
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DFT calculation-inspired Rh(I)-catalyzed reaction via suppression of α-H shift in α-alkyldiazoacetates†
Shunying
Liu‡
 a,
Jun
Jiang‡
b,
Jianghui
Chen
a,
Qinghua
Wei
a,
Wenfeng
Yao
a,
Fei
Xia
*ac and
Wenhao
Hu
*a
a,
Jun
Jiang‡
b,
Jianghui
Chen
a,
Qinghua
Wei
a,
Wenfeng
Yao
a,
Fei
Xia
*ac and
Wenhao
Hu
*a
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, Shanghai, 200062, China. E-mail: fxia@chem.ecnu.edu.cn; whu@chem.ecnu.edu.cn
bSchool of Chemistry and Chemical Engineering, Guangxi University, Nanning, 530004, China
cNYU-ECNU Center for Computational Chemistry at NYU Shanghai, Shanghai, 200062, China
First published on 22nd March 2017
Abstract
Metal-associated carbenes from diazo compounds promote many useful chemistry transformations in modern organic chemistry. However, compared to α-aryldiazoacetate-derived carbenes (ArDCs), the synthetic application of α-alkyldiazoacetate-derived carbenes (AlDCs) is greatly limited due to intramolecular α-H transfer (elimination) that results in alkenes as the main by-products. An intriguing α-alkyldiazoacetate-involved three-component reaction has been developed following DFT calculation inspiration to provide β-hydroxyl α-alkyl-α-amino acid derivatives in good yields. The intramolecular α-H shift of an α-alkyldiazoacetate-derived carbene was successfully suppressed by the association of a Rh(I) complex to form the corresponding active ammonium ylide, which was trapped before the fast 1,2-H transfer process. A Rh(I)-chiral diene complex was identified as an effective catalyst to give an asymmetric version of the reaction with good enantioselectivity. This reaction provides insight into extending the efficient transformation of α-alkyldiazoacetate-derived carbenes and their synthetic application.
Introduction
Metal-associated carbenes from diazo compounds play important roles in modern organic chemistry as active intermediates in useful chemistry transformations including X–H (X = C, O, N, etc.) insertions,1 cyclopropanations,2 ylides,3 1,2-migration,4etc.5 Among these transformations, efficiently developed transformations of metal-associated diazo carbenes are mainly established from α-aryldiazoacetate-derived carbenes (ArDCs) without α-protons (Scheme 1a). Compared to ArDCs, the synthetic application of α-alkyldiazoacetate-derived carbenes (AlDCs) is greatly limited. Their limitation in synthetic application is mainly attributed to the fact that AlDCs can readily undergo intramolecular α-H transfer (elimination), which results in alkenes as the main by-products (Scheme 1b). Even though there are several elegant examples of AlDCs for X–H insertion that were developed by Zhou,6 Feng7 and others,8 the development of AlDC chemistry is still greatly in demand.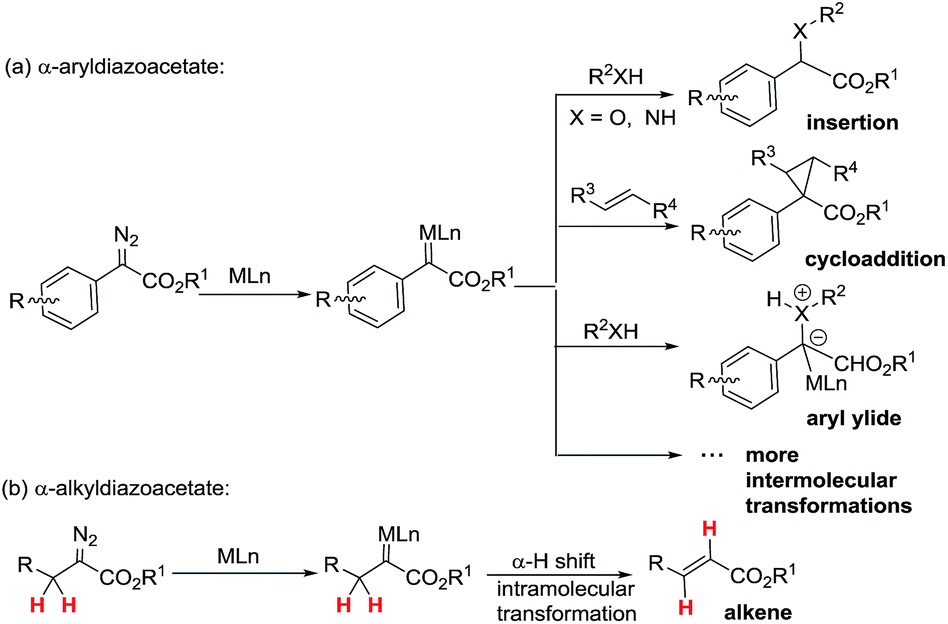 | ||
| Scheme 1 The main transformations of ArDCs (a) vs. AlDCs (b). | ||
Very recently, a Rh(I)-associated three-component reaction of α-phenyldiazoacetate, aniline and β-nitroacrylates was successfully developed by our group.9 For this reaction, we found that Rh2(OAc)4 only afforded a trace amount of the desired product even though the diazo compounds were completely decomposed. This result indicated that there was an obvious difference in the activation energies of Rh(I)- and Rh(II)-associated carbenes, and this prompted us to perform density functional theory (DFT) calculations. The investigations were conducted using the M06/Lanl2dz+6-31G* method10,11 in Gaussian 09 software,12 which is commonly used for describing metal carbene reactions.11,13 The frequency analyses were performed on optimized structures in the gas phase to verify whether they were transition states or stable structures. The solvent effect of CH2Cl2 was evaluated using the integral equation formalism model (IEFPCM).14 More computational details are provided in the section on DFT calculations in the ESI.†
We firstly conducted a conformation search on the generated Rh carbenes A3, A4 and A5, and their most stable conformers are shown in Scheme 2 and in the ESI.† It was found that the energy of Rh(II)-associated PhDC A3 was much higher than that of Rh(I)-associated PhDC A5 by 13.1 kcal mol−1. The calculated barriers of nitrogen extrusion viaTS-1-a, TS-1-b and TS-2-a are 8.0, 5.7 and 3.2 kcal mol−1, respectively, relative to their reactants, and are consistent with the values reported in the literatures13a,b for other reactions. These results confirm that the catalyst’s metal centre has an obvious effect on the activation energies of the reactions.
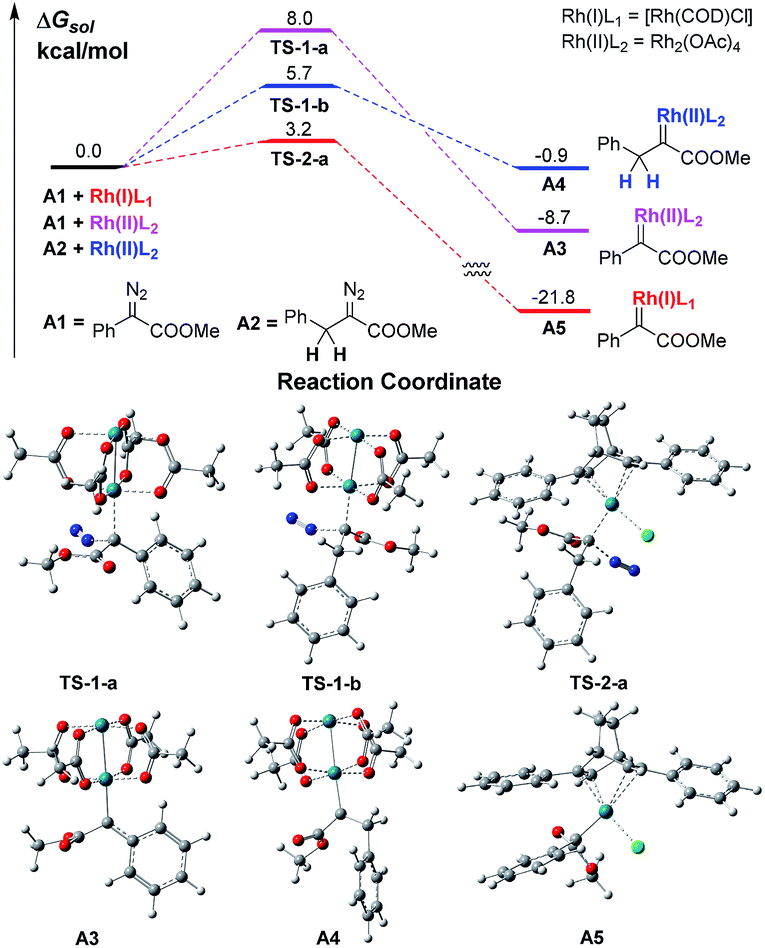 | ||
| Scheme 2 The free energy profiles of Rh(I)- and Rh(II)-associated PhDCs. | ||
A further DFT calculation revealed that when using Rh2(OAc)4 as the decomposition catalyst, the energy of a benzyldiazoacetate-derived carbene (BnDC), one of the most easily-prepared AlDCs, is actually much higher than that of a PhDC by 7.8 kcal mol−1, denoted as A4 and A3 in Scheme 2, respectively, which indicates that a BnDC is a more active intermediate than a PhDC. These results highly inspired us to hypothesize that Rh(I) complexes could possibly lead to lower energy metal-associated AlDCs that are relatively stable, to provide useful transformations before α-H transfer.
Results and discussion
The in situ generation of active ylide intermediates is one of the most important transformations of diazo carbenes, and can lead to further new transformations.15 We have been interested in discovering multicomponent reactions (MCRs) via a strategy of trapping Rh(II)-associated active ylides with appropriate electrophiles before rapid 1,2-proton transfer16 to construct α-aryl-α-amino acids.15b,17 For instance, trapping oxonium ylides with imines provided rapid access to β-hydroxyl α-aryl-α-amino acids.17 MCRs are among the most powerful transformations for constructing complex molecules from simple starting materials,18 thus we decided to develop a Rh(I)-catalyzed MCR by trapping alkyl ylides via AlDCs to validate our hypothesis.A preliminary investigation using DFT calculations of a Rh(I)-associated BnDC and its transformation into an α-H transfer product vs. a Rh(II)-associated BnDC was conducted (Scheme 3). As expected, the energy of the Rh(I)-associated BnDC is much lower than that of the Rh(II)-associated BnDC by 18.0 kcal mol−1. This suggests that the Rh(I)-associated BnDC possibly favors an ylide generation pathway over an α-H transfer pathway. The energy barriers of the transition states from the Rh(II)- and Rh(I)-associated BnDCs (TS-3 and TS-4, respectively) to their corresponding α-H transfer products were further investigated. The energy barrier of TS-3 is 11.1 kcal mol−1, illustrating a very fast α-H transfer process. The energy barrier of TS-4 is 21.2 kcal mol−1, which suggests the possibility that a Rh(I)-associated BnDC can be transformed into an ylide before α-H transfer. Thus, it is promising to develop MCR involving α-alkyldiazo compounds. To promote the desired MCR by trapping the alkyl ylide with a third component, both of the two very fast intramolecular processes, the α-H transfer in the metal carbene and the 1, 2-H transfer in the resulting ylide, should be overcome.
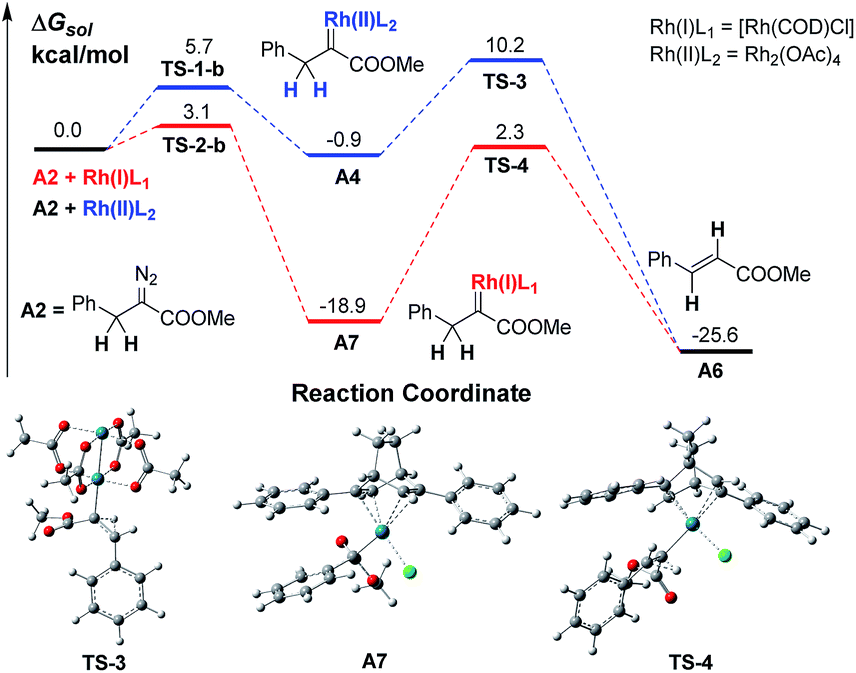 | ||
| Scheme 3 The free energy profile of the transformation of Rh(I)- and Rh(II)-associated α-H transfer in BnDCs. | ||
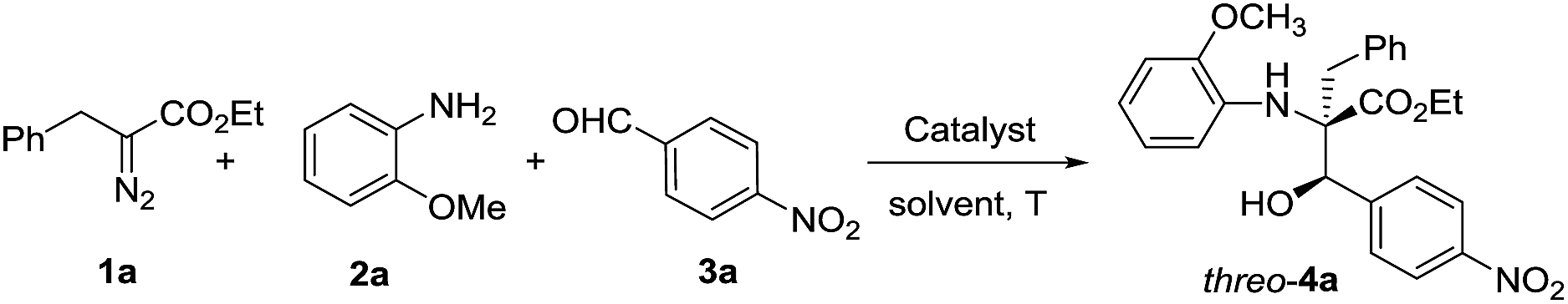
To confirm the theoretical results from the DFT investigations, our investigation of transformations began with the reaction of 2-diazo-3-phenylpropanoate (1a), 2-methoxyaniline (2a) and 4-nitrobenzaldehyde (3a) with 10 mol% catalyst (Table 1). The common catalyst for diazo decomposition, Rh2(OAc)4, was initially used to catalyze the reaction. A desired product 4a was obtained in a moderate 50% isolated yield but with poor diastereoselectivity (1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, entry 1). Rh2(TFA)4 and [RuCl2(p-cymene)]2 were also employed but no desired products were observed (entries 2–3). Significant amounts of imines from the condensation of 2a and 3a were observed as the main side products. [{PdCl(η3-C3H5)}2] catalyzed this reaction in only 10% yield and with no diastereoselectivity (entry 4). Then, [Rh(COD)Cl]2 was used and, as expected, was found to be a much more effective catalyst to give product 4a in 75% yield and with a 7:1 dr favoring the threo isomer (entry 5). Reducing the amount of [Rh(COD)Cl]2 seriously decreased the yield (entries 6–7). Dichloromethane (DCM) was identified as a superior solvent over dichloroethane (DCE) and toluene to improve the diastereoselectivity and retain a good yield, even with a smaller amount of catalyst (65% yield, 12:1 dr, entry 9 vs. 5 and 6). A higher temperature was better for obtaining a higher yield but unfavorable to the diastereoselectivity (entry 10 vs. 11).
:1 dr, entry 1). Rh2(TFA)4 and [RuCl2(p-cymene)]2 were also employed but no desired products were observed (entries 2–3). Significant amounts of imines from the condensation of 2a and 3a were observed as the main side products. [{PdCl(η3-C3H5)}2] catalyzed this reaction in only 10% yield and with no diastereoselectivity (entry 4). Then, [Rh(COD)Cl]2 was used and, as expected, was found to be a much more effective catalyst to give product 4a in 75% yield and with a 7:1 dr favoring the threo isomer (entry 5). Reducing the amount of [Rh(COD)Cl]2 seriously decreased the yield (entries 6–7). Dichloromethane (DCM) was identified as a superior solvent over dichloroethane (DCE) and toluene to improve the diastereoselectivity and retain a good yield, even with a smaller amount of catalyst (65% yield, 12:1 dr, entry 9 vs. 5 and 6). A higher temperature was better for obtaining a higher yield but unfavorable to the diastereoselectivity (entry 10 vs. 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | Yieldb (%) | drc |
|
a Unless otherwise noted, all of the reactions were carried out at a 0.1 mmol scale, 10 mol% catalyst and 1a:2a:3a = 1.0:1.2:2.0.
b Isolated yields.
c Detected using 1H NMR.
d N.R. = no reaction.
e Carried out using 4 mol% [Rh(COD)Cl]2.
f Carried out using 2 mol% [Rh(COD)Cl]2.
|
|||||
| 1 | Rh2(OAc)4 | Toluene | rt | 50 | 1.6:1 |
| 2 | Rh2(TFA)4 | Toluene | rt | N.R.d | — |
| 3 | [RuCl2(p-cymene)]2 | Toluene | rt | N.R. | — |
| 4 | [{PdCl(η3-C3H5)}2] | Toluene | rt | 10 | 1:1 |
| 5 | [Rh(COD)Cl]2 | Toluene | rt | 75 | 7:1 |
| 6e | [Rh(COD)Cl]2 | Toluene | rt | 55 | 7:1 |
| 7f | [Rh(COD)Cl]2 | Toluene | rt | 34 | 4:1 |
| 8e | [Rh(COD)Cl]2 | DCE | rt | 57 | 10:1 |
| 9e | [Rh(COD)Cl]2 | DCM | rt | 65 | 12:1 |
| 10e | [Rh(COD)Cl]2 | DCM | 0 | 58 | 13:1 |
| 11 | [Rh(COD)Cl]2 | DCM | 40 | 80 | 2:1 |
The efficiency of [Rh(COD)Cl]2 in promoting the alkyldiazoacetate-involved MCR compared to that of Rh2(OAc)4 was further illustrated using ethyl 2-diazopropanoate and ethyl 2-diazopentanoate, both of which gave a higher yield and much better diastereoselectivity (Table 2).
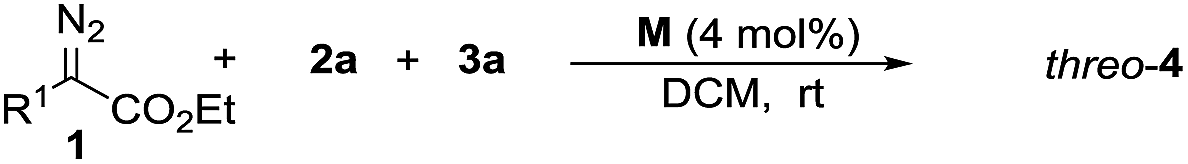
Under the optimized reaction conditions, a wide range of aldehydes, amines and diazo compounds was evaluated (Table 3). In most cases, a more electron-withdrawing aromatic aldehyde gave a higher product yield (entries 1–5), while a low yield of the three-component product was obtained with benzaldehyde and only a trace amount of product was obtained with electron-rich anisaldehyde (entries 6 and 7). These results are possibly due to the relatively weak electrophilic features of the electron-rich compounds to the active ammonium ylide. This process was also tolerant to other aromatic amines with different substituents on the aromatic rings to give a moderate yield and good diastereoselectivity up to >20:1 dr (entries 8–14). The diazo compounds were further extended to include tert-butyl-2-diazoacetate, giving the corresponding product in 41% yield and 2:1 dr (entry 15). The relative configuration of (2R*, 3R*)-threo-4o was established using X-ray single crystal analysis (see the figure in Table 3 and the ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1/R2 | Ar1 | Ar2 | Yield (%) | dr |
| a The reactions were carried out in the same way as those in Table 1. | |||||
| 1 | Bn/Et | o-MeOC6H4 | p-NO2C6H4 | 4a, 65 | 12:1 |
| 2 | Bn/Et | o-MeOC6H4 | o-NO2C6H4 | 4d, 50 | 2:1 |
| 3 | Bn/Et | o-MeOC6H4 | p-CNC6H4 | 4e, 30 | 7:1 |
| 4 | Bn/Et | o-MeOC6H4 | p-BrC6H4 | 4f, 50 | 6:1 |
| 5 | Bn/Et | o-MeOC6H4 | m-BrC6H4 | 4g, 56 | 3:1 |
| 6 | Bn/Et | o-MeOC6H4 | C6H5 | 4h, 18 | 4:1 |
| 7 | Bn/Et | o-MeOC6H4 | p-MeOC6H4 | Trace | — |
| 8 | Bn/Et | 2,4,6-CH3C6H2 | p-NO2C6H4 | 4i, 31 | 3:1 |
| 9 | Bn/Et | C6H5 | p-NO2C6H4 | 4j, 55 | >20:1 |
| 10 | Bn/Et | p-EtOC6H4 | p-NO2C6H4 | 4k, 45 | 2:1 |
| 11 | Bn/Et | 3,4,5-MeOC6H2 | p-NO2C6H4 | 4l, 44 | 4:1 |
| 12 | Bn/Et | p-MeOC6H4 | p-NO2C6H4 | 4m, 45 | 10:1 |
| 13 | Bn/Et | o-EtOC6H4 | p-NO2C6H4 | 4n, 60 | 10:1 |
| 14 | n-Pr/Et | 2,4,6-CH3C6H2 | p-NO2C6H4 | 4o, 52 | >20:1 |
| 15 | H/But | o-MeOC6H4 | p-NO2C6H4 | 4p, 41 | 2:1 |
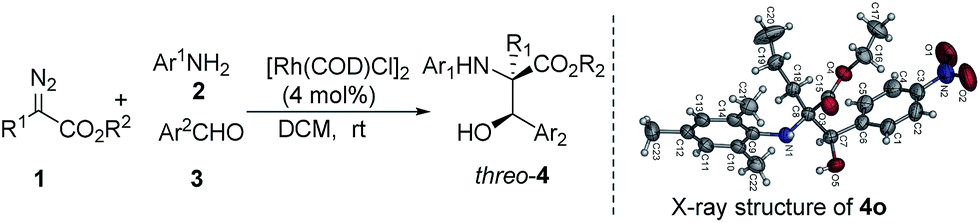
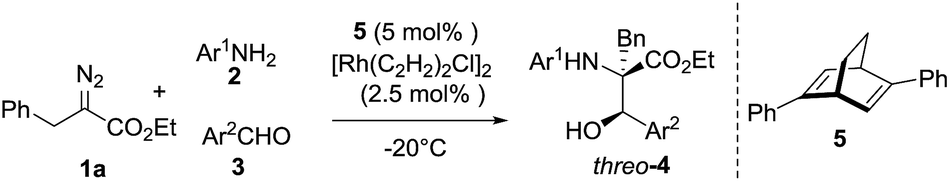
Our next efforts were focused on achieving enantioselective control of the reaction. Chiral Rh2(S-NTTL)4 and Rh2(S-DOSP)4, which are widely employed for the decomposition of diazo compounds, gave products that were almost completely racemic in very low yields in this case (3% ee and 4% ee, respectively). Using a synergistic catalytic system comprising Rh2(OAc)4 and chiral Zr/binol/molecular sieves17b or phosphoric acids,17c three-component products of skeletal β-hydroxyl α-alkyl-α-amino acid were obtained in less than 10% ee. Inspired by the success of using chiral diene ligands in Rh(I) catalysis as reported by Hayashi and Carreira,19 the Rh(I)/diene complexes were used to catalyze the reaction. Excitingly, the enantioselective three-component reaction was effectively accomplished to afford chiral β-hydroxyl α-alkyl-α-amino acid derivatives with moderate to good diastereoselectivity and enantiostereoselectivity (3:1 to 4:1 dr, 86:14 to 91:9 er, Table 4). β-Hydroxyl-α-amino acid derivatives including the β-hydroxyl-α-alkyl-α-amino acid moiety have been widely found in peptides (such as droxidopa, cyclosporin, vancomycin, etc.), enzyme inhibitors and other physiologically active compounds.20 Numerous efforts have been made to develop synthetic approaches for β-hydroxyl-α-amino acid core structures.21 Herein, the established method provides a facile construction strategy for these compounds from simple starting materials under mild conditions.
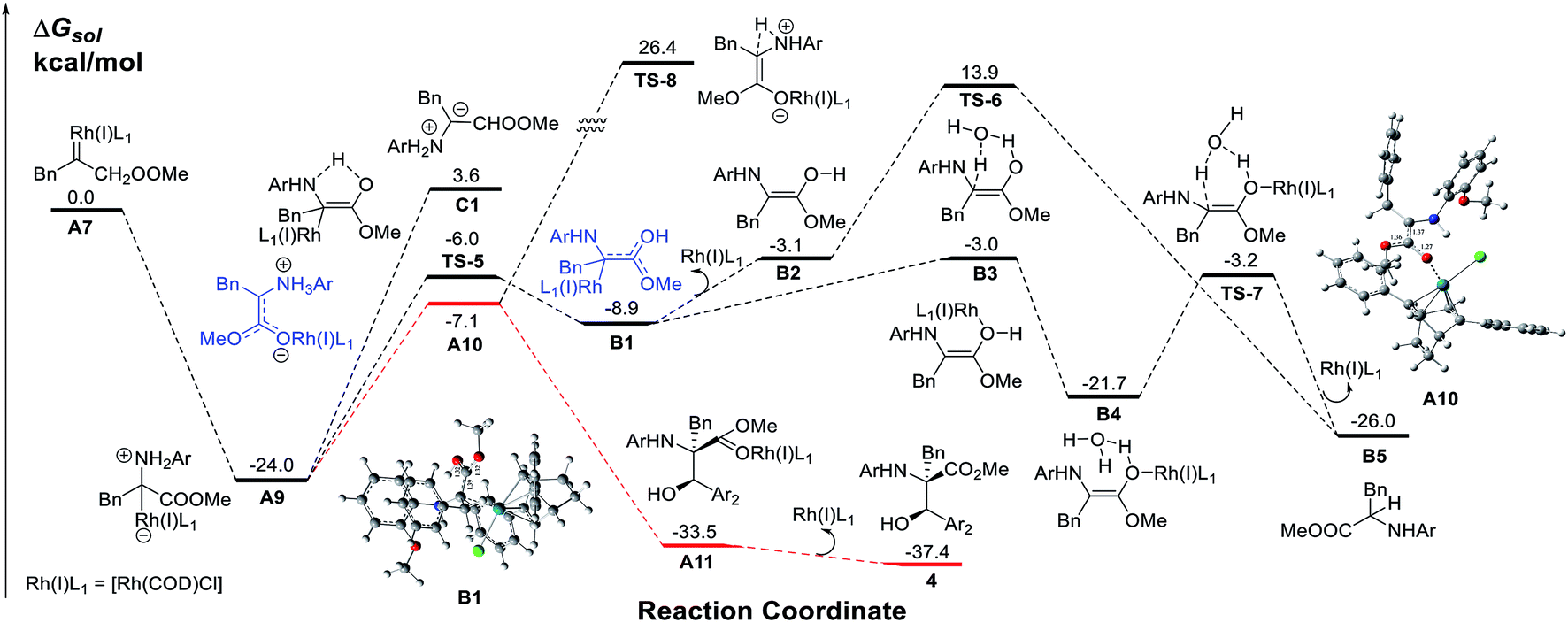
To obtain insight into the reaction mechanism, a detailed investigation using DFT calculations on the N–H insertion process via 1, 2-H transfer and the MCR process was conducted (Scheme 4). The results show that the nucleophilic attack of amines on the Rh(I) carbene is very facile with no energy barrier while the free ylide C1 pathway has an energy barrier as high as 27.6 kcal mol−1. A Rh(I)-associative enol intermediate B1 was formed via the transition state TS-5. The water-associated transition states TS-6 (metal-dissociative pathway) and TS-7 (metal-associative pathway)22 were well placed to be involved in the N–H insertion process. These results are consistent with those from previous theoretical studies on X–H bond insertion mechanisms performed by Yu and co-workers.23
 | ||
| Scheme 4 The calculated free energy profiles for the Rh(I) carbene in the N–H process and the multi-component process. | ||
Then, we examined the trapping of ammonium ylides by aldehydes. Interestingly, for the trapping pathway, the precursor for the attack of A9 by aldehydes is preferred to be the enolate intermediate A10, where the Rh(I)-ligand is attached to the oxygen atom of the carbonyl group of the carbene, rather than the enol intermediate B1. The DFT calculations (Fig. S1 in the ESI†) indicate that the MCR process has no energy barrier, with the release of a considerable amount of energy of 33.5 kcal mol−1 due to the C–C bond formation, concomitant with a spontaneous proton transfer from the amide group to the carbonyl oxygen atom on the aldehyde. The release of the Rh(I)-ligand from A11 yields the stable MCR product 4, with an exothermicity of 37.4 kcal mol−1. A comparison of the calculated pathways for N–H bond insertion and MCR clearly reveals that the associative pathway of N–H bond insertion is still less favorable in terms of kinetics and thermodynamics than that of MCR. These calculated results are in good agreement with our observations in experiments.
The proposed reaction mechanism is shown in Scheme 5. The reaction proceeds through Rh(I)-associated ammonium ylide intermediates A9/A10, which are generated from Rh(I)-associated carbene A7 and 2. The intermediates A9/A10 are trapped by electrophilic aldehyde 3via a favored attack model, leading to the addition intermediate A11. The dissociation of the Rh(I)-ligand from A11 with simultaneous 1, 2-proton transfer gives rise to the desired product 4.
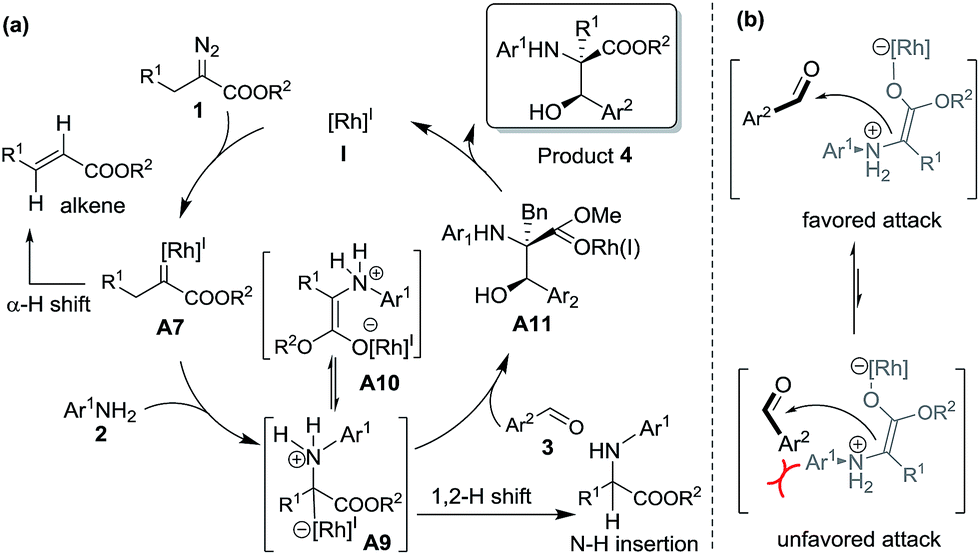 | ||
| Scheme 5 Plausible reaction mechanism and transition states. | ||
We then investigated the protein tyrosine phosphatase 1B (PTP1B) inhibitory activity of the products from this novel Rh(I)-catalyzed reaction (4a–d), with the aim of searching for new ways to treat type 2 diabetes mellitus (T2DM) and obesity.244a and 4c showed significant inhibitory activity against PTP1B, and the IC50 values were 10.04 and 5.73 μg mL−1, respectively.
Conclusions
In summary, we have discovered the first DFT-calculation inspired Rh(I)-catalyzed three-component reaction of α-alkyldiazoacetates via trapping of ammonium ylides before two rapid intramolecular processes: α-H transfer in alkyl carbenes and 1,2-H transfer in the resulting alkyl ylide. The α-H transfer in the α-alkyldiazoacetate carbene precursor was efficiently suspended by the association of the Rh(I) complex. An attempt to develop an asymmetric version of the Rh(I)/diene system was also successful with good enantioselectivity, and the biological activity of the products was primarily validated. The developed method provides an insight into extending the efficient transformation of α-alkyldiazoacetate-derived carbenes and affords β-hydroxyl α-alkyl-α-amino acids in moderate to good yields with diastereoselectivity.Acknowledgements
We thank the NSFC (No. 21332003, 21473056 and 21672066) and the STCSM (No. 15ZR1411000) for financial support. We thank Prof. Jia Li and Lixin Gao for the PTP1B inhibitory assay.Notes and references
- (a) K. B. Liao, S. Negretti, D. J. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230 CrossRef CAS PubMed; (b) D. Chen, D. X. Zhu and M. H. Xu, J. Am. Chem. Soc., 2016, 138, 1498 CrossRef CAS PubMed; (c) D. Chen, X. Zhang, W. Y. Qi, B. Xu and M. H. Xu, J. Am. Chem. Soc., 2015, 137, 5268 CrossRef CAS PubMed; (d) S. F. Zhu, Y. Can, H. X. Mao, J. H. Xie and Q. L. Zhou, Nat. Chem., 2010, 2, 546 CrossRef CAS PubMed.
- J. J. Shen, S. F. Zhu, Y. Cai, H. Xu and X. L. Xie, Angew. Chem., 2014, 126, 13404 ( Angew. Chem., Int. Ed. , 2014 , 53 , 13188 ) CrossRef.
- A. Padwa and M. D. Weingarten, Chem. Rev., 1996, 96, 223 CrossRef CAS PubMed.
- N. Jiang, Z. H. Qu and J. B. Wang, Org. Lett., 2001, 3, 2989 CrossRef CAS PubMed.
- M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed.
- (a) B. Liu, S. F. Zhu, W. Zhang, C. Chen and Q. L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834 CrossRef CAS PubMed; (b) S. F. Zhu, C. Chen, Y. Cai and Q. L. Zhou, Angew. Chem., 2008, 120, 946 ( Angew. Chem., Int. Ed. , 2008 , 47 , 932 ) CrossRef.
- W. Li, X. Liu, X. Hao, Y. Cai, L. Lin and X. Feng, Angew. Chem., 2012, 124, 8772 ( Angew. Chem., Int. Ed. , 2012 , 51 , 8644 ) CrossRef.
- S. Bachmann, D. Fielenbach and K. A. Jørgensen, Org. Biomol. Chem., 2004, 2, 3044 CAS.
- X. C. Ma, J. Jiang, S. Y. Lv, W. F. Yao, Y. Yang, S. Y. Liu, F. Xia and W. H. Hu, Angew. Chem., Int. Ed., 2014, 53, 13136 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS; (b) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS; (c) V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Chem. Phys., 2001, 22, 976 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) E. Nakamura, N. Yoshikai and M. Yamanaka, J. Am. Chem. Soc., 2002, 124, 7181 CrossRef CAS PubMed; (b) H. T. Bonge and T. Hansen, J. Org. Chem., 2010, 75, 2309 CrossRef CAS PubMed; (c) S. Y. Liu, W. F. Yao, Y. Liu, Q. H. Wei, J. H. Chen, X. Wu, F. Xia and W. H. Hu, Sci. Adv., 2017, 3, e1602467 CrossRef PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- (a) Contemporary Carbene Chemistry, John Wiley & Sons, ed. R. A. Moss and M. P. Doyle, 2013 Search PubMed; (b) X. Guo and W. H. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed.
- (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (b) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC.
- (a) W. H. Hu, X. F. Xu, J. Zhou, W. J. Liu, H. X. Huang, J. Hu, L. P. Yang and L. Z. Gong, J. Am. Chem. Soc., 2008, 130, 7782 CrossRef CAS PubMed; (b) X. Zhang, H. X. Huang, X. Guo, X. Y. Guan, L. P. Yang and W. H. Hu, Angew. Chem., 2008, 120, 6749 CrossRef; (c) J. Jiang, H. Xu, J. Xi, B. Ren, F. Lv, X. Guo, L. Jiang, Z. Zhang and W. H. Hu, J. Am. Chem. Soc., 2011, 133, 8428 CrossRef CAS PubMed; (d) D. Zhang, J. Zhou, F. Xia, Z. H. Kang and W. H. Hu, Nat. Commun., 2015, 6, 5801 CrossRef CAS PubMed; (e) L. Qiu, X. Guo, Y. Qian, C. C. Jing, C. Q. Ma, S. Y. Liu and W. H. Hu, Chem. Commun., 2016, 52, 11831 RSC.
- (a) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (b) M. González-López and J. T. Shaw, Chem. Rev., 2009, 109, 164 CrossRef PubMed; (c) J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371 CrossRef CAS PubMed; (d) J. P. Zhu, Q. Wang and M. X. Wang, Multicomponent Reaction in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed.
- (a) T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508 CrossRef CAS PubMed; (b) C. Defieber, J. F. Paquin, S. Serna and E. M. Carreira, Org. Lett., 2004, 6, 3873 CrossRef CAS PubMed.
- A. V. R. Rao, M. K. Gurjar, K. L. Reddy and A. S. Rao, Chem. Rev., 1995, 95, 2135 CrossRef CAS.
- (a) G. G. Li, H. T. Chang and K. B. Sharpless, Angew. Chem., Int. Ed., 1996, 35, 451 CrossRef CAS; (b) T. Kimura, V. P. Vassilev, G. J. Shen and C. H. Wong, J. Am. Chem. Soc., 1997, 119, 11734 CrossRef CAS.
- (a) Y. Liu, Z. Yu, J. Z. H. Zhang, L. Liu, F. Xia and J. L. Zhang, Chem. Sci., 2016, 7, 1988 RSC; (b) Y. Liu, Z. Yu, J. Z. H. Zhang, L. Liu and F. Xia, J. Phys. Chem. A, 2016, 120, 1925 CrossRef CAS PubMed.
- (a) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z. X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS PubMed; (b) Y. Liang, H. Zhou and Z. X. Yu, J. Am. Chem. Soc., 2009, 131, 17783 CrossRef CAS PubMed.
- Z. Y. Zhang and S. Y. Lee, Expert Opin. Invest. Drugs, 2003, 12, 223 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: DFT calculation details, optimized structures, experimental data and biological activity tests. CCDC 984118. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00257b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |