
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic control and empirical prediction of redox potentials for Co4O4 cubanes over a 1.4 V range: implications for catalyst design and evaluation of high-valent intermediates in water oxidation†
Andy I.
Nguyen
 ab,
Jianing
Wang
a,
Daniel S.
Levine
a,
Micah S.
Ziegler
ab and
T. Don
Tilley
*ab
ab,
Jianing
Wang
a,
Daniel S.
Levine
a,
Micah S.
Ziegler
ab and
T. Don
Tilley
*ab
aDepartment of Chemistry, University of California, Berkeley, California 94720-1460, USA. E-mail: tdtilley@berkeley.edu
bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
First published on 7th April 2017
Abstract
The oxo-cobalt cubane unit [Co4O4] is of interest as a homogeneous oxygen-evolution reaction (OER) catalyst, and as a functional mimic of heterogeneous cobalt oxide OER catalysts. The synthesis of several new cubanes allows evaluation of redox potentials for the [Co4O4] cluster, which are highly sensitive to the ligand environment and span a remarkable range of 1.42 V. The [CoIII4O4]4+/[CoIII3CoIVO4]5+ and [CoIII3CoIVO4]5+/[CoIII2CoIV2O4]6+ redox potentials are reliably predicted by the pKas of the ligands. Hydrogen bonding is also shown to significantly raise the redox potentials, by ∼500 mV. The potential-pKa correlation is used to evaluate the feasibility of various proposed OER catalytic intermediates, including high-valent Co-oxo species. The synthetic methods and structure–reactivity relationships developed by these studies should better guide the design of new cubane-based OER catalysts.
Introduction
Research on catalysts for the oxygen evolution reaction (OER), motivated by the goal of creating an artificial photosynthesis system, has generated a number of hypotheses that challenge the traditional limits of transition-metal oxidation states and bonding. Thus, catalytic OER cycles often invoke unusually high and rare oxidation states for the metal centers, which are bound to reactive terminal oxo ligands.1–9 In particular, for intensely studied cobalt-based OER catalysts, a CoIV-oxo intermediate is commonly invoked.3,6,9 However, the CoIV oxidation state is quite rare, and ligand field considerations seem to suggest a high instability for a terminal oxo ligand bound to CoIV.10 Recently, several experiments have confirmed the presence of CoIV species during water oxidation catalysis, but evidence for a discreet, terminal CoIV oxo species remains indirect.3,9,11,12 Remarkably, many density functional theory (DFT) calculations of cobalt-catalyzed OER suggest an even more unconventional species, a CoV-oxo intermediate that is sometimes described as a CoIV-oxyl radical.4,5 Experimental evidence for this type of intermediate is provided by kinetic analysis of OER mediated by a molecular Co4O4 cubane, which is a rare example of a structural and functional model for the active site of OER catalysis.6a For the oxo cubane Co4(μ-O)4(OAc)4(py)4 a singly oxidized state, formally CoIII3CoIV, is firmly established by isolation and spectroscopy.3,6a The doubly oxidized state, formally CoIII2CoIV2, is observed at highly positive potentials, by cyclic voltammetry.13 While the kinetic analysis points to involvement of the doubly oxidized state in the oxygen evolution mechanism, the nature of intermediates associated with this oxidation state remains largely unknown.For evaluation of possible CoIII2CoIV2 (or alternatively CoIII3CoV) intermediates, it would be quite useful to establish reliable methods for predicting the redox potentials associated with particular coordination environments. For example, the Co4O4 cubane Co4(μ-O)4(OAc)4(py)4 has been shown to form the hydroxide complex [Co4O4(OAc)3(OH)2(py)4]− by displacement of acetate, and kinetic studies on OER indicate that this species is oxidized to high-valent species.6 The resulting, transient intermediates have not been directly observed; therefore, alternative, indirect experimental methods for evaluation of candidate structures are useful. As shown here, a strategy for estimation of CoIV/CoV redox potentials for transitory intermediates is based on extrapolation of linear free-energy relationships (LFERs). This analysis requires a large and diverse set of related cubanes with various ligand sets and redox potentials, to provide a useful LFER from which redox properties can be confidently predicted.
In this report, we demonstrate that the Co4O4 core is readily and precisely manipulated to tune its chemical and electronic properties over an unprecedented range. While the parent cluster Co4O4(OAc)4(py)4 has been well-studied, its controlled structural modification has never before been demonstrated; such clusters are generally prepared by “self-assembly” methods rather than by rational syntheses.14 Synthetic methods were used to obtain electron-rich or electron poor cubanes, cubanes with mixed-carboxylate ligand sets, and cubanes possessing secondary-sphere hydrogen-bond donors. It is noteworthy that this synthetic methodology allows introduction of secondary-sphere hydrogen bonding into the cubane structure since the role of hydrogen bonding in electron transfer and water oxidation (especially in the OEC of photosystem II) has been an important topic for many years.1b–h
Analysis of a substantial library of oxo cubanes provides empirical linear correlations between ligand pKa values and redox potentials for singly and doubly oxidized species, over a range of 1.42 V. This analysis also quantifies the effect of hydrogen bonding on redox properties in this cubane system. These relationships offer a useful predictive tool for evaluating potential intermediates in water-splitting mechanisms. They should also provide important guidance in catalyst design studies for OER.
Results and discussion
Synthesis of new cobalt oxo cubanes
The synthesis of Co4O4 cubanes has previously been accomplished by the “self-assembly” route.14b While this method is simple to execute, its harshly oxidizing conditions and less predictable nature prevents the targeted synthesis of many structurally diverse cubane clusters. Notably, the “self-assembly” route only leads to symmetrically ligated cubanes, and has only been demonstrated with ligands that are not very electron rich or electron poor. Given potentially important applications of the Co4O4 cubane unit (vide supra), general synthetic routes that allow ready access to a diverse library of cubanes, including unsymmetrically substituted, electron poor and electron rich examples, are desired.The various cubane complexes to be discussed in this study possess a Co4O4 cubane core with different ligand sets that are described in a concise manner by a convenient descriptor. Compounds of the general formula [CoIII4O4XxLy](4−x)+ are abbreviated [xX–yL](4−x)+, where x and y are integers equal or greater than 0, and denote the stoichiometry of X (anionic ligand) and L (neutral ligand), respectively. If multiple types of X or L are present in the same complex, then the additional xX or yL is appended in the fashion [x1X1,x2X2,…xnXn–y1L1,y2L2,…ynLn](4−x)+. If the [Co4O4]4+ unit is oxidized to [Co4O4]5+ or [Co4O4]6+, the notation changes to [xX–yL](5−x)+ and [xX–yL](6−x)+, respectively. For example, a cubane with the formula [Co4O4(OAc)2(OH)(H2O)3(py)4]+ is abbreviated [2OAc,OH-3H2O,4py]+, and its successive oxidation products are [2OAc,OH-3H2O,4py]2+ and [2OAc,OH-3H2O,4py]3+.
Three general methods were used to synthesize the new cubane complexes, shown in Chart 1. The “self-assembly” route (method A, eqn (1)), patterned after the Das synthesis of Co4O4(OAc)4(py)4 from Co(NO3)2(H2O)6, NaX, L, and H2O2, has been used to synthesize a small number of closely related cubanes such as Co4O4(OAc)4(p-cyanopyridine)4 (4-OAc-4CNpy).14b,20 However, attempts to extend this method to many of the cubane derivatives described below were unsuccessful. Thus, this does not appear to be a generally successful synthetic strategy for the synthesis of new cubane complexes.
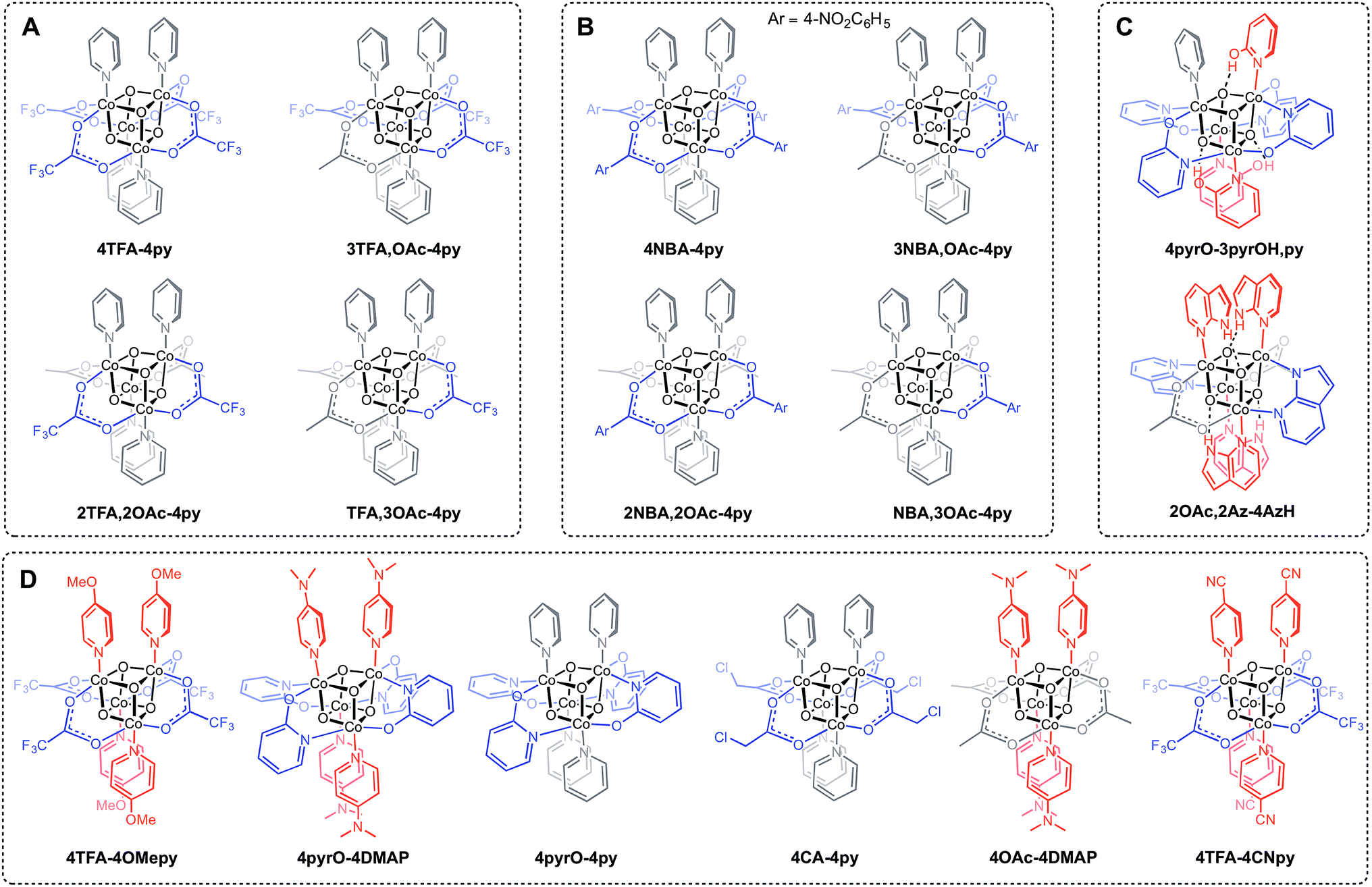 | ||
| Chart 1 New cubane complexes. (A) Trifluoroacetate (TFA) series synthesized by method C; (B) p-nitrobenzoate (NBA) series synthesized by method C; (C) hydrogen-bonded cubanes; (D) cubanes synthesized by a methods B and/or C. | ||
Exchange of neutral ligands (method B, eqn (2)) is useful in the synthesis of cubanes with L = DMAP. The cubane Co4O4(OAc)4(DMAP)4 (4-OAc-4DMAP) was produced in this way, in 67% recrystallized yield. Method B works well in other cases (vide infra), under conditions where pKa(L′H+) > pKa(LH+). Similarly, synthesis of the cubane with X′ = CF3COO− was achieved by heating 4OAc-4py with a slight excess of CF3CO2H to give Co4O4(O2CCF3)4(py)4 (4TFA-4py) in 56% yield. This procedure (method C, eqn (3)) is driven by release of HX; thus, it works well when pKa(HX') < pKa(HX). Methods B and C provide predictable and divergent routes for cubane diversification (Chart 1), and in contrast to method A, give crude products that are mostly free of Co(II) impurities, as determined by thin-layer chromatography. Remarkably, substitution of ligands by methods B and C is highly stereoselective, always placing the X ligands around equatorial faces and the L ligands on the “top” and “bottom” faces, as evidenced by NMR spectroscopy and crystallography (Fig. 1).
 | (1) |
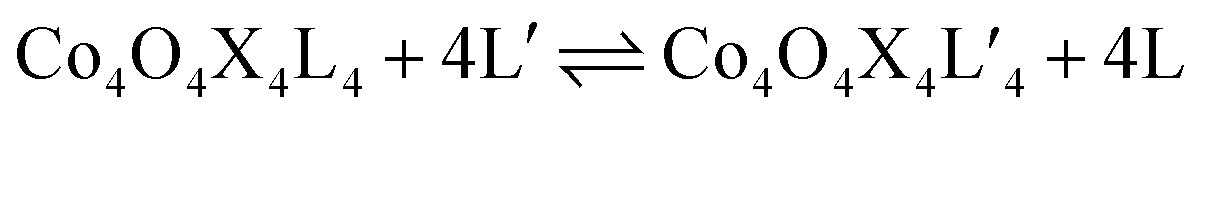 | (2) |
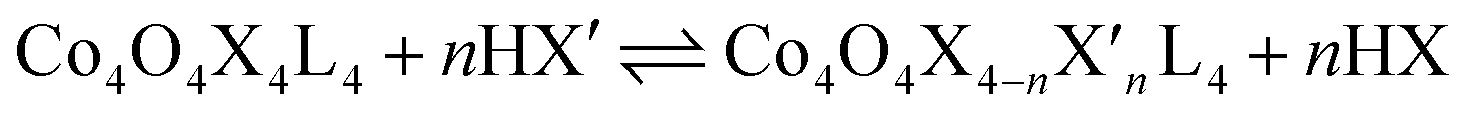 | (3) |
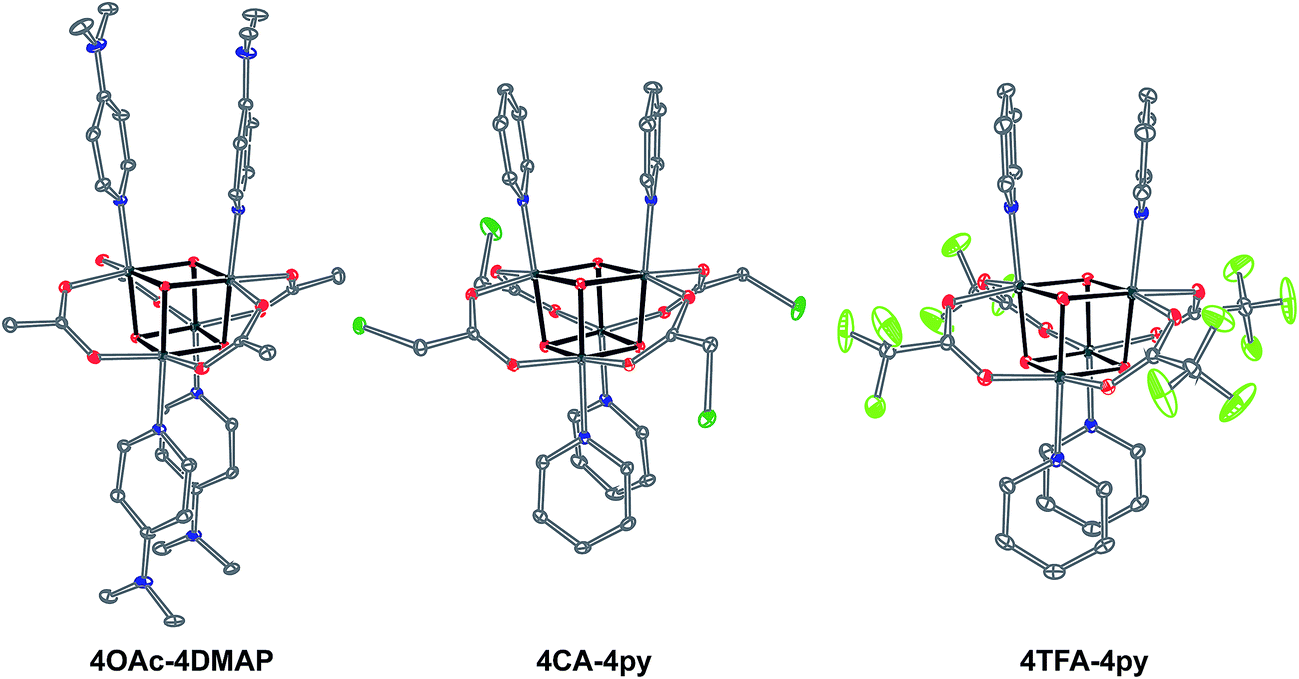 | ||
| Fig. 1 Single-crystal XRD structures of cubane complexes. Thermal ellipsoids are at 30% probability, with hydrogen atoms and solvent molecules omitted for clarity. | ||
Significantly, method C allows for the synthesis of mixed-carboxylate cubanes. Mono-, di-, or tri-substituted carboxylate cubanes were produced using two RCO2H equivalents per cubane. This mixture of mono-, di-, tri-, and tetra-substituted cubanes is generally separable by column chromatography since the compounds are highly colored and exhibit significantly different Rf values. By this method, Co4O4(OAc)4−n(CF3CO2)n(py)4 (nTFA,(4-n)OAc-4py) and Co4O4(OAc)4−n(p-nitrobenzoate)n(py)4 (nNBA,(4-n)OAc-4py) complexes, where n = 1, 2, and 3, have been synthesized and fully characterized. Interestingly, the formation of disubstituted cubanes is stereoselective for cis-substitution – that is the two carboxylate (X′) ligands are on adjacent faces of the cubane, as revealed by X-ray crystallography of 2TFA,2OAc-4py (Fig. 2). The 1H NMR spectra reveal the presence of only one product, suggesting that the crystal structures represent the bulk material.
 | ||
| Fig. 2 Single-crystal X-ray structure of 2TFA,2OAc-4py. Thermal ellipsoids shown at 30% probability. Hydrogen atoms were omitted for clarity. | ||
Cubanes bearing electron-rich X ligands 2-pyridonate (pKa(O–H) = 8.05)15 and 7-azainodolate (pKa(N1–H) ∼ 15)16 were synthesized in a manner similar to that of method C, but the products have a somewhat different composition (Scheme 1). Thus, heating 4OAc-4py with excess 2-hydroxypyridine in acetonitrile gave Co4O4(2-pyridonate)4(2-hydroxypyridine)3(py) (4pyrO-3pyrOH,py) in 37% yield, after column chromatography. Elemental analysis and mass spectrometry are consistent with the stoichiometries determined by NMR spectroscopy. Similarly, heating 4OAc-4py with an excess of 7-azaindole in acetonitrile gave Co4O4(OAc)2(7-azaindolate)2(7-azaindole)4 (2OAc,2Az-4AzH) in 48% yield. Though the pKa values of 2-hydroxypyridine and 7-azaindole are higher than those of HOAc, the reaction is driven by use of excess ligand and the insolubilities of 4pyrO-3pyrOH,py and 2OAc,2Az-4AzH in acetonitrile. These reactions provide different types of cubane structures, in that protonated versions of the X ligands serve as the L ligands (2-hydroxypyridine and 7-azaindole possess pyridyl nitrogen atoms), and substantial substitution of the L-positions on the cubane is observed. Interestingly, the protonated, L-type ligands engage in hydrogen bonding with their O–H or N–H bonds interacting with the μ3-oxo ions of the [Co4O4] core. The 2-hydroxypyridine ligand can be substituted for other L ligands by method B to produce 4pyrO-4py and 4pyrO-4DMAP. The latter two compounds were characterized by 1H NMR spectroscopy and single-crystal X-ray diffraction (Fig. 3). 1H NMR spectroscopy suggests that 4pyrO-3pyrOH,py coexists as C1-symmetric species in solution at room temperature, suggesting that hydrogen-bonds slow the rotation about the Co–N bonds. For 2OAc,2Az-4AzH, two C2-symmetric isomers co-crystallized, and differ by the relative orientation of the 7-azin ligands. The O(X)⋯O(X) distances of 2.44 Å in 4pyrO-3pyrOH,py, and the N(X)⋯O(X) distances of 2.69 Å in 2OAc,2Az-4AzH are consistent with strong to moderate hydrogen bonding.17 Thus, these cubanes possess an additional secondary coordination sphere that affects the structural properties of the cubane core. Secondary coordination sphere effects are highly important in biology, and mimicry of this feature in inorganic complexes is a long-standing challenge that often involves special ligand design.1c
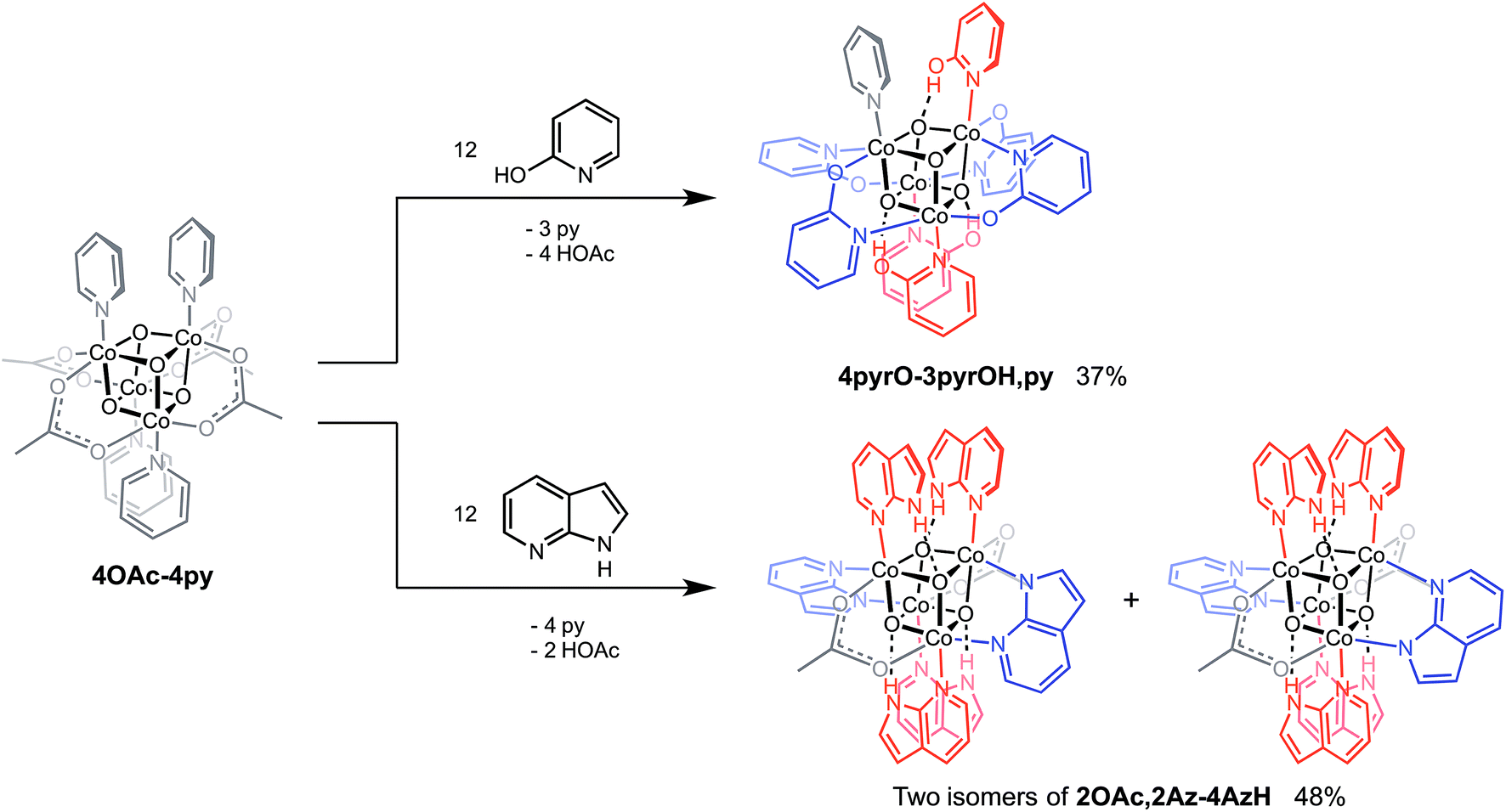 | ||
| Scheme 1 Synthesis of cubane complexes containing intramolecular hydrogen-bonding. | ||
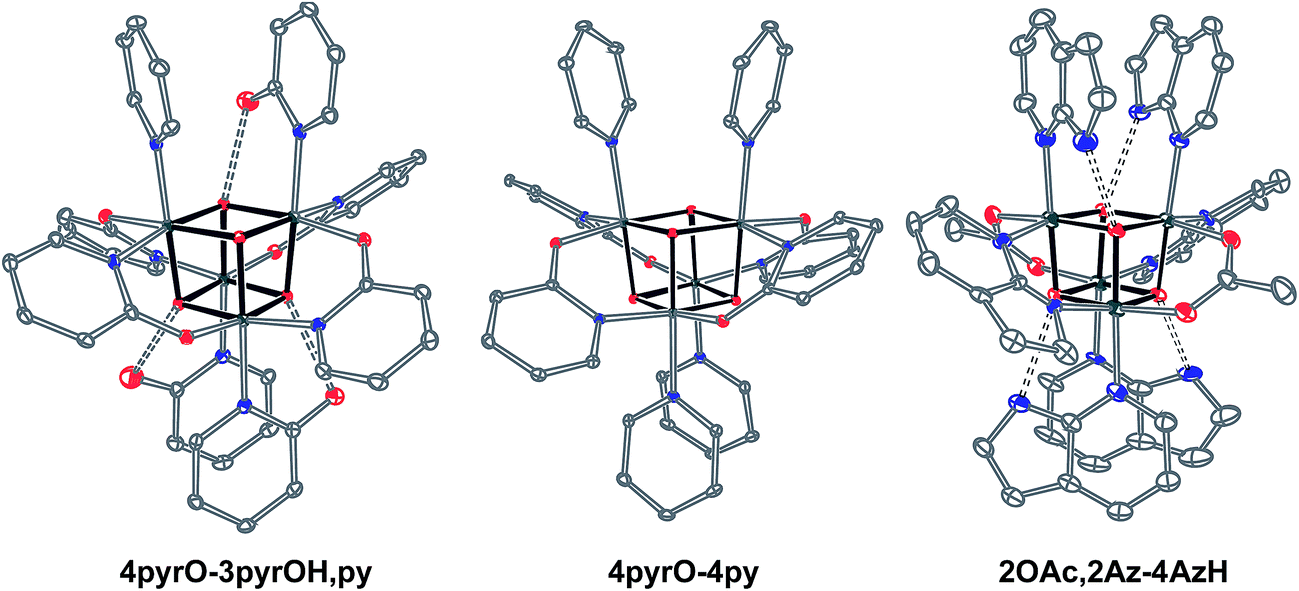 | ||
| Fig. 3 Single-crystal XRD structures of cubanes containing pyrindonate or 7-azaindolate ligands. Intramolecular hydrogen-bond donor–acceptor distances are distinguished as dashed-bonds. Thermal ellipsoids shown at 30% probability. Hydrogen atoms and solvent are omitted for clarity. | ||
Electrochemistry
All cyclic voltammograms of the cubanes exhibit a reversible event for the [CoIII4O4]4+/[CoIVCoIII3O4]5+ couple (E1), and depending on the ligand set, either a reversible or irreversible event for the [CoIVCoIII3O4]5+/{[CoVCoIII3O4]6+ ↔ [CoIV2CoIII2O4]6+} couple (E2, Fig. 4). The reversibility of E2 seems to depend on the combination of X and L, rather than the individual identities of X and L. In general, when there is a large asymmetry in the electron-donating properties of X and L (ΔpKa > 4), E2 is irreversible. Four examples that illustrate this trend are 4OAc-4DMAP, 4pyrO-4py, 4pyrO-4DMAP, and 4OAc-4py. The first two cubanes have two sets of ligands with ΔpKa > 4, and their cyclic voltammograms indicate an irreversible oxidation process at all measured scan rates (E2). However, the two sets of ligands in 4pyrO-4DMAP and 4OAc-4py have a ΔpKa < 2, and reversible E2 events. This observation may suggest that the E2 redox couple for cubanes with a ligand ΔpKa > 4 may involve some degree of ligand non-innocence. For example, if there is a large difference between the donating properties of X and L, electron–hole character may concentrate on the more donating ligand, leading to oxidative degradation. Conversely, when the X and L ligands are both good donors, the electron–hole character is distributed more evenly across all eight ligands to give a more stable complex. The electronic structure of the species resulting from this second oxidation will be discussed in later sections.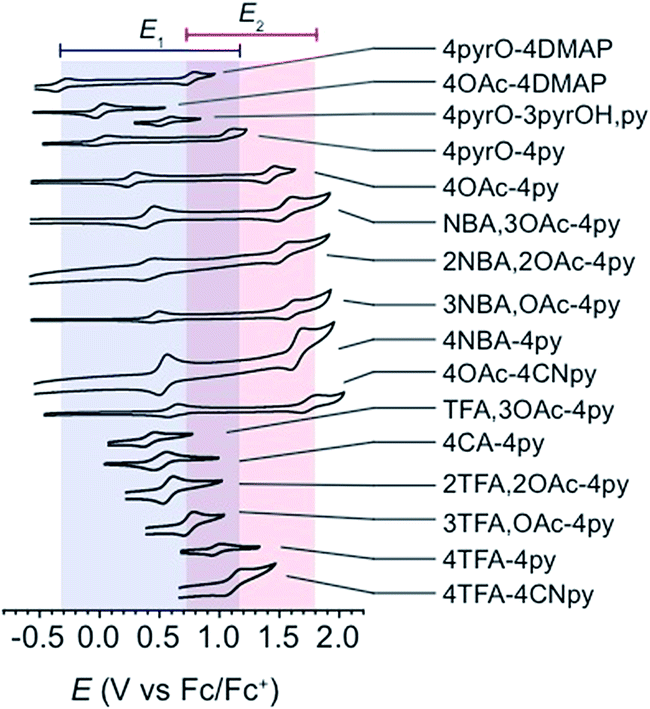 | ||
| Fig. 4 Cyclic voltammograms of cubane complexes, synthesized in this work, in organic solvents (see Table 1 for details, 100 mV s−1 scan rate, 0.1 M [nBu4N]PF6 electrolyte). The blue and red boxes indicate the range of E1 and E2 potentials, respectively. For some cubanes, the E2 values lie beyond the solvent's electrochemical window. | ||
In general, and as expected, the observed redox couples reflect a dependence on the donor properties of the cubane ligand set, such that they shift anodically with electron-withdrawing ligands and cathodically with electron-donating ligands. To evaluate the influence of the L ligands on redox potentials, sixteen cubanes were analyzed holding X = OAc constant while varying L (4OAc-4L, series 1), and conversely holding L = py constant while varying X (4X-4py, series 2). The redox potentials for both series (1 and 2, respectively) correlate linearly with a convenient descriptor for the ligand donating ability, the average pKa (aqueous)15,16,18,19 of HX and HL+ per cobalt center (eqn (4); Fig. 5A). This average pKa value descriptor will herein be referred to as the effective basicity of the ligand set. Here, pKa is employed as a free-energy representation for the electron-donating ability of the ligand. A previous report used Hammett parameters to correlate ligand donor ability with Co4O4 redox activity, but this analysis is limited to only substituted aryl-based ligands, whereas pKa is a much more widely useful parameter.20
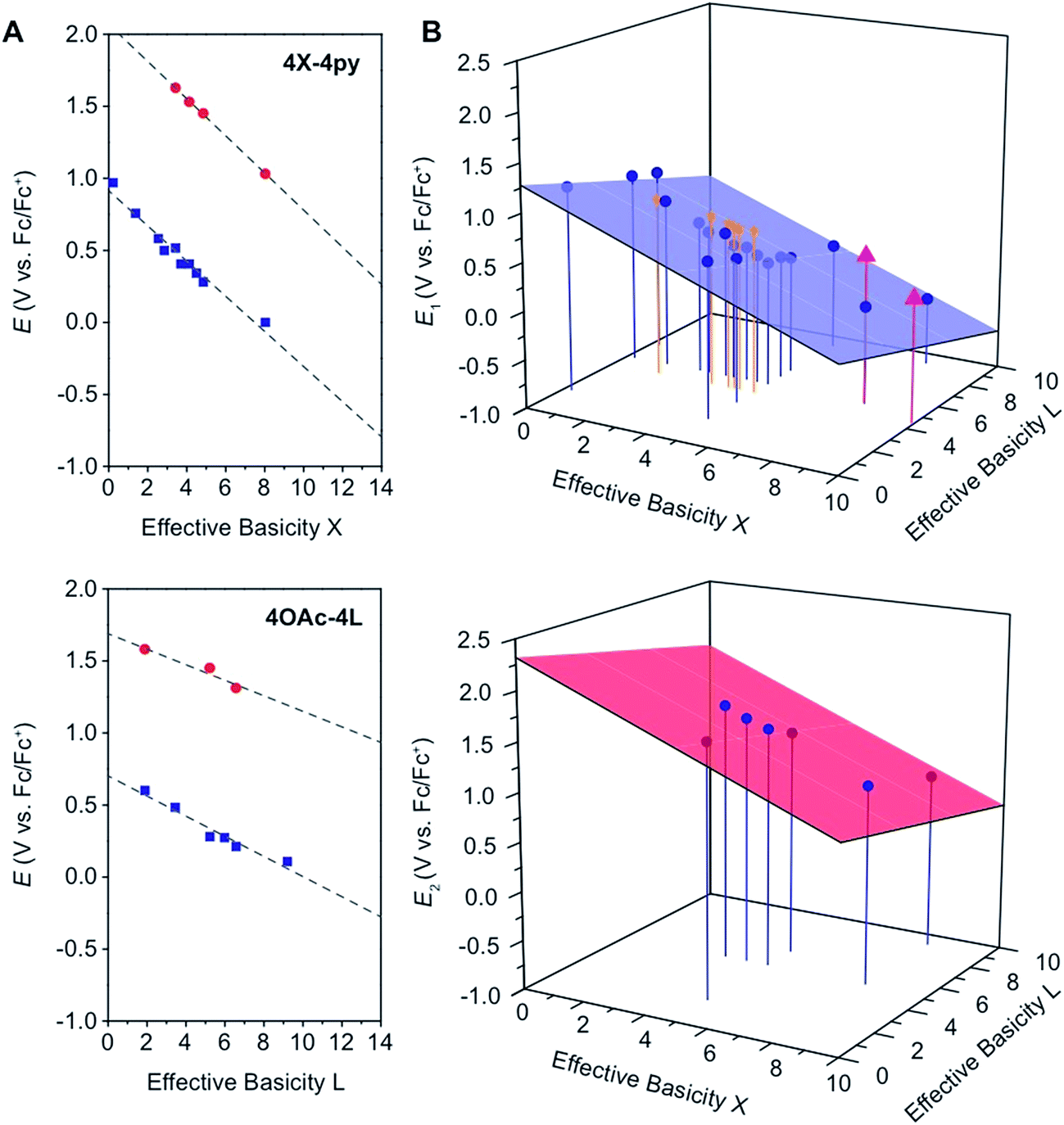 | ||
Fig. 5 (A) Plots and fits of E1 and E2 potentials versus effective basicity for 4X-4py (series 1, top) and 4OAc-4L (series 2, bottom). The blue squares ( ) are the E1 values and the red circles ( ) are the E1 values and the red circles ( ) are the E2 values. The dashed lines are the linear fits. (B) Surface fits of E1 (eqn (5), top) and E2 (eqn (6), bottom) for all cubane complexes. The blue circles ( ) are the E2 values. The dashed lines are the linear fits. (B) Surface fits of E1 (eqn (5), top) and E2 (eqn (6), bottom) for all cubane complexes. The blue circles ( ) are redox potentials for 4X-4L cubanes, the orange stars ( ) are redox potentials for 4X-4L cubanes, the orange stars ( ) are E1 values for [2X-4L]2+ from ref. 21, and the purple triangles ( ) are E1 values for [2X-4L]2+ from ref. 21, and the purple triangles ( ) are potentials for the hydrogen-bonded cubanes. Vertical drop lines are presented as visual guides. ) are potentials for the hydrogen-bonded cubanes. Vertical drop lines are presented as visual guides. | ||
A plot of E1vs. effective basicities for the complexes in series 1 is linear with a slope of −120 ± 7 mV dec−1, and series 2 also provides a linear relationship with a slope of −69 ± 9 mV dec−1. Similarly, the slopes of lines derived from the E2 values are very similar, at −128 ± 9 mV dec−1 (series 1) and −51 ± 9 mV dec−1 (series 2). The magnitudes of these slopes demonstrate that the redox couples for the cubanes are about twice as sensitive to the X vs. the L ligand. A possible cause for this effect will be discussed in the next section. Most surprising is that the redox potentials of unsymmetrical, mixed-carboxylate cubanes could be predicted simply by considering the summation of the pKa values. This fact would seem to suggest that the Co4O4 cubane core levels out the electronic effects of coordination asymmetry through an electronically communicative mechanism. In this context, the four cobalt ions in the Co4O4 unit essentially behave as a single “superion” entity. Both series of potentials fit to planar surfaces (eqn (5) and (6), Fig. 5B) with R2 values of 96% and 98%, respectively.
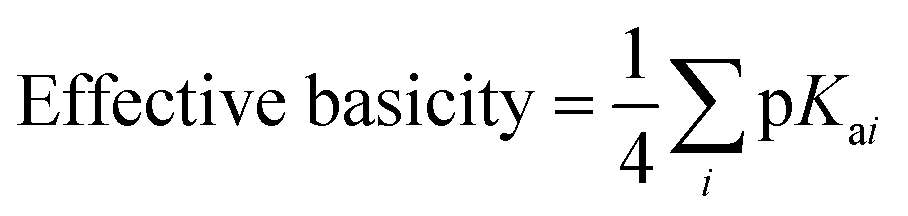 | (4) |
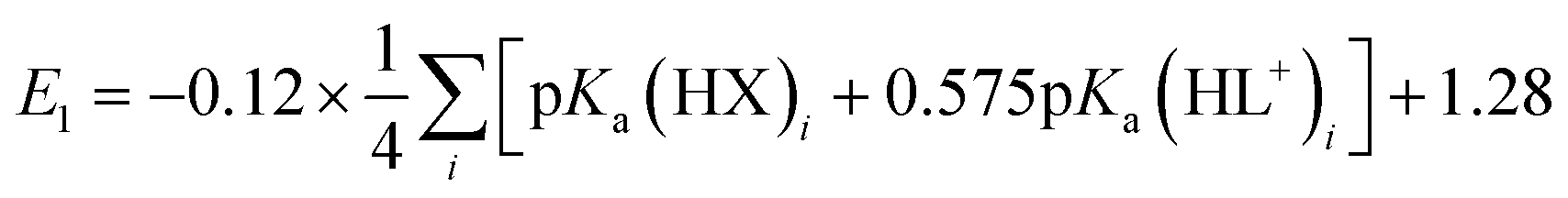 | (5) |
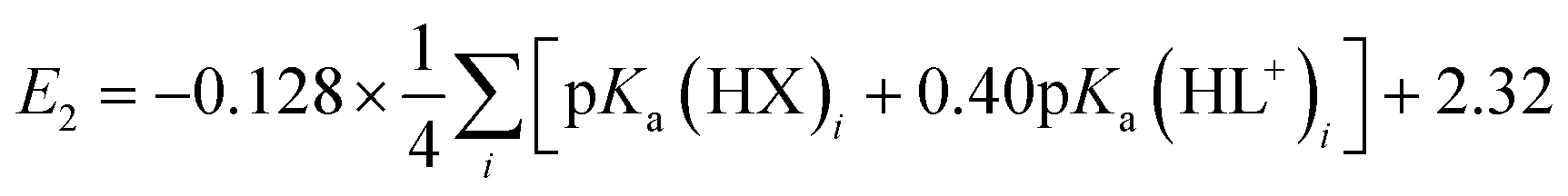 | (6) |
To explore the generality of eqn (5) and (6) derived from compound series 1 and 2, the E1 and E2 values for cubanes based on additional combinations of X and L were measured and compared to the predicted values. Including these additional combinations of X and L, the E1 redox potentials observed for tetracobalt oxo cubanes range from −0.30 to 1.12 V (a span of 1.42 V; Table 1). A smaller range of 830 mV was observed for E2, perhaps because some of the corresponding oxidation events exist outside the solvent's electrochemical window. Satisfyingly, the redox potentials of all the cubanes (without intramolecular hydrogen-bonds) are predicted by eqn (5) and (6) with maximum absolute errors of 0.099 and 0.050 V, respectively. In addition, reported redox potentials for cubanes of the type [Co4O4X2L24]2+ (L2 = 2,2′-bipyridine) follow the relationships described by eqn (5).21 This observation indicates that the relative number of X to L ligands does not affect the validity of eqn (5), and therefore the coefficients must reflect the intrinsic donor properties of X- versus L-type ligands. In summary, it appears that eqn (5) and (6) hold for (1) a wide range of X and L, (2) symmetric and asymmetric ligand coordination environments around the cubane, and (3) X ligands other than carboxylates.
| Cubane | Effective basicity X15,16,18,19 | Effective basicity L15,16,18,19 | E 1 (V) | E 2 (V) | Solvent | Reference |
|---|---|---|---|---|---|---|
| a bipy = 2,2-bipyridine, BA = benzoate, PFBA = pentafluorobenzoate, CBA = 4-chlorobenzoate, tBuBA = 4-tButylbenzoate. b Value obtained by differential pulse voltammetry (DPV). | ||||||
| 4TFA-4CNpy | 0.23 | 1.90 | 1.12 | CH2Cl2 | This work | |
| 4TFA-4OMepy | 0.23 | 6.39 | 0.90 | MeCN | This work | |
| 4TFA-4py | 0.23 | 5.25 | 0.97 | CH2Cl2 | This work | |
| 3TFA,OAc-4py | 1.37 | 5.25 | 0.76 | CH2Cl2 | This work | |
| 2TFA,2OAc-4py | 2.54 | 5.25 | 0.58 | CH2Cl2 | This work | |
| TFA,3OAc-4py | 3.70 | 5.25 | 0.40 | CH2Cl2 | This work | |
| 4CA-4py | 2.86 | 5.25 | 0.50 | MeCN | This work | |
| 4NBA-4py | 3.43 | 5.25 | 0.52 | 1.63 | MeCN | This work |
| 3NBA,OAc-4py | 3.79 | 5.25 | 0.47 | 1.59 | MeCN | This work |
| 2NBA,2OAc-4py | 4.15 | 5.25 | 0.41 | 1.53 | MeCN | This work |
| NBA,3OAc-4py | 4.50 | 5.25 | 0.34 | 1.48 | MeCN | This work |
| 4OAc-4py | 4.86 | 5.25 | 0.28 | 1.45 | MeCN | 13 |
| 4OAc-4CNpy | 4.86 | 1.90 | 0.60 | 1.58 | MeCN | 14 |
| 4OAc-4COOEtpy | 4.86 | 3.45 | 0.48 | MeCN | 20 | |
| 4OAc-4OMe | 4.86 | 6.39 | 0.21 | 1.31b | MeCN | 20 |
| 4OAc-4Mepy | 4.86 | 5.99 | 0.27 | MeCN | 20 | |
| 4OAc-4DMAP | 4.86 | 9.20 | 0.11 | 1.10b | MeCN | This work |
| 4pyrO-3pyrOH,py | 8.04 | 5.23 | 0.55 | CH2Cl2 | This work | |
| 4pyrO-4py | 8.04 | 5.25 | 0.0 | 1.03 | CH2Cl2 | This work |
| 4pyrO-4DMAP | 8.04 | 9.20 | −0.30 | 0.80 | CH2Cl2 | This work |
| 2OAc,2Az-4AzH | ∼10 | 4.45 | 0.30b | DMF | This work | |
| [2OAc-4bipy]2+ | 2.43 | 4.34 | 0.69 | MeCN | 21 | |
| [2NBA-4bipy]2+ | 1.71 | 4.34 | 0.77 | MeCN | 21 | |
| [2BA-4bipy]2+ | 2.10 | 4.34 | 0.70 | MeCN | 21 | |
| [2PFBA-4bipy]2+ | 0.80 | 4.34 | 0.86 | MeCN | 21 | |
| [2CBA-4bipy]2+ | 2.00 | 4.34 | 0.72 | MeCN | 21 | |
| [2tBuBA-4bipy]2+ | 2.19 | 4.34 | 0.68 | MeCN | 21 | |
Effects of protic solvent and hydrogen bonding
The redox potentials reported above were all measured in polar aprotic solvents (MeCN, DCM, or DMF). When measured in water, the E1 potentials experience an increase in the observed potential, by an average of +553 ± 26 mV (Table 2). This solvent dependence on redox potentials was previously noted for 4OAc-4py, but its origin has not been discussed.6,22 Unfortunately, only a few complexes are water-soluble enough to be examined in this solvent, and E2 potentials were not observed in water since these oxidation events occur beyond the electrochemical window for water. We propose that the observed increases in redox potentials in water originate from hydrogen-bonding interactions between the cubane μ3-oxo ligands and water, which reduces the μ3-oxo donation to cobalt. The relative electronic effects of the cubane ligands appear to persist in water, however, as indicated by a similar E1/pKa slope. The hydrogen-bonding effect is clearly demonstrated, even in aprotic solvent, by comparing 4pyrO-3pyrOH,py with 4pyrO-4py. The pKa values of 2-hydroxypyridinium cation and pyridinium are essentially identical (5.23 and 5.25, respectively), and according to eqn (5), should give nearly identical E1 values for the corresponding cubanes. However, the observed E1 value for 4pyrO-3pyrOH,py is 550 mV higher than that of 4pyrO-4py. In the case of 2OAc,2Az-4AzH, the experimental E1 value is 519 mV higher than predicted by eqn (5). The observed increases in redox potential with introduction of hydrogen bonding to the cubane (519 and 550 mV) are remarkably similar to that observed by changing the solvent to water (553 mV).DFT calculations of cubane electronic structures
To quantify the ligands' influence on the redox potentials of cubanes, an energy decomposition analysis (EDA) was performed. The results reveal that the X ligands stabilize the oxidized cubanes significantly better than L ligands (ESI and Table S1†). In 4OAc-4py, the overall interaction energy between the [CoIII4O4]4+ ion and acetate is −158 kcal mol−1, while the energy between [CoIII4O4]4+ and pyridine is only −43 kcal mol−1. The interaction energies for the two ligand types increase upon oxidation to [4OAc-4py]+: −243 kcal mol−1 for acetate and −46 kcal mol−1 for pyridine. As can be seen, the [Co4O4]–acetate bonding is greatly strengthened upon oxidation by −85 kcal mol−1, but the pyridine–[Co4O4] bonding only slightly increases, by −2 kcal mol−1. The frozen electron density term, (FRZ) which describes electrostatic and steric contributions to bonding, is at least an order of magnitude larger for the acetate ligand, and doubles in magnitude upon oxidation. The polarization and charge transfer terms are also generally larger for acetate than for pyridine; however, they are less than an order of magnitude different. The size of the FRZ term indicates that electrostatic forces dominate the stability of the oxidized cubane. This finding seems intuitive as CoIII and CoIV are both considered hard ions, and according to hard–soft acid–base theory should prefer ionic bonding interactions over dative interactions. To uncover trends in the bonding, EDA was also applied to 4TFA-4py and 4OAc-4CNpy. The energy difference in frozen components between [4X-4L]+ and 4X-4L (FRZox − FRZred = ΔFRZ) for the [Co4O4]–X and [Co4O4]–L bonds plot against pKa yields slopes of −0.8 kcal per (mol pKa) and −0.5 kcal per (mol pKa), respectively (Fig. S1†). Note that the energy values are within the error of DFT calculations, but there is a qualitative correlation between theory and the experimental redox trends.The doubly oxidized state, [4OAc-4py]2+, was analyzed by DFT calculations. These calculations determined that the S = 1 state is essentially degenerate with the S = 0 state (within 3.3 kcal mol−1). Interestingly, the calculations suggest two valence-trapped CoIV centers, as opposed to a valence delocalized system. There have been conflicting reports of hole delocalization in cobalt cubane systems by DFT, and delocalization has been suggested as a product of a self-interaction error.4,25 In support of a localized valence, UV-vis-NIR spectroelectrochemistry measurements (Fig. 6) on [4OAc-4py], [4OAc-4py]+, and [4OAc-4py]2+ did not show any bands typical of a delocalized (Robin-Day class III) intervalence charge-transfer. However, there is a strong increase in absorption in the visible region (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 cm−1), and growth of a low intensity, broad band centered around 5000 cm−1 upon oxidation from [4OAc-4py] to [4OAc-4py]+ to [4OAc-4py]2+; consistent with the Robin-Day Class II valence model. Recently, X-ray spectroscopic techniques provided additional support for a localized CoIV center that undergoes very fast self-exchange with the other Co ions in the cubane [4OAc-4py]+.13a
000–20000 cm−1), and growth of a low intensity, broad band centered around 5000 cm−1 upon oxidation from [4OAc-4py] to [4OAc-4py]+ to [4OAc-4py]2+; consistent with the Robin-Day Class II valence model. Recently, X-ray spectroscopic techniques provided additional support for a localized CoIV center that undergoes very fast self-exchange with the other Co ions in the cubane [4OAc-4py]+.13a
 | ||
| Fig. 6 (A) UV-vis-NIR plot of [4OAc-4py], [4OAc-4py]+, and [4OAc-4py]2+ produced by applying no potential, 0.68 V, and 1.88 V, respectively, in an spectroelectrochemical cell (6 mM [4OAc-4py], 1 mm path length, MeCN solvent, [nBu4N]PF6 electrolyte, Pt mesh working electrode, Pt wire counter electrode, and Ag/AgNO3 nonaqueous reference electrode). (B) DFT-calculated structure of [4OAc-4py]2+ with spin-densities indicated. | ||
Implications for cubane intermediates with hydroxide and oxide ligands
Perhaps the most important utility of the energy relationships of eqn (5) and (6) is their ability to predict electronic properties for cubane-based intermediates that might be considered in mechanistic investigations. For example, these relationships allow thermodynamic evaluations of proposed hydroxyl- and oxo-intermediates in the recently proposed mechanism for cobalt cubane-mediated OER.6 It is important to emphasize that the redox values predicted by eqn (5) and (6) are under the strict assumption that fundamental bonding interactions between carboxylates, pyridonates, hydroxide, or oxide with Co4O4 are similar, in the sense that they are primarily electrostatic (vide supra). However, hydroxide and oxide ligands are stronger π-donors than carboxylates and pyridonates, and this may cause the true redox potentials of cubanes bound to hydroxide or oxide to deviate somewhat from those derived by eqn (5) and (6). Nevertheless, from simple ligand-field arguments, the stronger π-donating properties should actually destabilize more the lower oxidation states of cobalt, which should result in negative deviations from eqn (5) and (6).A previous report from this laboratory described the aqueous behavior of Co4O4(OAc)4(py)4 (4OAc-4py) which reacts partially with hydroxide to produce [Co4O4(OAc)3(OH)2(py)4]− ([3OAc,2OH-4py]−), and the latter species may be in equilibrium with Co4O4(OAc)3(OH)(H2O)(py)4 (3OAc,OH-H2O,4py) (eqn (7)).6 The bis(hydroxide) was characterized by NMR spectroscopy, and is proposed to be the resting state of the cubane during OER catalysis. However, as noted in this study, the formation of [3OAc,2OH-4py]− is low yielding since the equilibrium strongly favors 4OAc-4py, which makes the isolation and direct electrochemical study of this species difficult.
 | (7) |
During catalysis, the Co(III)-hydroxide moiety undergoes a series of proton- and electron-transfer steps to form a putative [Co4O4]6+ oxidation state, which was proposed on the basis of kinetic studies6 and published DFT7 calculations to be a CoV-oxo intermediate. Note that no spectroscopic or structural evidence of a CoV-oxo exists, and thus, any evidence supporting or discrediting its existence is valuable to understanding OER catalysis. The proposed mechanism invokes a key electron transfer from the CoIV-oxo to [4OAc-4py]+ to generate the CoV-oxo intermediate ([Co4O4]6+ state) responsible for O–O bond formation.
No prior knowledge of the relative redox potentials contributed to the formulation of this disproportionation step; its existence is strongly inferred from the second-order rate dependence on [4OAc-4py]+. While this mechanistic study revealed that the [Co4O4]6+ state was achieved and only terminal oxygen ligands were involved, the level of water/hydroxide/oxo substitution and protonation states throughout the OER cycle are not known. For example, does the active species contain two terminal oxo-ligands, or only one, or none? The redox steps involved in this mechanism are undoubtedly coupled with proton transfers, as implicated by the first-order rate dependence on [OH−], but the ordering and extent of these processes are as yet unknown; in other words, kinetic data alone cannot definitively distinguish between a mechanism involving only aquo- and hydroxo-ligated cubanes and one involving the more unusual oxo ligand. Thus, if the redox potentials for corresponding oxo-, hydroxo-, and aquo-substituted Co4O4 clusters can be known (or well-estimated), it should be possible to rule out certain possible intermediates (those that cannot reach the [Co4O4]6+ state by disproportionation with [4OAc-4py]+), and discover which candidates are most plausible. The LFER equations (eqn (5) and (6)) allow simple evaluations of these candidate intermediates.
Using the conjugate acid pKa values of 15.7 for the OH− ligand, and ∼36 for O2−,18,26E1 and E2 redox potentials were calculated for the most likely catalytic intermediates (Scheme 2, Table 3). These E1 values suggest that oxidation of the catalytic intermediates in Table 3, from [CoIII4O4]4+ to [CoIII3CoIVO4]5+, by [4OAc-4py]+ occurs spontaneously. The E2 potentials for the hydroxo-ligated cubanes are significantly higher than the E1 of [4OAc-4py]+ which indicates that a simple electron transfer between cubanes to generate the [Co4O4]6+ state is not thermodynamically favored. However, if deprotonation of hydroxide to oxide occurred before electron-transfer, the oxidation to [Co4O4]6+ becomes favorable. Thus, these results suggest that the most likely active catalyst is [3OAc,2OH-4py]− (previously observed by NMR spectroscopy6), since it possesses redox couples consistent with cycling between [CoIII4O4]4+, [CoIII3CoIVO4]5+, and {[CoIII2CoIV2O4]6+ ↔ [CoIII3CoVO4]6+} states during OER catalysis. As previously reported,6 we favor the localized CoV formalism when a strong σ and π donor such as O2− is bound to a single Co ion, a viewpoint that is also supported by DFT. However, when multiple basic ligands (OH− and O2−) are present, it is reasonable to expect the more delocalized states (e.g. CoIV2) to significantly contribute.
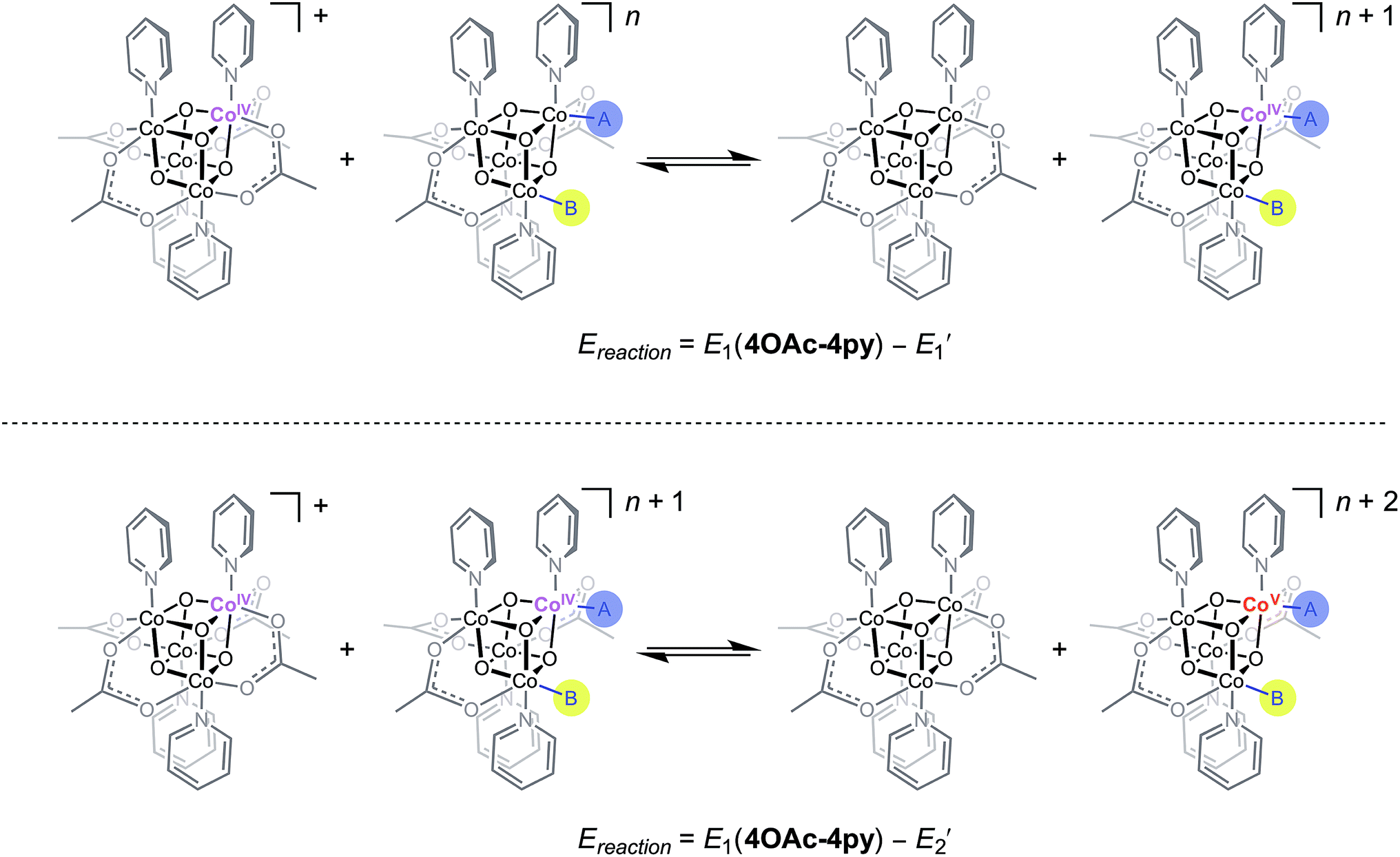 | ||
| Scheme 2 Redox disproportionation reactions with [4OAc-4py]+. A and B are either X or L ligands of the cubanes in Table 3. | ||
| Cubane | E′1a (V) | E′2a (V) | [E1(4OAc-4py) − E′1] (V) | [E1(4OAc-4py) − E′2] (V) |
|---|---|---|---|---|
| a Potential versus Fc/Fc+ in polar aprotic solvent. | ||||
| [4OAc,OH-4py]− | −0.09 | +0.86 | +0.37 | −0.58 |
| [4OAc,O-4py]2− | −0.70 | +0.21 | +0.96 | −0.07 |
| 3OAc,OH-H2O,4py | 0.01 | 0.96 | +0.27 | −0.69 |
| [3OAc,2OH-4py]− | −0.41 | +0.51 | +0.69 | −0.23 |
| [3OAc,OH,O-4py]2− | −1.02 | −0.14 | +1.30 | +0.42 |
| [3OAc,2O-4py]3− | −1.63 | −0.78 | +1.91 | +1.06 |
With this information, a revised OER catalytic cycle can be proposed (Scheme 3). This updated mechanism allows for two kinetically indistinguishable O–O coupling pathways, an acid–base mechanism or a radical-coupling pathway. Recently, Nocera and coworkers obtained direct evidence for the coupling of adjacent cobalt-oxygen species at pH 7 in the CoPi catalyst, and several DFT calculations have shown this pathway to be energetically reasonable.4,27,28 Nonetheless, these predicted redox values show that cubanes with hydroxo- and oxo-ligands can reach the a formal CoIV2 or CoV oxidation state by redox disproportionation reactions.
 | ||
| Scheme 3 Proposed mechanism of OER by Co4O4 with redox potentials (vs. Fc/Fc+) for each oxidation step shown. | ||
Conclusions
Cobalt oxo cubane complexes are readily manipulated by convenient syntheses that allow exchange of the nitrogen-donor and carboxylate ligands, the latter in a sequential, selective manner. This synthetic chemistry allows tuning of the cubane electronic properties over a wide range. Primary variables that influence the cubane's redox properties are the donor properties of the anionic and neutral ligands, and the incorporation of hydrogen-bond interactions. Two important principles that result from this work, that should guide OER catalyst design, are that (1) the [Co4O4] cubane is predictably modifiable over a remarkable range of electrochemical and chemical properties, and (2) the accessibility of a formal CoIV2/CoV-oxo intermediate by the cubane is energetically feasible given the appropriate ligand set. The strong influence of hydrogen bonding suggests that catalyst design must not only address the primary coordination sphere, but also second-sphere interactions between the catalytic core and the reaction medium. The fundamental reaction chemistry developed in this study provides new strategies for functionalization of the cubane with a vast array of chelating ligands and N-heterocyclic donors, and should allow incorporation of catalytic cubanes into more complex architectures involving biological molecules, surfaces, and 3-dimensional networks. Importantly, the linear free energy relationships described above are useful in predicting the redox properties of newly designed cubane-based materials.Conflict of interest
No competing financial interests have been declared.Acknowledgements
This work was primarily funded by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-05CH1123. We acknowledge the National Institutes of Health for funding the UC Berkeley CheXray X-ray crystallographic facility under Grant No. S10-RR027172 and funding the UC Berkeley College of Chemistry NMR facility under Grant No. S10 RR 03353-01. We acknowledge the National Science Foundation for also funding the UC Berkeley College of Chemistry NMR facility under Grant No. CHE-0130862, CHE-8703048, and HE79-26291-A02. We thank Philip C. Bunting and Prof. Jeffrey R. Long for help in obtaining UV-vis-NIR spectra. We thank Jaruwan Amtawong and Raúl Huerta-Lavorie for synthetic assistance.References
- (a) V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6(3), 1676–1695 RSC; (b) G. Brudvig, Philos. Trans. R. Soc., B, 2008, 363, 1211–1219 CrossRef CAS PubMed; (c) T. Taguchi, R. Gupta, B. Lassalle-Kaiser, D. W. Boyce, V. K. Yachandra, W. B. Tolman, J. K. Yano, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 1996–1999 CrossRef CAS PubMed; (d) P. H. Oyala, T. A. Stich, R. J. Debus and R. D. Britt, J. Am. Chem. Soc., 2015, 137, 8829–8837 CrossRef CAS PubMed; (e) C. Zhang, Biochim. Biophys. Acta, 2007, 1767, 439–499 Search PubMed; (f) U. Yasufumi, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 437, 55–60 Search PubMed; (g) B. C. Polander and B. A. Barry, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6112–6117 CrossRef CAS PubMed; (h) M. Shoji, H. Isobe, S. Yamanaka, Y. Umena, K. Kawakami, N. Kamiya, J.-R. Shen and K. Yamaguchi, Catal. Sci. Technol., 2013, 3, 1831–1848 RSC.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3(10), 807–813 CrossRef CAS PubMed.
- J. G. McAlpin, Y. Surendranath, M. Dincǎ, T. A. Stich, S. A. Stoian, W. H. Casey, D. G. Nocera and R. D. Britt, J. Am. Chem. Soc., 2010, 132(20), 6882–6883 CrossRef CAS PubMed.
- X. Li and P. E. M. Siegbahn, J. Am. Chem. Soc., 2013, 135(37), 13804–13813 CrossRef CAS PubMed.
- (a) A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2015, 119, 11072–11085 CrossRef CAS; (b) T. C. Davenport, H. S. Ahn, M. S. Ziegler and T. D. Tilley, Chem. Commun., 2014, 50, 6326–6329 RSC.
- (a) A. I. Nguyen, M. S. Ziegler, P. Oña-Burgos, M. Sturzbecher-Hohne, W. Kim, D. E. Bellone and T. D. Tilley, J. Am. Chem. Soc., 2015, 137(40), 12865–12872 CrossRef CAS PubMed; (b) P. F. Smith, L. Hunt, A. B. Laursen, V. Sagar, S. Kaushik, K. U. D. Calvinho, G. Marotta, E. Mosconi, F. De Angelis and G. C. Dismukes, J. Am. Chem. Soc., 2015, 137(49), 15460–15468 CrossRef CAS PubMed.
- X.-J. Su, M. Gao, L. Jiao, R.-Z. Liao, P. E. M. Siegbahn, J.-P. Cheng and M.-T. Zhang, Angew. Chem., Int. Ed., 2015, 54(16), 4909–4914 CrossRef CAS PubMed.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47(6), 1727–1752 CrossRef CAS PubMed.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133(36), 14431–14442 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, in Molecular Electronic Structures of Transition Metal Complexes I, Structure and Bonding, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer Berlin Heidelberg, 2011, pp. 17–28 Search PubMed.
- C. Costentin, T. R. Porter and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138(17), 5615–5622 CrossRef CAS PubMed.
- (a) A. I. Nguyen, R. G. Hadt, E. I. Solomon and T. D. Tilley, Chem. Sci., 2014, 5(7), 2874–2878 RSC; (b) M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- (a) R. G. Hadt, D. Hayes, C. N. Brodsky, A. M. Ullman, D. M. Casa, M. H. Upton, D. G. Nocera and L. X. Chen, J. Am. Chem. Soc., 2016, 138(34), 11017–11030 CrossRef CAS PubMed; (b) E. B. Clatworthy, X. Li, A. F. Masters and T. Maschmeyer, Chem. Commun., 2017, 52, 14412–14415 RSC.
- (a) J. K. Beattie, T. W. Hambley, J. A. Klepetko, A. F. Masters and P. Turner, Polyhedron, 1998, 17, 1343–1354 CrossRef CAS; (b) R. Chakrabarty, S. J. Bora and B. K. Das, Inorg. Chem., 2007, 46(22), 9450–9462 CrossRef CAS PubMed; (c) S. Mukherjee, J. A. Stull, J. Yano, T. C. Stamatatos, K. Pringouri, T. A. Stich, K. A. Abboud, R. D. Britt, V. K. Yacahndra and G. Christou, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2257–2262 CrossRef CAS PubMed; (d) F. Evangelisti, R. Güttinger, Moré, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2013, 135, 18734–18737 CrossRef CAS PubMed; (e) C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- X.-X. Zhou, C. J. Welch and J. Chattopadhyaya, Acta Chem. Scand., Ser. B, 1986, 40, 806–816 CrossRef CAS.
- N. Dufour, A.-M. Lebuis, M.-C. Corbeil, A. L. Beauchamp, P. Dufour, Y. Dartiguenave and M. Dartiguenave, Can. J. Chem., 1992, 70(12), 2914–2921 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, 1997 Search PubMed.
- Handbook of Chemistry and Physics, CRC Press, Florida, 87th edn, 2006 Search PubMed.
- R. M. Smith and R. J. Motekaitis, NIST Standard Reference Database 46, 3.0, National Institute of Standards and Technology, Washington, D.C., 1997 Search PubMed.
- S. Berardi, G. La Ganga, M. Natali, I. Bazzan, F. Puntoriero, A. Sartorel, F. Scandola, S. Campagna and M. Bonchio, J. Am. Chem. Soc., 2012, 134(27), 11104–11107 CrossRef CAS PubMed.
- K. Dimitrou, A. D. Brown, T. E. Concolino, A. L. Rheingold and G. Christou, Chem. Commun., 2001, 14, 1284–1285 RSC.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133(30), 11446–11449 CrossRef CAS PubMed.
- A. M. Ullman, Y. Liu, M. Huynh, D. K. Bediako, H. Wang, B. L. Anderson, D. C. Powers, J. J. Breen, H. D. Abruña and D. G. Nocera, J. Am. Chem. Soc., 2014, 136(50), 17681–17688 CrossRef CAS PubMed.
- J. Georges and S. Desmettre, Electrochim. Acta, 1984, 29(4), 521–525 CrossRef CAS.
- J. G. McAlpin, T. A. Stich, C. A. Ohlin, Y. Surendranath, D. G. Nocera, W. H. Casey and R. D. Britt, J. Am. Chem. Soc., 2011, 133(39), 15444–15452 CrossRef CAS PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, John Wiley & Sons, 6th edn, 1999 Search PubMed.
- A. M. Ullman, C. N. Brodsky, N. Li, S.-L. Zheng and D. G. Nocera, J. Am. Chem. Soc., 2016, 138(12), 4229–4236 CrossRef CAS PubMed.
- L.-P. Wang and T. Van Voorhis, J. Phys. Chem. Lett., 2011, 2(17), 2200–2204 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1532026–1532032. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00627f |
| This journal is © The Royal Society of Chemistry 2017 |