
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApoptosis-independent organoruthenium anticancer complexes that overcome multidrug resistance: self-assembly and phenotypic screening strategies†
Mun Juinn
Chow
 *a,
Mohammad
Alfiean
b,
Giorgia
Pastorin
cd,
Christian
Gaiddon
ef and
Wee Han
Ang
*ad
*a,
Mohammad
Alfiean
b,
Giorgia
Pastorin
cd,
Christian
Gaiddon
ef and
Wee Han
Ang
*ad
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543 Singapore. E-mail: a0037979@u.nus.edu
bSchool of Chemistry and Life Sciences, Nanyang Polytechnic, Singapore
cDepartment of Pharmacy, National University of Singapore, 3 Science Drive 3, 117543 Singapore
dNUS Graduate School for Integrative Sciences and Engineering, Singapore. E-mail: chmawh@nus.edu.sg; Tel: +65 6516 5131
eUniversité de Strasbourg, Strasbourg, France
fU1113 INSERM, 3 Avenue Molière, Strasbourg 67200, France
First published on 28th February 2017
Abstract
Multidrug resistance is a major impediment to chemotherapy and limits the efficacies of conventional anticancer drugs. A strategy to bypass multidrug resistance is to develop new drug candidates capable of inducing apoptosis-independent programmed cell death. However, cellular pathways implicated in alternative programmed cell death are not well-elucidated and multifactorial, making a target-based discovery approach a challenge. Here, we show that a coordination-directed three-component assembly and phenotypic screening strategy could be employed as a viable alternative for the identification of apoptosis-independent organoruthenium anticancer agents. Through an on-plate synthesis and screening of 195 organoruthenium complexes against apoptosis-sensitive and -resistant cancers, we identified two apoptosis-independent hits. Subsequent validation of the two hits showed a lack of induction of apoptotic biomarkers, a caspase-independent activity and an equal efficacy in both apoptosis-sensitive and -resistant colorectal cancers. This validated their apoptosis-independent modes-of-action, paving the way as potential candidates for the treatment of highly-refractory cancer phenotypes.
Introduction
Multidrug resistance (MDR) remains a major impediment to chemotherapy and limited effective treatments for drug-resistant cancers such as melanoma, glioblastoma or non-small cell lung cancer (NSCLC) are available.1–3 One of the main contributing mechanisms to MDR in such cancers is the defective or selective adaptation of the apoptotic pathway, leading to increased resistance to apoptotic cell death.4–7 This includes changes in p53 levels or function, abnormal expression of pro-/anti-apoptotic factors such as the Bcl-2 family proteins, or loss-of-expression of apoptosis executor caspases.8,9 Consequently, most clinical drugs are ineffective against such cancers because they act via the induction of apoptotic cell death.10,11 There is an urgent need for new first-in-class anticancer drug candidates that act primarily by inducing non-apoptotic cell death.In recent years, several other types of programmed cell deaths have been identified involving cellular pathways and morphological changes distinct to that of apoptosis.12 Concomitantly, apoptosis-independent anticancer drug candidates have been explored as treatment options for apoptosis-resistant cancers.13–16 Several metallodrugs have shown potential for such applications. Cu(I) and Cu(II) complexes have been known to induce a form of programmed necrosis known as paraptosis, characterized by mitochondria swelling and cytoplasmic vacuolation.17–20 Two Re(V) oxo complexes were recently reported to induce necroptosis, a distinct form of programmed necrosis mediated by RIP1/RIP3 kinase complex.21 Several Cu, Fe, Au and Pt complexes have also exhibited the ability to induce autophagic cell death.22–25 A number of Ru(II) complexes induced non-apoptotic cell death, although the exact cell death types have yet to be characterized.26–29 For example, RAS-1H and RAS-1T could bypass apoptosis-resistance in an in vitro system (Fig. 1).28
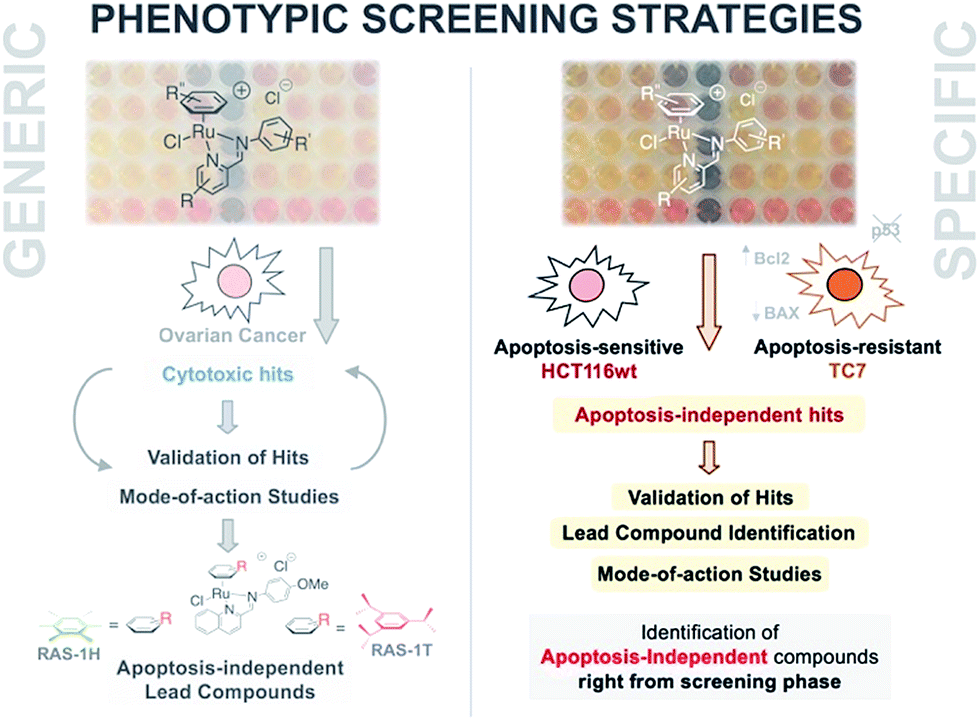 | ||
| Fig. 1 A phenotypic screening approach to identify apoptosis-independent Ru-arene Schiff-base complexes. (Left panel) Previous strategy with generic cytotoxicity screening approach; serendipitous discovery of apoptosis-independent compounds only after extensive mode-of-action studies.28,34,35 (Right panel) Phenotypic screening approach used for the current study; apoptosis-independent ‘hits’ could be identified right from the screening phase. Few steps required after to confirm validity of ‘hits’. | ||
In general, discovery of such compounds is sparse compared to the vast majority of apoptotic agents and few have been designed specifically for that goal. This is because, unlike the apoptosis pathway, which is extensively studied and contain many known druggable targets, cellular pathways implicated in other programmed cell deaths are less well-defined with fewer druggable targets. Thus, a target-based approach focused on the discovery of apoptosis-independent drugs is not entirely feasible. Furthermore, the efficiency of target-based drug discovery has been called into question, owing to the high attrition rates of candidates discovered through such an approach and the multifactorial nature of MDR.30–32 The low efficacy of these drug candidates has been attributed to false-positives in target validation and a failure to take into account redundancy and compensatory crosstalk in complex cellular signaling pathways.33
An alternative approach to target-based drug discovery would be the use of a phenotypic screening strategy for fast identification of hits. However, a generic phenotypic screening approach using only cytotoxicity as a standard-of-measure seldom yields drug candidates with novel mode-of-action.32 This means that identification of apoptosis-independent hits in this manner would rely on serendipitous discovery after extensive testing and mode-of-action studies (Fig. 1).28 Hence, a methodology combining the efficiency of the phenotypic screening strategy and the specificity of the target-based approach, coupled with the availability of a large compound library, would be required to accurately identify apoptosis-independent drug candidates in the early phases of the drug discovery process.
In the present study, we utilize a phenotypic screening strategy that would allow us to identify apoptosis-independent candidates right from the screening phase (Fig. 1). We coupled the use of (i) a previously established Coordination-Directed Three-Component Assembly (C3A) protocol as a convenient way to prepare a new library of 195 Ru-arene Schiff-base (RAS) complexes for screening,34 and (ii) the screening against two colorectal cancer cell lines with different sensitivity towards apoptosis i.e. apoptosis-sensitive HCT116 colorectal carcinoma and apoptosis-resistant TC7.36 We investigated the structural factors involved in self-assembly of RAS complexes and shed light on the structural-dependence of the C3A assembly process. Further analysis and comparison of the differential killing in both cell lines allowed for the identification of apoptosis-independent hits, which were validated by investigating their efficacy in resistant cell lines, caspase dependence and induction of apoptotic biomarkers. Although, similar phenotypic screening strategies exploiting differential cytotoxicity in paired cell lines for hit identification are known,37–39 this is the first one (to the best of our knowledge) developed specifically for the identification of apoptosis-independent anticancer drug candidates.
Results and discussion
Self-assembly of RAS complexes by C3A is highly dependent on steric encumbrance around Ru metal center
C3A is an ideal strategy for generating compound libraries for phenotypic screening strategies. We reported earlier environmental conditions such as solvent, pH suitability and functional group compatibility that impede RAS assembly,34 but detailed structural factors were not well-understood. A direct mechanistic study to glean structure–activity relationships could be revealing but would be herculean. However, since it was possible to access a large library of structurally-diverse RAS compounds via C3A, investigations into the effect of structural features, particularly the spatial arrangement of peripheral functional groups around the core RAS scaffold, on C3A assembly levels could be readily performed. Therefore, a total of 195 RAS complexes were prepared using C3A (Fig. 2b) using starting components as shown in Fig. 2.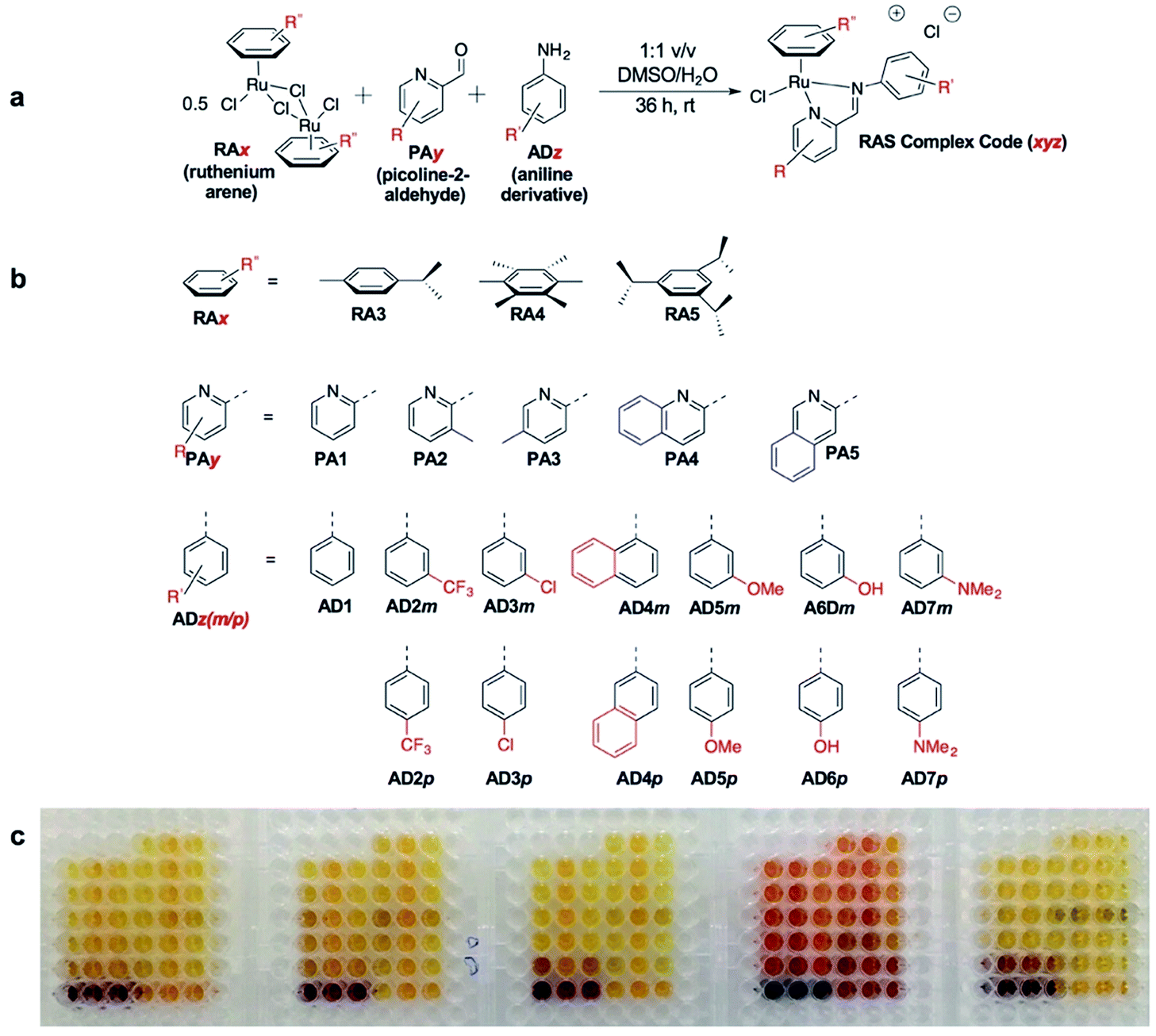 | ||
| Fig. 2 On-a-plate coordination-directed three-component assembly (C3A) of 195 RAS complexes. (a) General reaction scheme and condition. (b) The different functional moiety included in the current study. (c) A picture of the 195 complexes synthesized in a 96-well plate format. Each well contains a different RAS complex. | ||
Briefly, stock solutions containing RAx (10 mM), PAy (40 mM) and ADz (40 mM) were prepared in DMSO and added in a predetermined order to a 96-well plate containing equal volume of ddH2O, such that each well would contain one final RAS complex (Fig. 2c). The reaction mixtures were incubated with shaking at rt for 36 h before the crude products were analyzed by ESI-MS and the assembly yields ascertained using RP-HPLC. In general, 166 out of 195 (85%) RAS complexes assembled via C3A had high assembly yield/purity >70% with a significant proportion achieving >90% yields (Fig. 3a; ESI Table S1†). Only 9 (5%) RAS complexes were unsuccessful with assembly yields <50%.
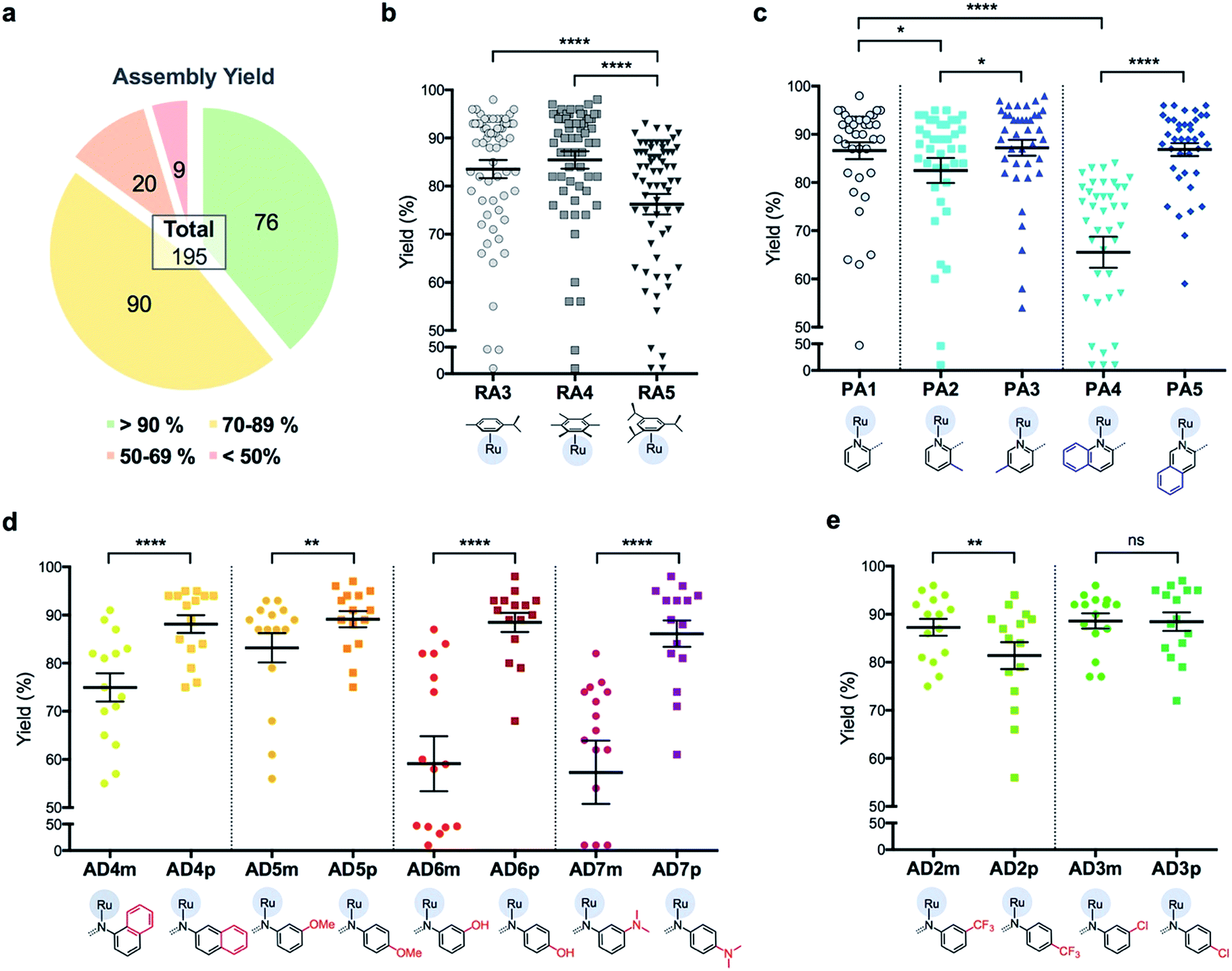 | ||
| Fig. 3 Analysis of assembly yield for 195 RAS complexes reveals a high dependence on steric environment around Ru-centre. (a) Summary of assembly yield, classified into four sub bands. (b) Comparison of assembly yield between the groups having different arene ligands. Mean ± S.E.M. (n = 65, ****p < 0.0001; paired one-way ANOVA test with Tukey post-hoc test). (c) Selected comparison of assembly yield between the groups having different picolinaldehyde (PA) ligands. Mean ± S.E.M. (n = 39, *p < 0.05, ****p < 0.0001; paired one-way ANOVA test with Tukey post-hoc test). (d and e) Comparison of assembly yield between the groups having different aniline derivative (AD) ligands. Mean ± S.E.M. (n = 15, ns – not significant, **p < 0.01, ****p < 0.0001; paired two-tailed t-test). | ||
The success of C3A assembly was influenced by both steric and electronic factors. First, components that contributed to increased steric bulk around Ru resulted in lower assembly yields. For instance, RAS complexes synthesized using more sterically-encumbered RA5 had significantly lower yields compared to less sterically hindered RA3 or RA4 (Fig. 3b). Similarly, the bulkier PA2 and PA4 had lower assembly yields compared to less bulky PA1 (Fig. 3c). Moreover, structural isomers with functional moieties positioned in closer proximity to Ru directly increased steric bulk and resulted in lower assembly yield. For example, less RAS assembly was obtained from PA4 compared to isomeric PA5 despite other conditions being maintained (Fig. 3c and 4a). Similarly, meta-aniline derivatives (ADm), which had functional groups positioned nearer to the Ru, also resulted in lower assembly yields compared to their para-analogues (ADp) (Fig. 3d and 4b). Furthermore, steric bulk that hindered imine formation during ligand assembly, as is the case for PA2 (Fig. 3c and 4c), also affected reaction yields.
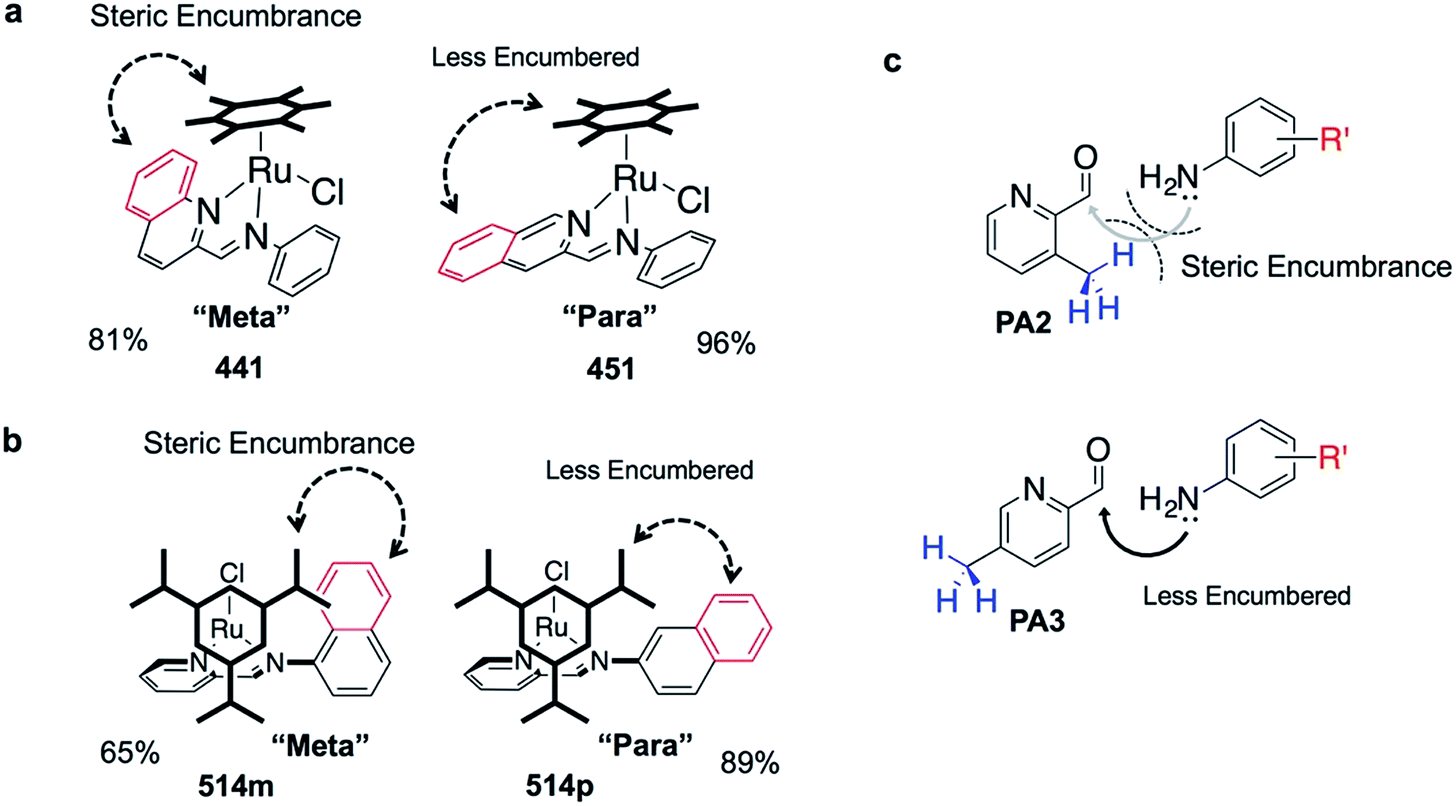 | ||
| Fig. 4 Diagrams illustrating how steric hindrance interferes with the assembly process, resulting in poorer assembly yield. (a) Comparing steric environment between 441 and 451. (b) Comparing steric environment between complexes 514m and 514p. (c) Comparing imine formation with PA2 and with PA3. Steric hindrance may make imine formation less favorable, which could hinder subsequent RAS formation. | ||
Beside the dominant steric factors, a secondary electronic effect also affected assembly yield. This was particularly evident in the case of an electron-donating R′ substituent on the AD component. R′ groups at the meta-positions resulted in steric hindrance that led to lower assembly while R′ groups at the para-positions had little influence on C3A. At the meta-position however, electron-donating R′ groups (AD6m, AD7m) consistently reduced assembly yields of RAS complexes (Fig. 3d) while electron-withdrawing R′ groups (AD2m, AD3m) improved yields marginally (Fig. 3e). The lower assembly levels by AD6m and AD7m were presumably due to competition of the substituents toward Ru-binding, which directly interfered with C3A. This was earlier observed for substituents comprising guanidine and thiol moieties which directly bound Ru. In the para-position (AD6p and AD7p), the substituents would be in conjugation with the aniline ring systems which reduced their nucleophilicity by mesomeric effect. In the meta-position however, the nucleophilicity of the substituents remained high and could compete for Ru-binding.
Taken together, C3A proved to be a highly reliable and efficient methodology for the synthesis of RAS complexes with highly predictable levels of RAS assembly. Complexes formed from the most favorable components gave the highest yield (e.g.437p, 98%) while those formed from the most steric encumbered components gave the lowest yield (e.g.547m, <10%).
Apoptosis-independent ‘hits’ identified using a phenotypic screening approach
In order to identify apoptosis-independent RAS complexes, we employed the use of two colorectal cancer cell lines with different sensitivity to drug treatment, namely wild type HCT116 and TC7. HCT116 is a cell lineage that has physiological expression of biomarkers associated with apoptosis such as p53, BAX and Bcl-2 and is therefore sensitive to drugs that act via the induction of apoptosis (apoptosis-sensitive). In contrast, TC7 is p53-null, has decreased expression of pro-apoptotic BAX and increased expression of anti-apoptotic Bcl-2, making it the least sensitive to apoptosis-inducing drugs in a panel of tested colorectal cancer cell lines (apoptosis-resistant).36 Apoptosis-inducing clinical drugs such as oxaliplatin, doxorubicin and 5-fluorouracil were shown be highly cytotoxic towards HCT116 but had significantly poorer activities against TC7. In contrast, compounds which exerted their efficacies via apoptosis-independent pathways were equipotent against HCT116 and TC7.28 Therefore, we screened RAS complexes against these 2 well-characterized cell lines by taking advantage of their phenotypic differences.First, 166 RAS complexes with assembly yield greater than 70% were selected for screening in HCT116. The RAS library was synthesized on-plate (96-well) without any further work-up or purification and exposed directly to cells at a fixed concentration of 25 μM. Cell viabilities were determined after 48 h (ESI Fig. S3 and Table S2†) and analyses were consistent with previous studies that showed a correlation between hydrophobicity and cytotoxicity (Fig. 5), despite the fact that the complexes were intrinsically hydrophilic as determined by log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) pow measurements.34,35 Variation in the position of the functional groups did not have any observable effect on cytotoxicities since RAS complexes prepared from PA4 and PA5 showed similar inhibition of cancer cell viabilities (Fig. 5c). The same was observed when comparing ADm and their ADp counterparts (Fig. 5d).
pow measurements.34,35 Variation in the position of the functional groups did not have any observable effect on cytotoxicities since RAS complexes prepared from PA4 and PA5 showed similar inhibition of cancer cell viabilities (Fig. 5c). The same was observed when comparing ADm and their ADp counterparts (Fig. 5d).
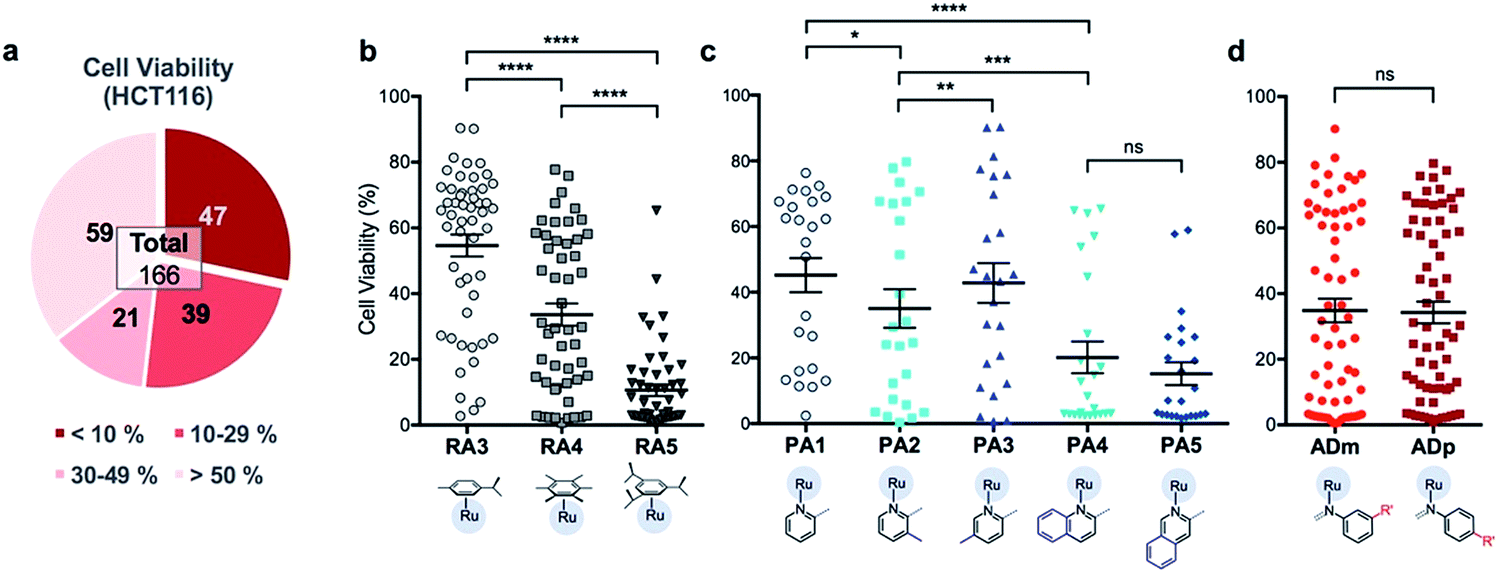 | ||
| Fig. 5 Analysis of HCT116 cell viability% screened with 166 RAS complexes reveal that structural isomerism do not affect efficacy of RAS complexes. (a) Summary of cell viabilities, classified into four sub bands. Selected comparison of cell viability% between the groups treated with RAS complexes (b) having different arene ligands. Mean ± S.E.M. (n = 51, ****p < 0.0001; paired one-way ANOVA test with Tukey post-hoc test); (c) having different picolinaldehyde (PA) ligands. Mean ± S.E.M. (n = 24, ns – not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; paired one-way ANOVA test with Tukey post-hoc test); and (d) having aniline derivative (AD) ligands with R′ moieties in the meta or para position. Mean ± S.E.M. (n = 65, ns – not significant; paired two-tailed t-test). | ||
Next, efficacious RAS complexes (107 out of 166) were evaluated against TC7 under the same experimental conditions (ESI Fig. S4 and Table S3†). The differences in cell viabilities between TC7 and HCT116 treatment were determined and 22 initial hits with a difference <10% were chosen to ascertain dose-dependencies (Fig. 6a). Therefore, the estimated IC50 (eIC50) values of each initial hit were derived from their cell viability curves in the respective cell lines (ESI Fig. S5†) and resistance factors (RF) were calculated by taking the ratio of eIC50 in both cell lines (Fig. 6b). Two RAS complexes, 532m and 532p, with the lowest RF values were selected as lead candidates for apoptosis-independent compounds (Fig. 6c and d).
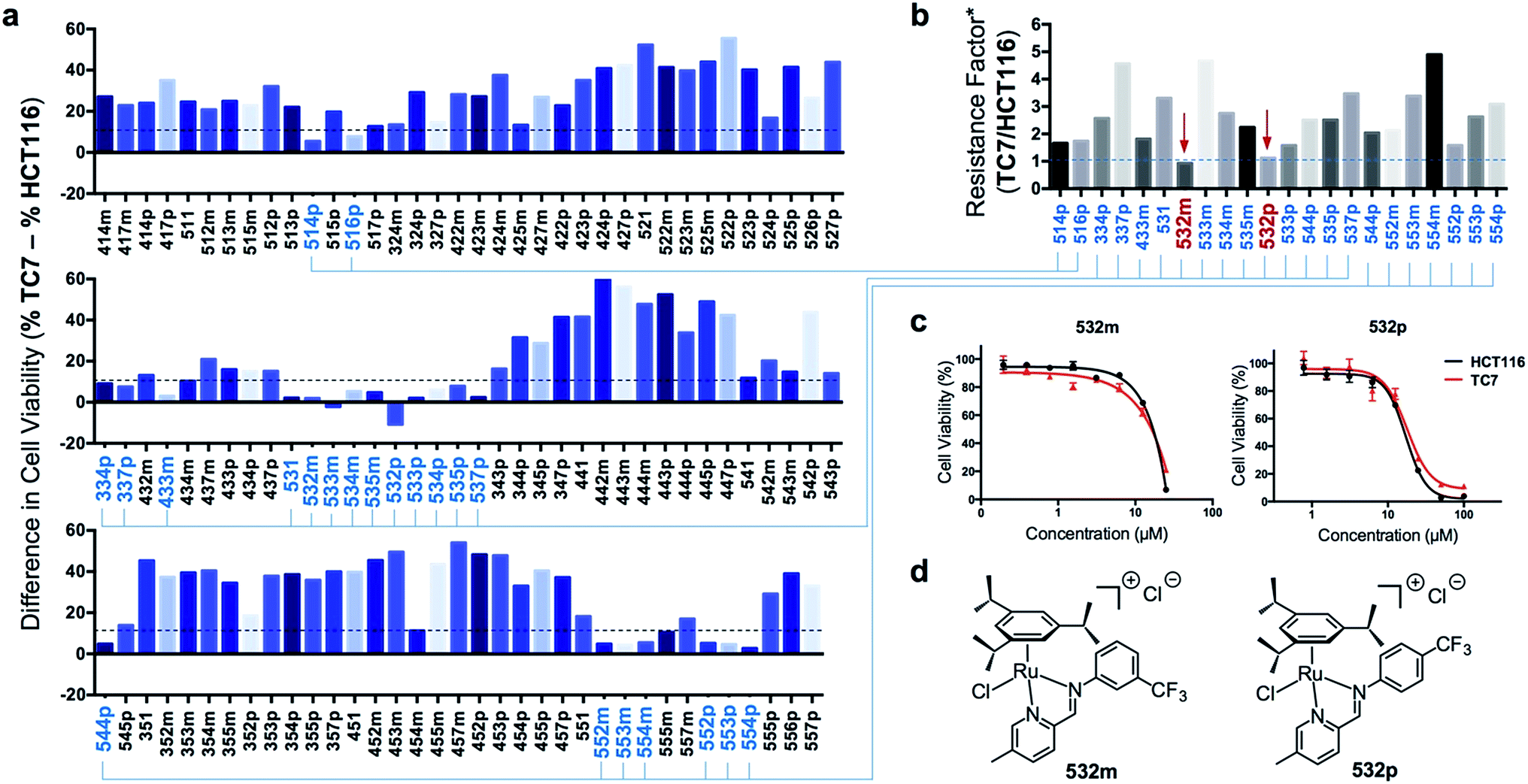 | ||
| Fig. 6 Screening and identification of two apoptosis-independent ‘hits’. (a) Summary of the difference in cell viabilities (%) between apoptosis-resistant TC7 and apoptosis-sensitive HCT116 treated with the corresponding RAS complexes (25 μM). A smaller difference indicated a greater ability to overcome apoptosis-resistance. 22 complexes (highlighted in blue) were selected for a secondary screening. (b) Summary of the resistance factors# (RF) of the 22 RAS complexes. A lower RF indicated a greater ability to overcome apoptosis resistance. (c) Cell viability curves of TC7 and HCT116 cells treated with various concentration of ‘hit’ compounds 532m and 532p. (d) Structures of the 2 ’hit’ compounds identified. #RF was calculated by taking the ratio of the eIC50 in TC7 and HCT116. The eIC50 was calculated based on the cell viability curves obtained from one experiment and three replicates for each concentration of drug treatment. | ||
Synthesis of 532m and 532p and validation of apoptosis-independence
To ensure accuracy in the hit-identification process, validation was carried out to eliminate false positives. First, 532m and 532p were synthesized in a preparative scale and characterized spectroscopically using 1H NMR, ESI-MS and UV-vis before elemental analyses for purity. Once the identity, purity and stability of both RAS complexes were established, we tested their activities in a panel of drug-sensitive and -resistant gastric and colorectal cancer cell lines. Finally, we investigated the caspase-dependence and induction of apoptotic biomarkers by both complexes.Preparative-scale syntheses of 532m and 532p were performed following similar reaction conditions to the earlier on-plate format (Fig. 2a). Crude products were purified using silica gel column chromatography and characterized by 1H NMR and ESI-MS. The 1H NMR spectra of 532m and 532p revealed resonances typical of RAS complexes (ESI Fig. S6 and S10†).34 The singlet resonance peaks corresponding to the imine proton at ca. 9.3–9.4 indicated chelation of the imino-pyridine ligands to Ru. ESI-MS spectra also showed the typical [M]+ molecular ion peaks and with isotopic distribution consistent with calculations (ESI Fig. S9 and S13†). The purity of the complexes was determined to be greater than 95% using elemental analyses, after accounting for residual solvent. UV-vis stability studies showed that the complexes were generally stable to ligand substitution in aqueous solution and that aquation of the Cl ligand could be suppressed in 0.9% w/v NaCl solution (ESI Fig. S14†).
The cytotoxic activities of 532m and 532p were determined in a panel of drug-sensitive (AGS, HCT116) and -resistant (HCT116 p53-/-, TC7) cell lines (Table 1, ESI Fig. S15†). Both compounds showed low micromolar IC50 values in AGS gastric cancer cells and were several times more cytotoxic than cisplatin, commonly used in gastric cancer treatment. In wild type HCT116 colorectal cancer cells, 532m and 532p had modest IC50 values and were less cytotoxic than oxaliplatin and 5-fluorouracil, which are clinical drugs used in the adjuvant treatment of colorectal cancers. However, 532m and 532p were more efficacious against p53-null HCT116 p53-/- and apoptosis-resistant TC7 cells. In particular, 532m was marginally more cytotoxic and 2 times more cytotoxic in HCT116 p53-/- cells than oxaliplatin and 5-fluorouracil, respectively. Against TC7 cells, the differences were even more evident, with 532m being 3 times and 68 times more active than oxaliplatin and 5-fluorouracil, respectively. The calculated RF values were also particularly revealing about the p53-independent and apoptosis-independent nature of these RAS complexes. For instance, when considering the p53-dependence variable in wild type HCT116 and HCT116 p53-/- cells, 532m and 532p exhibited RF of 0.9 and 0.8, respectively, compared to p53-dependent oxaliplatin and 5-fluorouracil with RF of 4.6 and 2.3. Similarly, taking into account the apoptosis-dependence when comparing wild type HCT116 and TC7 cells, 532m and 532p were equally efficacious against HCT116 and TC7 with RFs of 0.8, which was in good agreement with values obtained during initial screening. In contrast, apoptosis-inducing oxaliplatin and 5-fluorouracil were less potent in the apoptosis-resistant lineage, with RF values of 9.2 and 55, respectively (Fig. 7a).
| Complex | IC50a [μM] | Resistance factorb | ||||
|---|---|---|---|---|---|---|
| AGS | HCT116 (wild type) | HCT116 p53-/- | TC7 | p53 dependence [p53-/-/wild type] | Apoptosis [TC7/HCT116] | |
| a IC50 values is the concentration of Ru complexes required to inhibit 50% of cell growth with respect to control groups, measured by MTT assay after 48 h of incubation. Data obtained are based on the average of three independent experiments, and the reported errors are the corresponding standard deviations. The IC50 were corrected using actual [Ru] determined using ICP-OES. b Resistance factors were calculated by taking the ratio of IC50. | ||||||
| 532m | 4.13 ± 0.57 | 10.1 ± 0.3 | 9.20 ± 0.44 | 8.03 ± 0.78 | 0.9 | 0.8 |
| 532p | 6.75 ± 2.06 | 17.8 ± 1.5 | 14.5 ± 1.18 | 13.6 ± 1.3 | 0.8 | 0.8 |
| Oxaliplatin | n.d. | 2.62 ± 0.63 | 12.1 ± 1.8 | 24.0 ± 12.4 | 4.6 | 9.2 |
| 5-Fluorouracil | n.d. | 9.94 ± 2.3 | 22.6 ± 5.5 | 548 ± 218 | 2.3 | 55 |
| Cisplatin | 31.5 ± 3.5 | n.d. | n.d. | n.d. | n.d. | n.d. |
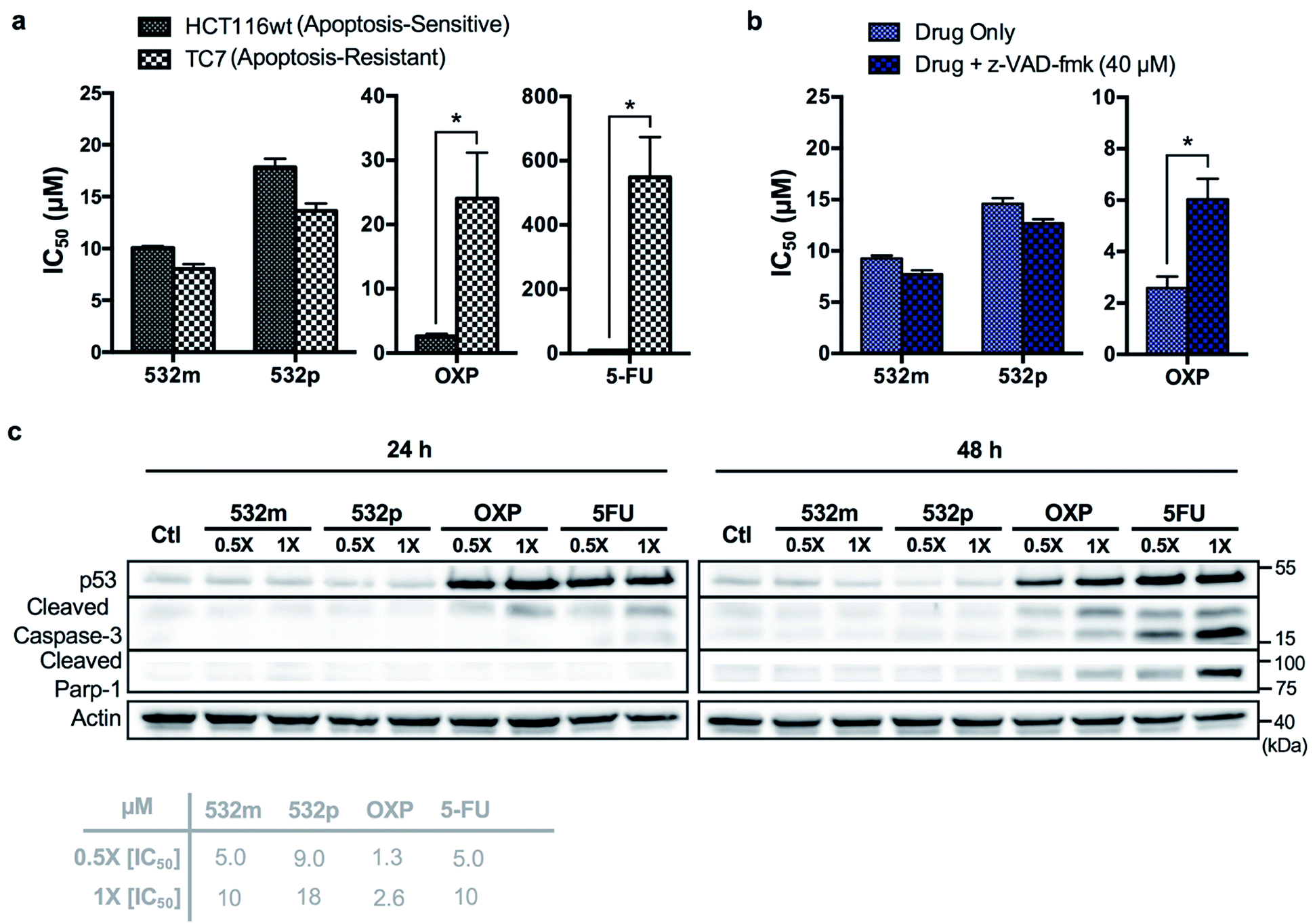 | ||
| Fig. 7 Complexes 532m and 532p induce p53-independent, non-apoptotic cell death. (a) IC50 values of 532m, 532p, oxaliplatin and 5-fluorouracil after 48 h treatment in apoptosis-sensitive (HCT116) and apoptosis-resistant (TC7) cell lines. Data represent mean ± S.E.M. of three independent experiments (*p < 0.05; two-tailed Student's t-test). (b) IC50 values of 532m, 532p and oxaliplatin after 48 h treatment in the absence and presence of apoptosis inhibitor, z-VAD-fmk (40 μM). Data represent mean ± S.E.M. of three independent experiments (*p < 0.05; two-tailed Student's t-test). (c) Western blot analysis of proteins related to the apoptosis pathway and in HCT116 cells after treatment with 532m, 532p, oxaliplatin and 5-fluorouracil at 0.5× and 1× [IC50] for 24 h and 48 h. Homogeneous protein loading determined with reference to actin. | ||
We validated the apoptosis-independent activity of 532m and 532p by examining several hallmarks of apoptosis. Anticancer drugs such as oxaliplatin and 5-fluorouracil that act via apoptosis are dependent on apoptosis-executor caspases and inhibition of these caspases would result in decreased efficacies. Treatment with these compounds would result in cleavage and activation of caspase, and increase expression of other related apoptotic biomarkers. Indeed, co-incubation with a pan caspase inhibitor, z-VAD-fmk, conferred a cytoprotective effect to HCT116 cells treated with oxaliplatin but had no effect on cells treated with 532m or 532p (Fig. 7b and ESI Fig. S16†). Western blot analysis of HCT116 cells treated with oxaliplatin and 5-fluorouracil revealed significant induction of p53 expression and cleaved caspase-3 at both 24 h and 48 h time-points, and downstream cleavage of PARP-1 at 48 h. In contrast, cells treated with 532m and 532p did not display such expression profile regardless of duration or concentration of treatment (Fig. 7c). Further investigation also showed a lack of induction of other apoptotic biomarkers such as cleaved caspase-7 or -9, or DNA-damage biomarker pH2AX by 532m or 532p (ESI Fig. S17†).
Taken together, the similar activities in both apoptosis-sensitive (HCT116) and apoptosis-resistant (TC7) cell lines, caspase-independent activity and the lack of induction of apoptotic biomarkers confirmed the apoptosis-independent activity of 532m and 532p, validating the accuracy and efficiency of the phenotypic screening strategy.
Conclusion
In this work, we demonstrated the successful development and use of a phenotypic screening assay for the identification of apoptosis-independent hits, 532m and 532p. We further validated their apoptosis-independent mode-of-action by showing the lack of induction of several apoptotic biomarkers, a lack of dependence on caspases and their equal efficacies in apoptosis-sensitive and -resistant cell lines. The initial screen at a fixed concentration (25 μM) revealed 22 preliminary hits but we recognized the need for a secondary validation, taking into account the non-linear nature of dose–response and the large variation in toxicities between different complexes. A secondary dose-dependent screen with a range of concentrations revealed 532m and 532p to be potential hits with a high degree of confidence. Hits identified via such focused phenotypic screening strategies should exhibit higher degrees of apoptosis independence compared to a generic screening methodology. Indeed, 532m and 532p both had RF values <1 alluding to their independence from the apoptotic pathway for cell-killing. This is in stark contrast to apoptosis-independent RAS-1H and RAS-1T had RFs of 4.5 and 2.8, which indicated some degree of apoptosis-dependence, although they were still significantly lower than that of clinical drugs such as etoposide, 5-fluorouracil and doxorubicin.28 We compared these results in a different cell-pair comprising HCT116 wt and HCT116 p53-/-, to determine their p53-dependence, since p53-dependence is an important hallmark of apoptosis. The results further revealed that both apoptosis-independent 532m and 532p acted via p53-independent pathways consistent with our hypotheses.As a first line of investigation for the identification of apoptosis-independent candidates, determination of the exact mode of cell death would be less relevant than ascertaining that cell death induction was indeed non-apoptotic. In that regard, phenotypic screening designed to delineated non-apoptotic from apoptotic mechanisms would be more revealing than a generic efficacy-based approach. In order to fully realized the potential and suitability of apoptosis-independent candidates such as 532m and 532p for the treatment of resistant cancer progenies, more throughout scrutiny including target identification, mode-of-action and in vivo studies would have to be applied. Nevertheless, the combined use of a convenient compound-library generation methodology, and a specific phenotypic screening strategy presented in this study is a reliable first step to identifying effective treatment for apoptosis-resistant cancers and development of similar strategies may be a way to accelerate the discovery of such compounds.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge financial support from Ministry of Education and the National University of Singapore (R143-000-680-114) and Ligue contre le Cancer, COST CM1105, Accociation pour la Recherche contre le Cancer (ARC). The authors also thank Associate Professor Han-ming Shen (NUS) for providing the HCT116 and HCT116 p53-/- cell lines and CMMAC for performing elemental analysis and ICP-OES experiments.Notes and references
- M. S. Soengas and S. W. Lowe, Oncogene, 2003, 22, 3138–3151 CrossRef CAS PubMed.
- C. Krakstad and M. Chekenya, Mol. Cancer, 2010, 9, 135 CrossRef PubMed.
- I. Paul and J. M. Jones, World J. Clin. Oncol., 2014, 5, 588–594 CrossRef PubMed.
- E. Hervouet, M. Cheray, F. M. Vallette and P. F. Cartron, Cells, 2013, 2, 545–573 CrossRef CAS PubMed.
- F. H. Igney and P. H. Krammer, Nat. Rev. Cancer, 2002, 2, 277–288 CrossRef CAS PubMed.
- S. Fulda, Int. J. Cancer, 2009, 124, 511–515 CrossRef CAS PubMed.
- S. W. Lowe and A. W. Lin, Carcinogenesis, 2000, 21, 485–495 CrossRef CAS PubMed.
- Y. Pommier, O. Sordet, S. Antony, R. L. Hayward and K. W. Kohn, Oncogene, 2004, 23, 2934–2949 CrossRef CAS PubMed.
- D. M. O'Gorman and T. G. Cotter, Leukemia, 2001, 15, 21–34 CrossRef.
- U. Fischer and K. Schulze-Osthoff, Cell Death Differ., 2005, 12, 942–961 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- G. Kroemer, L. Galluzzi, P. Vandenabeele, J. Abrams, E. S. Alnemri, E. H. Baehrecke, M. V. Blagosklonny, W. S. El-Deiry, P. Golstein, D. R. Green, M. Hengartner, R. A. Knight, S. Kumar, S. A. Lipton, W. Malorni, G. Nunez, M. E. Peter, J. Tschopp, J. Yuan, M. Piacentini, B. Zhivotovsky and G. Melino, Cell Death Differ., 2009, 16, 3–11 CrossRef CAS PubMed.
- A. Kornienko, V. Mathieu, S. V. Rastogi, F. Lefranc and R. Kiss, J. Med. Chem., 2013, 56, 4823–4839 CrossRef CAS PubMed.
- P. Kreuzaler and C. J. Watson, Nat. Rev. Cancer, 2012, 12, 411–424 CrossRef CAS PubMed.
- M. W. Robinson, J. H. Overmeyer, A. M. Young, P. W. Erhardt and W. A. Maltese, J. Med. Chem., 2012, 55, 1940–1956 CrossRef CAS PubMed.
- A. M. Wasik, S. Almestrand, X. Wang, K. Hultenby, A. L. Dackland, P. Andersson, E. Kimby, B. Christensson and B. Sander, Cell Death Dis., 2011, 2, e225 CrossRef CAS PubMed.
- A. A. Kumbhar, A. T. Franks, R. J. Butcher and K. J. Franz, Chem. Commun., 2013, 49, 2460–2462 RSC.
- S. Tardito, I. Bassanetti, C. Bignardi, L. Elviri, M. Tegoni, C. Mucchino, O. Bussolati, R. Franchi-Gazzola and L. Marchio, J. Am. Chem. Soc., 2011, 133, 6235–6242 CrossRef CAS PubMed.
- S. Tardito, C. Isella, E. Medico, L. Marchio, E. Bevilacqua, M. Hatzoglou, O. Bussolati and R. Franchi-Gazzola, J. Biol. Chem., 2009, 284, 24306–24319 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, D. Colavito, S. Alidori, G. G. Lobbia, V. Gandin, F. Tisato and C. Santini, J. Med. Chem., 2006, 49, 7317–7324 CrossRef CAS PubMed.
- K. Suntharalingam, S. G. Awuah, P. M. Bruno, T. C. Johnstone, F. Wang, W. Lin, Y. R. Zheng, J. E. Page, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2015, 137, 2967–2974 CrossRef CAS PubMed.
- C. Trejo-Solís, D. Jimenez-Farfan, S. Rodriguez-Enriquez, F. Fernandez-Valverde, A. Cruz-Salgado, L. Ruiz-Azuara and J. Sotelo, BMC Cancer, 2012, 12, 156 CrossRef PubMed.
- M. Librizzi, A. Longo, R. Chiarelli, J. Amin, J. Spencer and C. Luparello, Chem. Res. Toxicol., 2012, 25, 2608–2616 CrossRef CAS PubMed.
- S. Tian, F. M. Siu, S. C. Kui, C. N. Lok and C. M. Che, Chem. Commun., 2011, 47, 9318–9320 RSC.
- W. J. Guo, Y. M. Zhang, L. Zhang, B. Huang, F. F. Tao, W. Chen, Z. J. Guo, Q. Xu and Y. Sun, Autophagy, 2013, 9, 996–1008 CrossRef CAS PubMed.
- J. Yuan, Z. Lei, X. Wang, F. Zhu and D. Chen, Metallomics, 2015, 7, 896–907 RSC.
- J. J. Soldevila-Barreda, I. Romero-Canelon, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- M. J. Chow, C. Licona, G. Pastorin, G. Mellitzer, W. H. Ang and C. Gaiddon, Chem. Sci., 2016, 7, 4117–4124 RSC.
- B. Siewert, V. H. van Rixel, E. J. van Rooden, S. L. Hopkins, M. J. Moester, F. Ariese, M. A. Siegler and S. Bonnet, Chem.–Eur. J., 2016, 22, 10960–10968 CrossRef CAS PubMed.
- F. Sams-Dodd, Drug Discovery Today, 2005, 10, 139–147 CrossRef CAS PubMed.
- D. Swinney, Clin. Pharmacol. Ther., 2013, 93, 299–301 CrossRef CAS PubMed.
- J. G. Moffat, J. Rudolph and D. Bailey, Nat. Rev. Drug Discovery, 2014, 13, 588–602 CrossRef CAS PubMed.
- R. Williams, Expert Opin. Invest. Drugs, 2013, 22, 1627–1644 CrossRef CAS PubMed.
- M. J. Chow, C. Licona, D. Y. Q. Wong, G. Pastorin, C. Gaiddon and W. H. Ang, J. Med. Chem., 2014, 57, 6043–6059 CrossRef CAS PubMed.
- M. J. Chow, M. V. Babak, D. Y. Wong, G. Pastorin, C. Gaiddon and W. H. Ang, Mol. Pharm., 2016, 13, 2543–2554 CrossRef CAS PubMed.
- S. Violette, L. Poulain, E. Dussaulx, D. Pepin, A. M. Faussat, J. Chambaz, J. M. Lacorte, C. Staedel and T. Lesuffleur, Int. J. Cancer, 2002, 98, 498–504 CrossRef CAS PubMed.
- A. T. Shaw, M. M. Winslow, M. Magendantz, C. Ouyang, J. Dowdle, A. Subramanian, T. A. Lewis, R. L. Maglathin, N. Tolliday and T. Jacks, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8773–8778 CrossRef CAS PubMed.
- S. Dolma, S. L. Lessnick, W. C. Hahn and B. R. Stockwell, Cancer Cell, 2003, 3, 285–296 CrossRef CAS PubMed.
- M. Weiwer, J. A. Bittker, T. A. Lewis, K. Shimada, W. S. Yang, L. MacPherson, S. Dandapani, M. Palmer, B. R. Stockwell, S. L. Schreiber and B. Munoz, Bioorg. Med. Chem. Lett., 2012, 22, 1822–1826 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, assembly yield/purity of RAS complexes from the on-plate synthesis and cell viability data from phenotypic screening assay, 1H NMR, ESI-MS and UV-vis spectra for 532m and 532p, and western blot data in PDF format. See DOI: 10.1039/c7sc00497d |
| This journal is © The Royal Society of Chemistry 2017 |