
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the excitonic processes in polymeric photocatalysts†
Hui
Wang‡
,
Shenlong
Jiang‡
,
Shichuan
Chen
,
Xiaodong
Zhang
*,
Wei
Shao
,
Xianshun
Sun
,
Zhi
Zhao
,
Qun
Zhang
*,
Yi
Luo
and
Yi
Xie
 *
*
Hefei National Laboratory for Physical Science at the Microscale, Collaborative Innovation Center of Chemistry for Energy Materials, Synergetic Innovation Center of Quantum Information and Quantum Physics, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: zhxid@ustc.edu.cn; qunzh@ustc.edu.cn; yxie@ustc.edu.cn
First published on 24th March 2017
Abstract
Understanding the photoexcitation processes in semiconductors is critical for the design of advanced photocatalytic materials. Nevertheless, traditional viewpoints focus on photogenerated free charge carriers, which are somehow invalid once the many-body effects are taken into account, especially for polymeric photocatalysts. Here we systematically investigate the photoexcitation processes involved in the polymer matrix of graphitic carbon nitride (g-C3N4) by combining photoluminescence spectroscopy and ultrafast transient absorption spectroscopy, validating the strong excitonic effects in the well-known photocatalyst for the first time. The identification of the robust triplet–triplet annihilation process, in which two triplet excitons collide to produce a singlet exciton, highlights an important nonradiative depopulation pathway of excited species and thereby offers potential strategies to regulate the photocatalytic activities of polymeric g-C3N4. The work establishes a new understanding of the photocatalytic mechanism in the polymeric g-C3N4 matrix, and thus paves the way for designing effective polymeric photocatalysts through excitonic engineering.
Introduction
For decades, enormous efforts have been devoted to the development of photocatalysts with efficient artificial solar-driven catalytic processes owing to their great potential for solving the energy crisis and preventing environmental pollution.1–3 Among the large number of photocatalytic materials, polymeric semiconductors show intriguing prospects by virtue of their rich sources, high stability, and easy regulation.4–6 Nevertheless, an ambiguous understanding of the photoinduced processes in these fascinating materials would inevitably hamper further progress. In contrast to inorganic photocatalysts that dominate weak charge interactions due to their strong screening effects and large dielectric properties, polymeric photocatalysts usually possess much stronger interactions between electrons and holes.7,8 In this case, distinct photoexcitation processes would be expected once Coulomb interactions among charges are taken into account, resulting in neutral excitons or bound electron–hole pairs that play an important role in the charge transfer and energy transfer processes in polymeric semiconductors.9 However, the excitonic effects and relevant excitonic processes involved in polymeric photocatalysts have long been ignored. For instance, optimization of band structures and charge separation, which is currently accepted as one of the most efficient strategies for the regulation of hot-carrier generation in photocatalysts, is incomprehensive when the competitive exciton generation (with respect to hot-carrier generation) is taken into account. The robust excitonic effects in these materials would lead to strong interactions among excited species, and thus complicated photoexcitation processes. Furthermore, the excitonic effects are known to be closely related to the quantum efficiency of photoexcited systems, offering an alternative tool for the design of efficient functional materials.10–13 Therefore, it is necessary to take the relevant excitonic processes into account when dealing with strong excitonic systems. For the well-known graphitic carbon nitride g-C3N4, most arguments focused on photogenerated charge carrier regulation toward promoted photocatalytic performances,14–18 whereas the involved excitonic processes have seldom been explored. Recently we demonstrated the existence of singlet and triplet excitons in g-C3N4 and found that the involved excitonic processes can be effectively regulated by the incorporation of carbonyl groups, thereby facilitating the generation of triplet excitons and enhancing the production of singlet oxygen.19 The intriguing excitonic-effects-dominated photocatalytic molecular oxygen activation behavior in polymeric carbon nitride inspires us to further pursue excitonic aspects in polymeric photocatalysts.Herein, we demonstrated that excitonic effects are closely related to the quantum efficiency of the photocatalytic processes of the g-C3N4 system. By means of photoluminescence and ultrafast transient absorption spectroscopy, we verified the P-type delayed fluorescence associated with the robust triplet–triplet annihilation (TTA) process in the polymeric g-C3N4 matrix for the first time. The novel conversion from two triplet excitons to a singlet exciton confirms the existence of strong excitonic interactions among the excited species, and reveals the nonradiative depopulation pathway of excited species in the g-C3N4 matrix. A comprehensive understanding of the involved photophysical processes was gained, which enables us to pursue effective photocatalytic performances in polymeric systems via excitonic engineering. This work offers an in-depth understanding of the excitonic processes in the carbon nitride matrix, in which the photophysical processes and the strong correlations among excitons in the carbon nitride matrix are described in detail for the first time.
Results and discussion
The g-C3N4 samples were prepared through the thermal condensation of melamine. As shown in Fig. 1a, the X-ray diffraction (XRD) pattern shows two main diffraction peaks around 13.1° and 27.8°, which can be indexed to (100) interplanar structural packing and (002) interlayer stacking peaks, respectively. Fourier transform infrared (FT-IR) measurements were further performed to confirm the structure. As displayed in Fig. 1b, the bands at 810 and 1200–1650 cm−1 correspond to the out-of-plane bending modes of tri-s-triazine and the typical stretching modes of the C–N heterocycle, respectively, clearly revealing the tri-s-triazine-based structure of the g-C3N4 matrix. X-ray photoelectron spectroscopy (XPS) analyses indicate that the chemical composition is mainly carbon and nitride (Fig. 1c). Note that a tiny signal of O 1s was observed, arising as a result of the small number of absorbed oxygen molecules. The C 1s spectrum (inset of Fig. 1c) exhibits two main peaks centered at 284.8 and 288.2 eV, assignable to the standard reference carbon and the sp2-bonded carbon of the tri-s-triazine-based structure, respectively. The ultraviolet-visible (UV-vis) spectrum exhibits an optical absorption edge at around 430 nm (Fig. 1d). On the basis of the above structural characterizations, we conclude that a typical polymeric carbon nitride with a tri-s-triazine-based structure has been successfully obtained.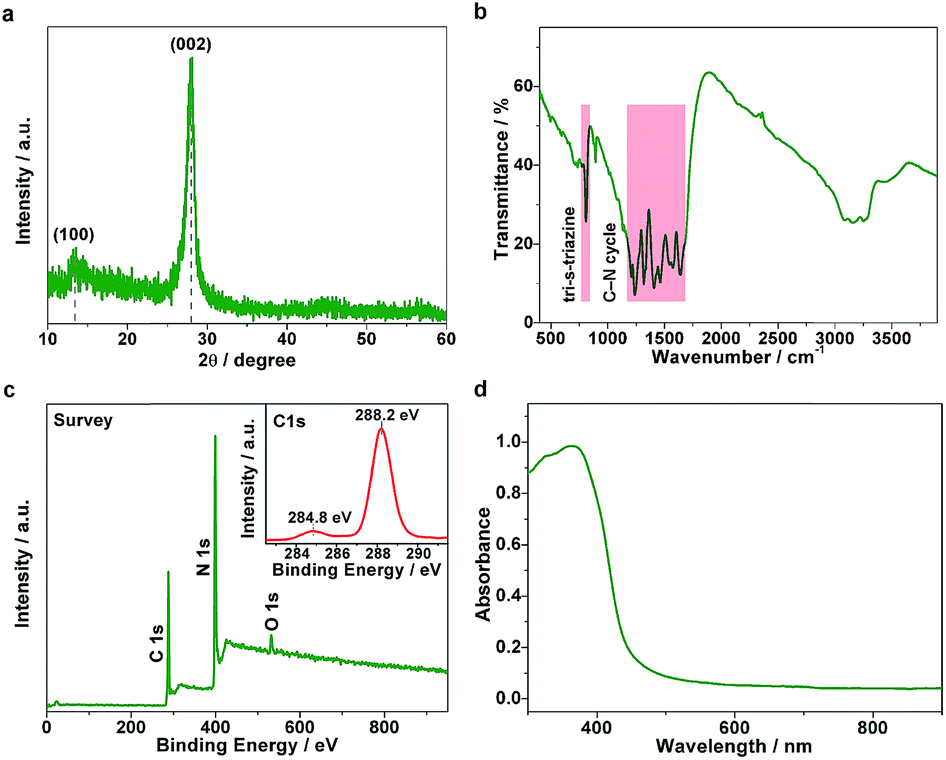 | ||
| Fig. 1 Structural characterizations. (a) XRD pattern, (b) FT-IR spectrum, (c) XPS survey spectrum (inset: C 1s spectrum), and (d) UV-vis absorption spectrum of the g-C3N4 sample. | ||
To understand the excitonic processes in the photoexcited g-C3N4 system, we performed photoluminescence measurements including steady-state and time-resolved prompt fluorescence (PF) and phosphorescence (PH) measurements. Different to PF which is responsible for the radiative decay of singlet excitons, PH comes from the radiative decay of triplet excitons and hence possesses a much longer lifetime than PF due to the spin-flip restriction.20,21 Thus it is possible to discriminate PH from PF in the spectra at certain delay times. Fig. 2a shows a representative PH spectrum (red) recorded at a very long delay time of 1 ms, which exhibits an obvious red-shift with respect to the PF profile (black). The PF and PH emissions were found to peak at 452 and 496 nm, respectively, suggesting a singlet–triplet energy gap of ∼0.243 eV, in accordance with our previous report.19 The tiny discrepancy is inevitable due to a few differences in the details of preparation for the two papers. Temperature-dependent fluorescence spectra suggest a significant enhanced emission at low temperature (see Fig. S1, ESI†). Given the richness of the structural defects in the g-C3N4 sample, the emission would be mainly assigned to excitonic decay rather than the recombination of charge carriers (which would be tightly trapped at defect sites at a low temperature).
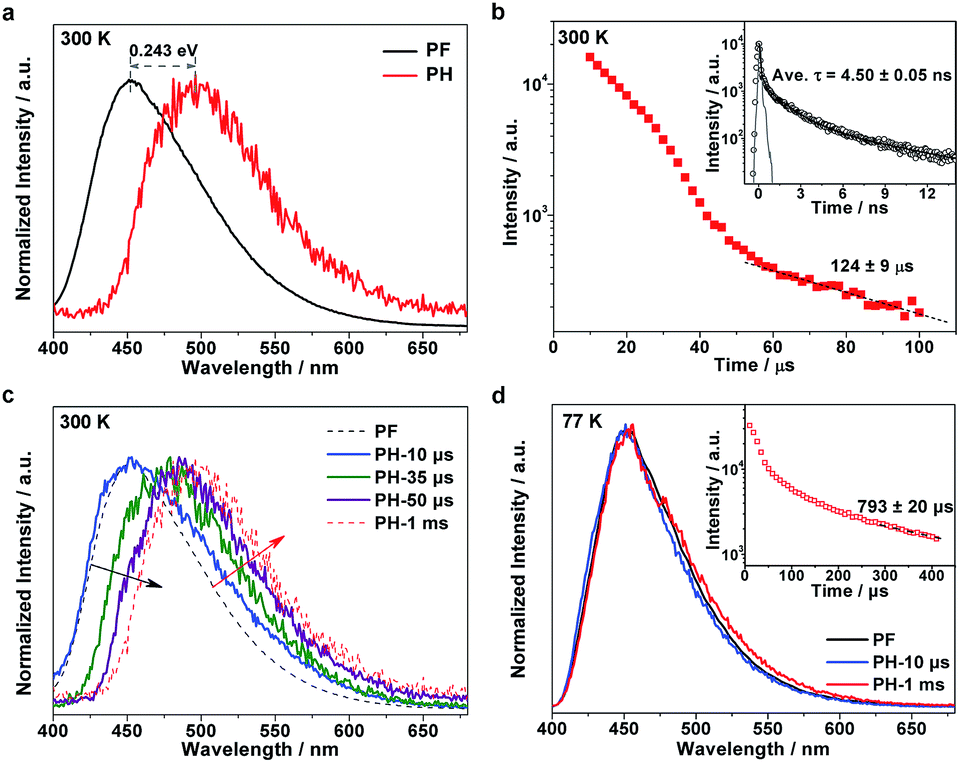 | ||
| Fig. 2 (a) Normalized steady-state PF and PH spectra at 300 K (delay time 1 ms). PH was recorded by accumulating 50 flash counts per point. (b) Time-resolved PH kinetics at 300 K. Inset: time-resolved PF kinetics at 300 K. (c) Normalized steady-state PH spectra at different delay times. (d) Normalized steady-state PF and PH spectra at 77 K. Inset: time-resolved PH kinetics at 77 K. | ||
To investigate the exciton kinetics involved in the system, we further performed time-resolved PF and PH measurements by monitoring their corresponding emission peaks. Given the heterogeneity of the carbon nitride matrix and possibly the fluorophore–fluorophore interactions,22 it turned out that a triple-exponential fitting can describe the PF decay well (inset in Fig. 2b; fitting results listed in Table S1†), yielding a mean lifetime of ∼4.5 ns. In terms of the PH kinetics, an interesting multi-stage decay profile was observed (Fig. 2b). The occurrence of early, fast stages most likely reflects bimolecular long-range triplet–triplet annihilation.22 With a decrease in the concentration of triplet excitons, the decay finally becomes a monomolecular exponential fit, with a triplet lifetime of ∼124 μs.
It is well known that in strongly confined systems like organic crystals, the excitation of nearby molecules can result in exciton–exciton annihilation due to strong interactions. To examine the spectral evolution associated with the early, fast stages, we conducted PH measurements at different delay times ranging from 10 to 50 μs. As shown in Fig. 2c, the 10 μs delayed emission (labeled PH-10 μs; blue, solid line) nearly coincides with the PF profile (black, dashed line) but its wing is broadened towards the red end of the spectrum. Given the huge difference between the PF lifetime (∼4.5 ns) and the delay time (10 μs), the dominant portion of the PH-10 μs profile should be attributed to delayed fluorescence, while its broadened wing reflects the PH contribution. Notably, the emergence of dominant delayed fluorescence implies the existence of a certain pathway leading to the longtime generation of singlet excitons. By increasing the delay times from 10 to 35 and further to 50 μs, the delayed fluorescence contribution is gradually reduced (black, oblique arrow) while the PH contribution increases (red, oblique arrow). When further delayed to 1 ms, the PH measurements yielded the nominal PH profile (red, dashed line). Apparently, the evolution of the delayed emission spectra discloses the unique photoexcitation processes in the g-C3N4 matrix, which involves the conversion between singlet and triplet excitons.
To investigate the involved photophysical processes for the above singlet–triplet conversion, we performed low-temperature (77 K) measurements with other conditions unchanged. Remarkably, the steady-state emissions monitored at 10 μs and 1 ms turned out to be nearly identical to the PF profile, as shown in Fig. 2d. Although the time-resolved PH profile also features multiple stages (inset of Fig. 2d), the observed emissions seemed independent of the adopted delay times. Thus it would be safe to assign such emissions to delayed fluorescence rather than PH. Moreover, low temperature prompt emission exhibits a narrowed wing, suggesting the promoted contribution of singlet-exciton radiative recombination towards prompt fluorescence under room temperature (see Fig. S2, ESI†). The fact that such delayed fluorescence emissions were observed at 77 K (instead of 300 K) indicates that they should be P-type delayed fluorescence emissions, which are induced by triplet–triplet annihilation (TTA), rather than thermally activated E-type delayed fluorescence.23–26 On the basis of the above observations and analyses, we can summarize the pertinent photophysical processes in the g-C3N4 system as follows:
(1) Photoexcitation:
| CN + hv → CNS, |
(2) PF:
| CNS → CN + hv, |
(3) Intersystem crossing:
| CNS → CNT, |
(4) PH:
| CNT → CN, |
(5) TTA:
| CNT + CNT → CNS + CN, |
(6) Delayed fluorescence:
| CNS → CN + hv, |
It is worth noting that the strong correlations (e.g. excitonic effects, triplet–triplet annihilation) among the photogenerated species tend to be intrinsic features of the g-C3N4 matrix, which are not dependent on defective structures (see Fig. S3, ESI†). Since the photoexcitation of g-C3N4 is strongly dependent on which precursor is used, we further evaluate the photoluminescence properties of two other carbon nitride samples obtained by the thermal condensation of urea and dicyandiamide, respectively. The observation of delayed fluorescence features clearly suggests inherent excitonic effects and triplet–triplet annihilation in these carbon nitride samples (see details in Fig. S4, ESI†), further verifying our conclusion of strong correlations among photogenerated species in the g-C3N4 matrix.
To further understand the excitonic processes involved in the system, we examined the g-C3N4 samples using femtosecond time-resolved transient absorption (TA) spectroscopy, a useful tool proven to be robust for tracking the ultrafast conversion between singlet and triplet excitons.27–30 A scheme featuring an ultraviolet pump/white-light continuum (WLC) probe was employed in our TA measurements (details in ESI†). A femtosecond pump laser was chosen with a 400 nm wavelength, suitable for exciting the samples (refer to the UV-vis absorption spectrum in Fig. 1d). The subsequent WLC probe (420–720 nm) monitored the spectral evolution (Fig. 3a) from the first singlet excited state (S1). Note that the relaxation kinetics were found to depend on the probing wavelength. In order to properly determine the characteristic relaxation time constants, we applied a global fitting procedure to a set of kinetic traces acquired from 510 to 560 nm (6 traces with a 10 nm interval), as shown in Fig. 3b. The tri-exponential global fitting results are τ1 = 3.4 ± 0.3 ps (50%), τ2 = 43 ± 5 ps (20%), and τ3 = 3270 ± 68 ps (30%). With the aid of the photoluminescence results discussed above, the three consecutive relaxation pathways can be understood as follows. Following an initial, fast (a few picoseconds) intersystem crossing (ISC) process (τ1) from S1 to T1 (the lowest triplet excited state), the resulting T1 excitons may undergo a TTA process to generate S1 excitons on a time scale of a few tens of picoseconds (τ2), and the formed S1 excitons subsequently decay on a nanosecond time scale (τ3). The relevant photophysical diagram is schematically depicted in Fig. 3c. Note that the above assignments are based on the following two facts: (i) within the nanosecond time window during the TA measurements, the early-time, high triplet-exciton concentration facilitates the TTA process, rationalizing its fast nature; (ii) the slowest relaxation time constants derived from the TA measurements (∼3.3 ns) reasonably match the corresponding time-resolved fluorescence results (∼4.5 ns, see Fig. 3b), indicating a transition from S1 to S0.
 | ||
| Fig. 3 Ultrafast TA characterizations. (a) Representative TA spectra at different probe delays and (b) TA kinetic traces at different probing wavelengths for g-C3N4 (0.68 mJ cm−2). The solid line shows the global fitting of the decay kinetics using a tri-exponential function. The TA signal is given in mOD where OD stands for optical density. (c) Schematic illustration of the photophysical processes involved in the g-C3N4 system. | ||
Considering that the TTA process is highly dependent on exciton density, we further examined the exciton kinetics of photoexcited g-C3N4 at different pump fluences. The TA spectra show similar profiles (Fig. 4a), whereas the exciton kinetics exhibit pronounced pump-fluence dependence (Fig. 4b): with an increase in pump fluence from 0.51 to 0.85 mJ cm−2, τ1, τ2, and τ3 vary from ∼3.5 to ∼3.3 ps, ∼60 to ∼26 ps, and ∼4.5 to ∼2.2 ns, respectively. The negligible change in the first component suggests that the ISC rate is independent of the exciton concentration. Nevertheless, the second component that represents the TTA process shows pronounced acceleration with increasing pump fluence. Such an accelerated TTA process arises as a result of the promoted collision probability due to the increased concentration of triplet excitons under higher pump fluences. Moreover, the third component that represents the S1 exciton decay also exhibits acceleration to a certain extent with an increase in pump fluence, which originates from the singlet exciton–exciton annihilation that is known to widely exist in strongly confined systems.31,32 This set of pump fluence dependence experiments supports the strong excitonic effects in the g-C3N4 matrix.
 | ||
| Fig. 4 Transient absorption spectra and dynamics under different conditions. (a) Representative TA spectra taken at a probe delay of 3 ps under different pump fluences (0.51 and 0.85 mJ cm−2). (b) TA kinetic traces at different probing wavelengths (510–560 nm, 6 traces with a 10 nm interval) under different pump fluences. (c) TA kinetic traces at different probing wavelengths (510–560 nm, 6 traces with a 10 nm interval) in different solvents. (d) Time-resolved PF kinetics at 300 K in different solvents. | ||
To gain deeper insights into the involved exciton kinetics, we conducted a comparison test in the following two situations: g-C3N4 samples in mixtures of water/2-bromoethanol and water/ethanol. On the basis of the prominently promoted spin–orbit coupling by an external bromine atom,33,34 the enhanced ISC process is expected to bring about a promoted triplet-exciton yield, thus resulting in a more effective TTA process in the presence of a bromine atom. As predicted, in the presence of a bromine atom the three decay processes are accelerated: τ1 from ∼3.1 to ∼2.0 ps, τ2 from ∼40 to ∼12 ps, and τ3 from ∼3554 to ∼102 ps, as shown in Fig. 4c. Also, it can be seen in the corresponding time-resolved PF spectra (Fig. 4d; fitting results are listed in Table S1, ESI†) that the heavy-atom effect results in the significant reduction of exciton lifetimes (from ∼3.24 to ∼0.50 ns on average), reflecting the enhanced exciton depopulation that correlates to the accelerated conversion between singlet and triplet excitons. Again, the existence of robust excitonic processes within the g-C3N4 matrix is strongly confirmed by the above ultrafast TA results in conjunction with the photoluminescence results. The above discussions clearly validate the proposed model which describes the involved photophysical processes well, and confirm the robust excitonic effects in the polymeric g-C3N4 matrix. Furthermore, the two verifications (that is, pump fluence and heavy-atom-effect regulations) provide us with excitonic engineering strategies to regulate the photocatalytic performances of the g-C3N4 matrix.
To further understand the excitonic effects on the photocatalytic performance of a g-C3N4 sample, 2-bromoethanol was introduced into the photocatalytic system of the g-C3N4 matrix. Hydrogen evolution measurements were carried out, and it was found that the g-C3N4 sample possesses an average H2 evolution rate of ∼6.7 μmol h−1 in a methanol/ethanol/water system (as shown in Fig. 5a). Nevertheless, H2 generation is significantly reduced with the addition of 2-bromoethanol, to only ∼0.4 μmol h−1. Electron spin resonance (ESR) measurements were then performed to investigate the molecular oxygen activation behaviors of a g-C3N4 sample in the presence/absence of 2-bromoethanol. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was first employed as a trapping agent for the superoxide radical (O2˙−). As illustrated in Fig. 5c, the sextet ESR signal is in accordance with that of DMPO–OOH, a spin adduct derived from DMPO–O2˙−,35 verifying the generation of O2˙−. Furthermore, the ESR spectra of the 2,2,6,6-tetramethylpiperidine (TEMP) and g-C3N4 mixture display typical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet signals with a g-value of 2.0056 (as shown in Fig. 5d), which are in accordance with those of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO),36 suggesting the generation of singlet oxygen (1O2) in the photocatalytic molecular oxygen activation process. However, the generation of both O2˙− and 1O2 was suppressed in the presence of a Br atom. As depicted in Fig. 5d, the generation of H2 and O2˙− occurs mainly through the charge transfer process, in which the H2O and O2 molecules are reduced by photogenerated electrons. On the other hand, the exciton energy transfer process between long-lived triplet excitons and ground state oxygen molecules is feasible for 1O2 generation. The simultaneous suppression of the photocatalytic processes is ascribed to the drastic consumption of photoexcited species (both charge carriers and excitons) through the promoted TTA process caused by the additional 2-bromoethanol. Moreover, the excitonic effects in g-C3N4 nanosheets on the optical excitation processes were also investigated (see details in Fig. S5, ESI†), as the close relationship between thickness and photocatalytic performances. Herein, the significant suppressed intersystem crossing rate in carbon nitride nanosheets was first demonstrated, leading to a low triplet exciton concentration and faint triplet–triplet annihilation, which not only echoes to the extremely high PL quantum yield and photocatalytic performance,37–40 but also confirms the crucial role of the excitonic effect in the photoexcitation process in g-C3N4. In view of the above analyses, we conclude that the excitonic effect in g-C3N4 matrix is crucial for achieving efficient photoexcitation applications.
:1:1 triplet signals with a g-value of 2.0056 (as shown in Fig. 5d), which are in accordance with those of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO),36 suggesting the generation of singlet oxygen (1O2) in the photocatalytic molecular oxygen activation process. However, the generation of both O2˙− and 1O2 was suppressed in the presence of a Br atom. As depicted in Fig. 5d, the generation of H2 and O2˙− occurs mainly through the charge transfer process, in which the H2O and O2 molecules are reduced by photogenerated electrons. On the other hand, the exciton energy transfer process between long-lived triplet excitons and ground state oxygen molecules is feasible for 1O2 generation. The simultaneous suppression of the photocatalytic processes is ascribed to the drastic consumption of photoexcited species (both charge carriers and excitons) through the promoted TTA process caused by the additional 2-bromoethanol. Moreover, the excitonic effects in g-C3N4 nanosheets on the optical excitation processes were also investigated (see details in Fig. S5, ESI†), as the close relationship between thickness and photocatalytic performances. Herein, the significant suppressed intersystem crossing rate in carbon nitride nanosheets was first demonstrated, leading to a low triplet exciton concentration and faint triplet–triplet annihilation, which not only echoes to the extremely high PL quantum yield and photocatalytic performance,37–40 but also confirms the crucial role of the excitonic effect in the photoexcitation process in g-C3N4. In view of the above analyses, we conclude that the excitonic effect in g-C3N4 matrix is crucial for achieving efficient photoexcitation applications.
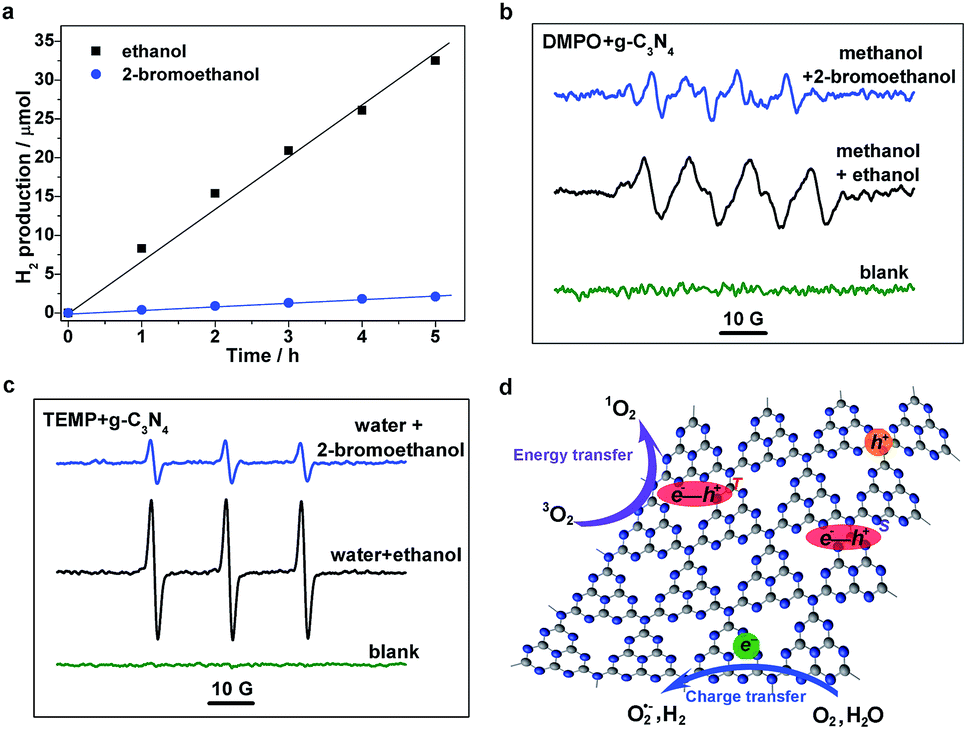 | ||
| Fig. 5 Photocatalytic evaluation of g-C3N4 samples. (a) Time-dependent H2 evolution. (b) DMPO- and (c) TEMP-trapped ESR measurements in different solutions. (d) Schematic illustration for various photocatalytic processes. | ||
Conclusions
To summarize, the strong excitonic effects and relevant excitonic processes involved in polymeric photocatalysts have been systematically investigated for the first time using polymeric g-C3N4 as an example. On the one hand, the results from photoluminescence spectroscopy indicate that the involved P-type delayed fluorescence originates from triplet–triplet annihilation at room temperature, revealing strong excitonic effects and highlighting the important depopulation pathway of excited species that is closely related to the quantum efficiency of photoexcitation applications in the g-C3N4 matrix. On the other hand, the results from ultrafast transient absorption spectroscopy elucidate the involved photophysical processes, offering effective research approaches as well as regulation strategies for exciton kinetics in a polymer matrix. This work not only establishes a comprehensive understanding of the photocatalytic processes in a polymer matrix of graphitic carbon nitride, but also provides valuable guidance from a fundamental perspective for the design of other polymeric photocatalysts based on excitonic engineering.Acknowledgements
The work was supported by the National Natural Science Foundation of China (U1532265, U1632149, 21331005, 11321503, 21401181, 91422303, 21573211, 21633007, 21421063), the Youth Innovation Promotion Association of CAS (2017493), the National Key R & D Program on Nano Science & Technology of the MOST (2016YFA0200602), the Strategic Priority Research Program of the CAS (XDB01020000), and the Fundamental Research Funds for the Central Universities (WK2060190027, WK2340000063, WK6030000020).Notes and references
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206 RSC.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- N. Serpone and A. V. Emeline, J. Phys. Chem. Lett., 2012, 3, 673 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505 CrossRef CAS PubMed.
- Z. Zhang, J. Long, L. Yang, W. Chen, W. Dai, X. Fu and X. Wang, Chem. Sci., 2011, 2, 1826 RSC.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354 RSC.
- S. Brazovskii and N. Kirova, Chem. Soc. Rev., 2010, 39, 2453 RSC.
- J. L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971 CrossRef PubMed.
- Y. Cao, I. D. Parker, G. Yu, C. Zhang and A. J. Heeger, Nature, 1999, 397, 414 CrossRef CAS.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334 CrossRef CAS PubMed.
- O. E. Semonin, J. M. Luther, S. Choi, H. Y. Chen, J. B. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530 CrossRef CAS PubMed.
- H. Wang, X. Sun, D. Li, X. Zhang, S. Chen, W. Shao, Y. Tian and Y. Xie, J. Am. Chem. Soc., 2017, 139, 2468 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893 RSC.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763 CrossRef CAS.
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970 CrossRef CAS PubMed.
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li, Z. H. Wang, J. S. Liu and X. C. Wang, Chem. Soc. Rev., 2014, 43, 5234 RSC.
- H. Wang, S. Jiang, S. Chen, D. Li, X. Zhang, W. Shao, X. Sun, J. Xie, Z. Zhao, Q. Zhang, Y. Tian and Y. Xie, Adv. Mater., 2016, 28, 6940 CrossRef CAS PubMed.
- Y. V. Romanovskii, A. Gerhard, B. Schweitzer, U. Scherf, R. I. Personov and H. Bässler, Phys. Rev. Lett., 2000, 84, 1027 CrossRef CAS PubMed.
- A. P. Monkman, H. D. Burrows, L. J. Hartwell, L. E. Horsburgh, I. Hamblett and S. Navaratnam, Phys. Rev. Lett., 2001, 86, 1358 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirts and C. L. Fraser, Nat. Mater., 2009, 8, 747 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580 RSC.
- C. A. Parker and T. A. Joyce, Chem. Commun., 1966, 185, 234 RSC.
- J. Partee, E. L. Frankevich, B. Uhlhorn, J. Shinar, Y. Ding and J. T. Barton, Phys. Rev. Lett., 1999, 82, 3673 CrossRef CAS.
- X. Sun, X. Wang, X. Li, J. Ge, Q. Zhang, J. Jiang and G. Zhang, Macromol. Rapid Commun., 2015, 36, 298 CrossRef CAS PubMed.
- J. Ge, Q. Zhang, J. Jiang, Z. Geng, S. Jiang, K. Fan, Z. Guo, J. Hu, Z. Chen, Y. Chen, X. Wang and Y. Luo, Phys. Chem. Chem. Phys., 2015, 17, 13129 RSC.
- B. Wu, J. Hu, P. Cui, L. Jiang, Z. Chen, Q. Zhang, C. Wang and Y. Luo, J. Am. Chem. Soc., 2015, 137, 8769 CrossRef CAS PubMed.
- J. Hu, Q. Zhang and Y. Luo, J. Phys. Chem. Lett., 2016, 7, 3908 CrossRef CAS PubMed.
- Y. Z. Ma, L. Valkunas, S. L. Dexheimer, S. M. Bachilo and G. R. Fleming, Phys. Rev. Lett., 2005, 94, 157402 CrossRef PubMed.
- I. B. Martini, A. D. Smith and B. J. Schwartz, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 35204 CrossRef.
- S. P. McGlynn, R. Sunseri and N. Christodouleas, J. Chem. Phys., 1962, 37, 1818 CrossRef CAS.
- A. Gorman, J. Killoran, C. O’Shea, T. Kenna, W. M. Gallagher and D. F. O’Shea, J. Am. Chem. Soc., 2004, 126, 10619 CrossRef CAS PubMed.
- S. Belkin, R. J. Mehlhorn, K. Hideg, O. Hankovsky and L. Packer, Arch. Biochem. Biophys., 1987, 256, 232 CrossRef CAS PubMed.
- R. Konaka, E. Kasahara, W. C. Dunlap, Y. Yamamoto, K. C. Chien and M. Inoue, Free Radical Biol. Med., 1999, 27, 294 CrossRef CAS PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18 CrossRef CAS PubMed.
- Y. Chen, B. Wang, S. Lin, Y. Zhang and X. Wang, J. Phys. Chem. C, 2014, 118, 29981 CAS.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures, tables, and experimental information. See DOI: 10.1039/c7sc00307b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |