate the pKa of the resulting α-proton in the aryl ketone is a critical component to the successful development of this method. Furthermore, the dialkylamine-substituted cyanohydrin facilitates the regio- and stereospecific construction of α-ternary β,γ-unsaturated ketones, which can either participate in the Kumada cross-coupling via the requisite trimethylammonium salts or be retained to facilitate a formal synthesis of trichostatic acid and thereby establish the stereochemical course of this process. Overall, this study constitutes a novel approach to the construction of enantiomerically enriched α-ternary β,γ-unsaturated aryl ketones, which represent important intermediates for target-directed synthesis, in addition to providing a strategy for controlling the isomerization of β,γ-unsaturated aryl ketones.Acknowledgements
We sincerely thank the National Sciences and Engineering Research Council (NSERC) for a Discovery Grant and Queen's University for generous financial support. NSERC is also thanked for supporting a Tier 1 Canada Research Chair (PAE). We also acknowledge the Royal Society for a Wolfson Research Merit Award (PAE) and the EPSRC and AstraZeneca (Alderley Park) for a PhD Studentship (SO). We are also grateful offer to Dr Paul Kemmitt (AZ) for his support and helpful discussions.Notes and references
- For an excellent review and tome on umpolung reactions, see: (a) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239 CrossRef; (b) Umpoled Synthons: A Survey of Sources and Uses in Synthesis, ed. T. A. Hase, Wiley, New York, 1987 Search PubMed.
- For an example of the palladium-catalyzed allylic alkylation with a cyanohydrin pronucleophile, see: J. Tsuji, I. Shimizu, I. Minami, Y. Ohashi, T. Sugiura and K. Takahashi, J. Org. Chem., 1985, 50, 1523 CrossRef CAS.
- For related palladium-catalyzed allylic substitution reactions with acyl silanes and stannanes, see: (a) Y. Obora, Y. Ogawa, Y. Imai, T. Kawamura and Y. Tsuji, J. Am. Chem. Soc., 2001, 123, 10489 CrossRef CAS PubMed; (b) Y. Obora, M. Nakanishi, M. Tokunaga and Y. Tsuji, J. Org. Chem., 2001, 67, 5835 CrossRef.
- For palladium-catalyzed allylic substitution reactions with carbon monoxide, see: (a) Y. Tamaru, K. Yasui, H. Takanabe, S. Tanaka and K. Fugami, Angew. Chem., Int. Ed., 1992, 31, 645 CrossRef; (b) H. Sakurai, K. Tanabe and K. Narasaka, Chem. Lett., 1999, 309 CrossRef.
- For related metal-catalyzed allylic substitution reactions with acyl anion equivalents, see: (a) B. M. Trost, G.-H. Kuo and T. Benneche, J. Am. Chem. Soc., 1988, 110, 621 CrossRef CAS; (b) B. M. Trost, O. Dirat, J. Dudash Jr and E. J. Hembre, Angew. Chem., Int. Ed., 2001, 40, 3658 CrossRef CAS; (c) C. Gnamm, S. Förster, N. Miller, K. Brödner and G. Helmchen, Synlett, 2007, 790 CAS; (d) S. Förster, O. Tverskoy and G. Helmchen, Synlett, 2008, 2803 Search PubMed; (e) B. M. Trost, M. Osipov, P. S. J. Kaib and M. T. Sorum, Org. Lett., 2011, 13, 3222 CrossRef CAS PubMed; (f) S. Breitler and E. M. Carreira, J. Am. Chem. Soc., 2015, 137, 5296 CrossRef CAS PubMed.
- For a recent review on enantioselective enolate alkylation reactions, see: B. M. Stoltz and J. T. Mohr, in Science of Synthesis: Stereoselective Synthesis 3, ed. J. G. de Vries, P. A. Evans and G. A. Molander, Thieme, Stuttgart, Germany, 2011, pp. 615–674 Search PubMed.
- For recent reviews on the transition metal-catalyzed allylic alkylation of enolates and enolate equivalents, see: (a) M. Braun and T. Meier, Synlett, 2006, 661 CrossRef CAS; (b) J. T. Mohr and B. M. Stoltz, Chem.–Asian J., 2007, 2, 1476 CrossRef CAS PubMed; (c) S. Oliver and P. A. Evans, Synthesis, 2013, 45, 3179 CrossRef CAS.
- For recent examples of asymmetric vinylation of enolate and enolate equivalents, see: (a) H. Kim and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 398 CrossRef CAS PubMed; (b) A. M. Taylor, R. A. Altman and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 9900 CrossRef CAS PubMed; (c) S. Lou and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 5010 CrossRef CAS PubMed and pertinent references cited therein.
- For the challenges associated with controlling isomerization in the synthesis of β,γ-unsaturated ketones, see: (a) E. Piers and P. C. Marais, J. Org. Chem., 1990, 55, 3454 CrossRef CAS; (b) D. Solé, E. Peidró and J. Bonjoch, Org. Lett., 2000, 2, 2225 CrossRef; (c) D. Solé, F. Diaba and J. Bonjoch, J. Org. Chem., 2003, 68, 5746 CrossRef PubMed.
- P. A. Evans and D. K. Leahy, in Modern Rhodium-Catalyzed Organic Reactions, ed. P. A. Evans, Wiley-VCH, Weinheim, Germany, 2005, pp. 191–214 Search PubMed.
- For mechanistic studies, see: P. A. Evans and J. D. Nelson, J. Am. Chem. Soc., 1998, 120, 5581 CrossRef CAS.
- (a) P. A. Evans, S. Oliver and J. Chae, J. Am. Chem. Soc., 2012, 134, 19314 CrossRef CAS PubMed; (b) P. A. Evans and S. Oliver, Org. Lett., 2013, 15, 5626 CrossRef CAS PubMed; (c) B. W. H. Turnbull, S. Oliver and P. A. Evans, J. Am. Chem. Soc., 2015, 137, 15374 CrossRef CAS PubMed.
- N. Kurono, M. Yamaguchi, K. Suzuki and T. Ohkuma, J. Org. Chem., 2005, 70, 6530 CrossRef CAS PubMed.
- For an alternative approach involving reductive acyl cross-coupling, see: A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442 CrossRef CAS PubMed.
- For a related divergent approach that forms racemic α-ternary β,γ-unsaturated and trisubstituted α,β-unsaturated ketones, see: Q.-A. Chen, F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2015, 137, 3157 CrossRef CAS PubMed.
- The deprotection was conducted at low temperature to suppress competing side reactions derived from isomerization and the addition of the liberated cyanide to the α,β-unsaturated aryl ketone.
- Interestingly, trimethylphosphite provided inferior selectivity and efficiency irrespective of the stoichiometry, which is in sharp contrast to our earlier studies.10,11.
- The term conservation of enantiomeric excess (cee) = (ee of product/ee of starting material) × 100, P. A. Evans, J. E. Robinson and J. D. Nelson, J. Am. Chem. Soc., 1999, 121, 6761 CrossRef CAS.
- (a) A. Hirao, S. Itsuno, S. Nakahama and N. Yamazaki, J. Chem. Soc., Chem. Commun., 1981, 315 RSC; (b) E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551 CrossRef CAS.
- For seminal examples, see: (a) E. Wenkart, A.-L. Han and C.-J. Jenny, J. Chem. Soc., Chem. Commun., 1988, 975 RSC; (b) S. B. Blakey and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 6046 CrossRef CAS PubMed; (c) L.-G. Xie and Z.-X. Wang, Angew. Chem., Int. Ed., 2011, 50, 4901 CrossRef CAS PubMed; (d) H. Zhang, S. Hagihara and K. Itami, Chem.–Eur. J., 2015, 21, 16796 CrossRef CAS PubMed.
- For the Ni-catalyzed reduction of aryl trimethylammonium salts, see: Y.-Q.-Q. Yi, W.-C. Yang, D.-D. Zhai, X.-Y. Zhang, S.-Q. Li and B.-T. Guan, Chem. Commun., 2016, 52, 10894 RSC.
- J. T. Reeves, D. R. Fandrick, Z. Tan, J. J. Song, H. Lee, N. K. Yee and C. H. Senanayake, Org. Lett., 2010, 12, 4388 CrossRef CAS PubMed.
- M. Yoshida, M. Kijima, M. Akita and T. Beppu, J. Biol. Chem., 1990, 265, 17174 CAS.
- (a) R. Pappo, D. S. Allen Jr, R. U. Lemieux and W. S. Johnson, J. Org. Chem., 1956, 21, 478 CrossRef CAS; (b) W. Yu, Y. Mei, Y. Kang, Z. Hua and Z. Jin, Org. Lett., 2004, 6, 3217 CrossRef CAS PubMed.
- J. T. Markiewicz, D. J. Schauer, J. Löfstedt, S. J. Corden, O. Wiest and P. Helquist, J. Org. Chem., 2010, 75, 2061 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc05705e |
| This journal is © The Royal Society of Chemistry 2017 |