
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedented selective homogeneous cobalt-catalysed reductive alkoxylation of cyclic imides under mild conditions†
Jose R.
Cabrero-Antonino
 ,
Rosa
Adam
,
Veronica
Papa
,
Mattes
Holsten
,
Kathrin
Junge
and
Matthias
Beller
*
,
Rosa
Adam
,
Veronica
Papa
,
Mattes
Holsten
,
Kathrin
Junge
and
Matthias
Beller
*
Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de
First published on 12th June 2017
Abstract
The first general and efficient non-noble metal-catalysed reductive C2-alkoxylation of cyclic imides (phthalimides and succinimides) is presented. Crucial for the success is the use of [Co(BF4)2·6H2O/triphos (L1)] combination and no external additives are required. Using the optimal cobalt-system, the hydrogenation of the aromatic ring of the parent phthalimide is avoided and only one of the carbonyl groups is selectively functionalized. The resulting products, N- and aryl-ring substituted 3-alkoxy-2,3-dihydro-1H-isoindolin-1-one and N-substituted 3-alkoxy-pyrrolidin-2-one derivatives, are prepared under mild conditions in good to excellent isolated yields. Intramolecular reductive couplings can also be performed affording tricyclic compounds in a one-step process. The present protocol opens the way to the development of new base-metal processes for the straightforward synthesis of functionalized N-heterocyclic compounds of pharmaceutical and biological interest.
Introduction
Catalytic reductive transformations of carboxylic acid derivatives are of importance and a current hot topic in catalysis.1 These reactions are already applied in industry for the formation of bulk products and intermediates. Moreover, they offer interesting possibilities for valorization of biomass-derived building blocks and to apply new strategies in organic synthesis. Hence, the design of improved catalysts, and also the development of new methodologies for the selective reduction of this class of compounds, continues to attract the interest of academic and industrial researchers.Cyclic imides, and in particular phthalimides,2 are an important type of carboxylic acid derivative and several of these compounds show interesting biological activities. Among the possible products obtained from the reduction of phthalimides, isoindolinones and substituted derivatives are the most desired as they are valuable scaffolds in pharmaceuticals and agrochemicals, as well as relevant building blocks for organic synthesis (Fig. 1).3
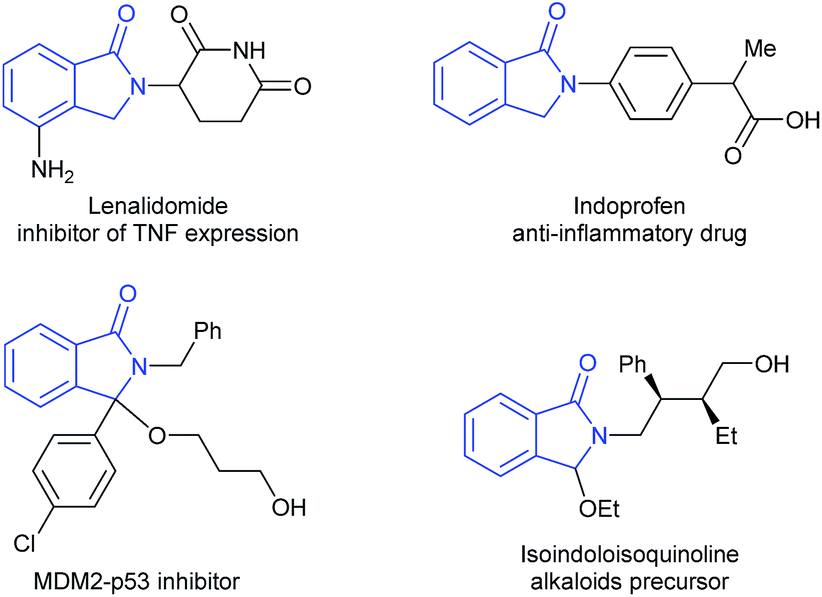 | ||
| Fig. 1 Examples of relevant isoindolinone derivatives. | ||
As a consequence, in the last years several – often multi-step – organic methodologies have been reported for their synthesis.3q,4 Clearly, the selective mono-reduction of readily available phthalimides represents the most suitable and direct approximation to these compounds. Traditionally, procedures for this reduction required the use of over-stoichiometric amounts of Zn or Sn in the presence of strong acids or organometallic hydrides (NaBH4, B2H6 and LiAlH4). Despite the usefulness of these methods on laboratory scale, they have drawbacks due to their limited functional group tolerance, the generation of over-reduction products and significant amounts of waste.3f,t,4g,k
As a greener approach to the reduction of phthalimides, hydrogenations using heterogeneous catalysts have been applied (i.e. RANEY® nickel), although they require harsh reaction conditions.1d,f To overcome these limitations, in the last decade also methodologies based on molecularly-defined complexes have been developed proceeding at milder reaction conditions (Fig. 2). After the original report by Patton and Drago dealing with the hydrogenation of N-methylsuccinimide with a ruthenium catalyst,5 alternative procedures were reported by the groups of Bruneau,6 Ikariya,7 Bergens,8 García,9 Agbossou-Niedercorn,10 Xie,11 Zhang12 and our group.13 These protocols afforded valuable products such as aliphatic lactams, 2-hydroxymethylbenzamides, ω-hydroxylactams, benzamides, aliphatic cyclic amines, 1,4-diols and isoindolinone derivatives directly from phthalimides. Despite all these advancements, still significant limitations exist, especially related with narrow substrate scope, the use of precious catalysts and/or the employment of hydrosilanes as reducing agents. Moreover, from the point of view of obtaining the desired isoindolinone derivatives, some of these protocols present drawbacks as the concomitant hydrogenation of the aromatic ring, the lack of selectivity in the reduction of one of the carbonyl groups or the occurrence of C–N bond cleavage.
 | ||
| Fig. 2 (Up) Described examples of homogeneous catalytic reduction of phthalimides using hydrogen or silanes as reductor. (Bottom) General [Co/triphos]-catalysed inter- and intramolecular selective reductive alkoxylation of cyclic imides (this work). (Bn = benzyl). | ||
Recently, our group developed a direct protocol for the selective one-pot C2-alkoxylation and amination of cyclic imides to give 3-substituted-2,3-dihydro-1H-isoindolinones.14
In this Ru-catalysed methodology, aromatic ring hydrogenations were completely avoided and aryl ring-substituted phthalimides showed selective monoalkoxylation of one of the carbonyl groups (Fig. 2). Crucial for the catalytic activity was the presence of methanosulfonic acid (MSA) as an additive.
In the last years, (1,1,1-tris(diphenylphosphinomethyl)-ethane), so-called triphos, became a privileged ligand for the hydrogenation of carboxylic acids and related derivatives.14,15 Basically, in all these cases, active Ru catalysts are generated. In the last decade, the replacement of precious metals by inexpensive and widely abundant first-row base metals such as Fe,16 Co16h,i and Mn17 has gained increasing importance in hydrogenation chemistry. For example, several cobalt-based systems have shown interesting activity for reductions of C–O,18 C–N,18b,19 C–C18b,c,20 multiple bonds and N-heterocycles.21 Notably in 2015, the groups of de Bruin and Elsevier18f achieved for the first time the hydrogenation of carboxylic acids and esters using [Co(BF4)2·6H2O/triphos (L1)]. In addition, our group reported the CO2 hydrogenation to methanol using a modified related catalyst [Co(acac)3/triphos (L1)/HNTf2].18i
Inspired by these works, we envisaged the possibility to perform the selective reduction of imides using a cobalt-based catalyst system. Here, we show for the first time, a general and efficient methodology for the non-noble metal-catalysed reductive C2-functionalization of cyclic imides (phthalimides and succinimides).
Results and discussion
At the start of this project the reductive methoxylation of N-methylphthalimide 1a using methanol as solvent was selected as benchmark reaction (Table 1). Initially, the reaction was performed with Co(BF4)2·6H2O using similar conditions (150 °C, 60 bar H2, 18 h) known for Ru catalysts. However, no activity was observed (Table 1, entry 1). When the same reaction was conducted in the presence of 5 mol% of triphos L1, a quantitative yield of 3-methoxy-2-methylisoindolin-1-one 2a was obtained with excellent selectivity (Table 1, entry 2). Gratifyingly, no traces of products coming from reduction of both carbonyl groups or aromatic ring hydrogenation were observed.|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | T (°C) | H2 (bar) | [Co] | [L1] | Conv.b (%) | 2a (%) |
| a Standard reaction conditions: N-methylphthalimide 1a (82.2 mg, 0.5 mmol), Co(BF4)2·6H2O (0.5 to 5 mol%), triphos L1 (1 to 10 mol%), H2 (10–60 bar), MeOH (2 mL), 90–150 °C and 3–18 h. [Co] = [Co(BF4)2·6H2O] and [L1] in mol% with respect to 1a. b Conversion of 1a and yields of 2a were calculated by GC using hexadecane as internal standard. c The reaction was carried out without hydrogen using a pressure tube. d Run at 3 h. | ||||||
| 1 | 150 | 60 | 2.5 | — | — | — |
| 2 | 150 | 60 | 2.5 | 5 | >99 | >99 |
| 3 | 130 | 60 | 2.5 | 5 | >99 | >99 |
| 4 | 130 | 30 | 2.5 | 5 | >99 | >99 |
| 5 | 110 | 60 | 2.5 | 5 | >99 | >99 |
| 6 | 110 | 30 | 2.5 | 5 | >99 | >99 |
| 7 | 90 | 50 | 2.5 | 5 | >99 | >99 |
| 8 | 90 | 30 | 2.5 | 5 | >99 | >99 |
| 9 | 90 | 20 | 2.5 | 5 | >99 | >99 |
| 10 | 90 | 10 | 2.5 | 5 | 86 | 84 |
| 11 | 70 | 60 | 2.5 | 5 | 91 | 90 |
| 12 | 70 | 40 | 2.5 | 5 | 87 | 84 |
| 13 | 70 | 20 | 5 | 10 | 90 | 89 |
| 14c | 90 | — | 2.5 | 5 | — | — |
| 15d | 90 | 20 | 2.5 | 5 | 53 | 52 |
| 16 | 90 | 20 | 2.5 | 3.75 | 77 | 75 |
| 17 | 90 | 20 | 2.5 | 2.5 | 60 | 58 |
| 18 | 90 | 20 | 5 | 5 | >99 | 99 |
| 19 | 90 | 20 | 1.5 | 3 | 93 | 93 |
| 20 | 90 | 20 | 0.5 | 1 | 40 | 38 |
| 21 | 90 | 30 | 1.5 | 3 | >99 | 96 |
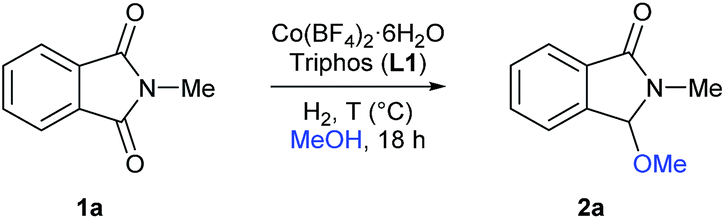
Then, the effect of pressure and temperature was evaluated in more detail (Table 1, entries 3–13). To our delight, the reaction proceeds efficiently at much milder conditions, and excellent yields of methoxylated product 2a were obtained at 90 °C and 20 bar of hydrogen (Table 1, entry 9). In addition, the catalytic system also showed high activities at 70 °C, albeit higher pressures of hydrogen or catalyst loadings were required in these cases (Table 1, entries 11–13). To demonstrate the need of hydrogen, we performed the reaction at 90 °C in a pressure tube and no conversion was detected (Table 1, entry 14).
At this point, the effect of the relative amounts of ligand L1 with respect to the cobalt precursor was investigated in more detail (Table 1, entries 16–18). While for 2.5 mol% of cobalt pre-catalyst, two equivalents of ligand L1 were required to perform the reaction efficiently (Table 1, entries 9, 16 and 17), at higher cobalt catalyst loadings (5 mol%) only one equivalent of L1 was enough for an equally efficient methoxylation of N-methylphthalimide 1a (Table 1, entry 18). Finally, the catalytic system also afforded high yields of product 2a at 1.5 mol%, but using 30 bar of hydrogen (Table 1, entry 21).
Next, the catalytic activity of different metal pre-catalysts was evaluated (Table S1†). Among all the different cobalt precursors tested (Table S1,† entries 1–12), [Co(BF4)2·6H2O] and [Co(ClO4)2·6H2O] (Table S1,† entries 1 and 10) afforded the highest activity. In contrast, Co(acac)3, Co(acac)2 and also Ru(acac)3 in combination with ligand L1 (triphos) were not active under the optimal reaction conditions (Table S1,† entry 2, 3 and 13, respectively). The non-activity of the ruthenium pre-catalyst is notworthy, since [Ru(acac)3/L1/MSA] has been recently described as an active catalytic system for the same reaction. Apparently, for the formation of our active non-noble catalyst no extra acid additive is necessary. Taking into account the important role of the tetrafluoroborate anion (−BF4) in present system, tetrafluoroborate salts of copper(II), iron(II) and zinc(II) were also tested in the benchmark reaction (Table S1,† entries 14–16). However, no activity was detected for any of them, indicating that cobalt is unique for this reaction.
Regarding the ligand, we tested, besides L1, several tridentate (L2–L5), tetradentate (L6), bidentate (L7–L11) and monodentate (L12) ligands (Scheme 1) for the reductive methoxylation of N-methylphthalimide (1a). Apart from triphos (L1), which exhibited the best yield (>99%), only the tridentate ligand L2 afforded 2a, albeit in low yields (11%).
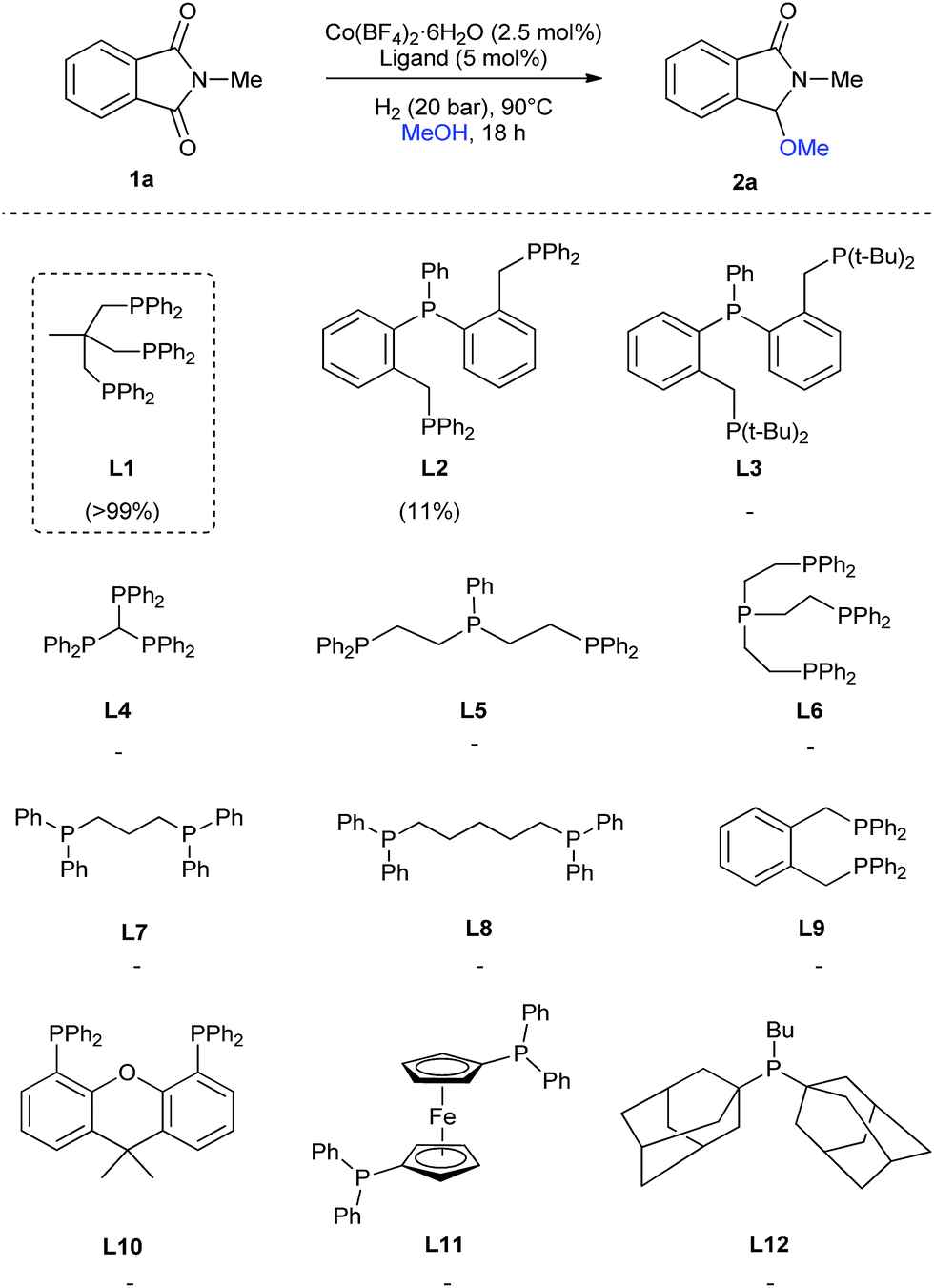 | ||
| Scheme 1 Cobalt-catalysed reductive methoxylation of N-methylphthalimide (1a): influence of the ligand. Standard reaction conditions: N-methylphthalimide 1a (82.2 mg, 0.5 mmol), Co(BF4)2·6H2O (4.3 mg, 0.0125 mmol, 2.5 mol%), ligand (0.025 mmol, 5 mol%, 2.5 eq. to Co), H2 (20 bar), MeOH (2 mL), 90 °C and 18 h. Yields of product 2a were calculated by GC using hexadecane as internal standard. | ||
Having established the [Co(BF4)2·6H2O/triphos (L1)] system as the best catalyst, we decided to explore its activity for the reductive methoxylation of more than 20 symmetrical substituted cyclic imides (Table 2). In general, all the reactions were conducted using 2.5 mol% of cobalt pre-catalyst, 5 mol% of ligand L1 under 90 °C and 20 bar of hydrogen.
|
|
|||
|---|---|---|---|
| Entrya | Cyclic imide 1 | [Co] (mol%) | 2 [%] |
| a Standard reaction conditions: cyclic imide (0.5 mmol), Co(BF4)2·6H2O (4.25 mg, 0.0125 mmol, 2.5 mol%), triphos L1 (15.6 mg, 0.025 mmol, 5 mol%, 2 eq. to Co), H2 (20 bar), MeOH (2 mL), 90 °C and 18 h. When the reaction was carried out using 4 to 6 mol% of cobalt precatalyst, 1.5 eq. of L1 respect to the metal was added. b Isolated yield of the product after purification by column chromatography on silica are given between brackets. c Run at 110 °C. | |||
| 1 |
|
2.5 | 2a [95] |
| 2 |
|
4 | 2b [89] |
| 3 |
|
4 | 2c [90] |
| 4 |

|
4 | 2d [96] |
| 5 |
|
2.5 | 2e [97] |
| 6 |
|
2.5 | 2f [93] |
| 7 |
|
2.5 | 2g [88] |
| 8 |
|
6 | 2h [91] |
| 9 |
|
4 | 2i [98] |
| 10c |

|
2.5 | 2j [95] |
| 11 |
|
2.5 | 2k [96] |
| 12 |
|
2.5 | 2l [94] |
| 13 |
|
4 | 2m [99] |
| 14 |

|
5 | 2n [99] |
| 15c |
|
2.5 | 2o [86] |
| 16 |
|
2.5 | 2p [89] |
| 17 |

|
2.5 | 2q [81] |
| 18 |
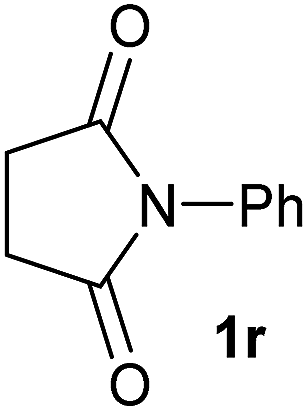
|
2.5 | 2r [89] |
| 19 |
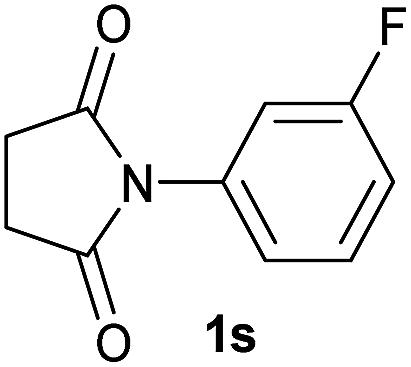
|
2.5 | 2s [94] |
| 20 |
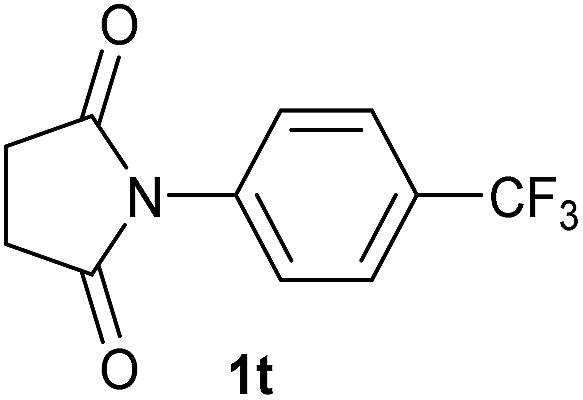
|
2.5 | 2t [85] |
| 21 |
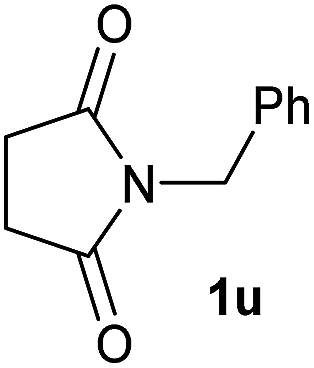
|
4 | 2u [80] |

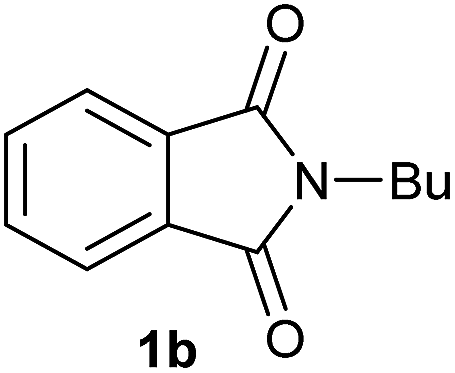
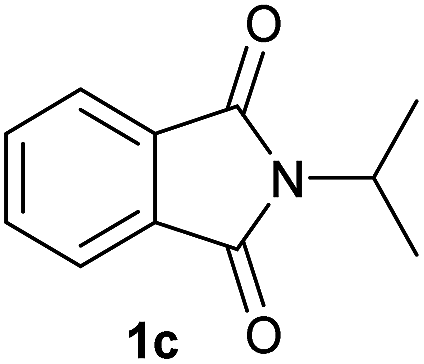
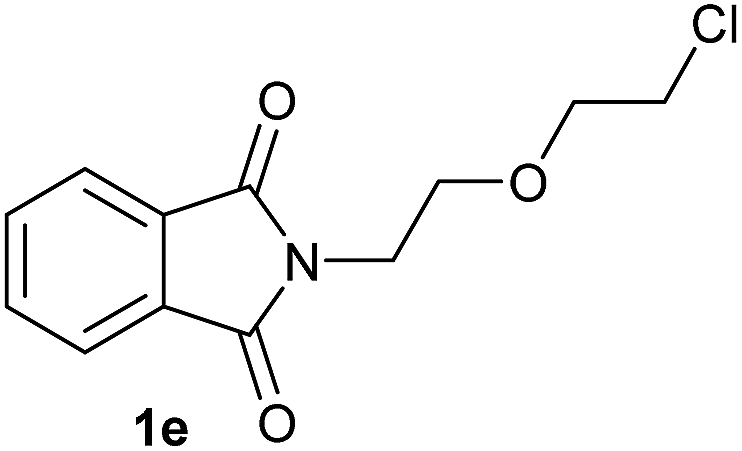
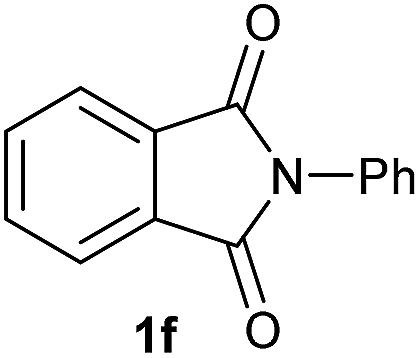
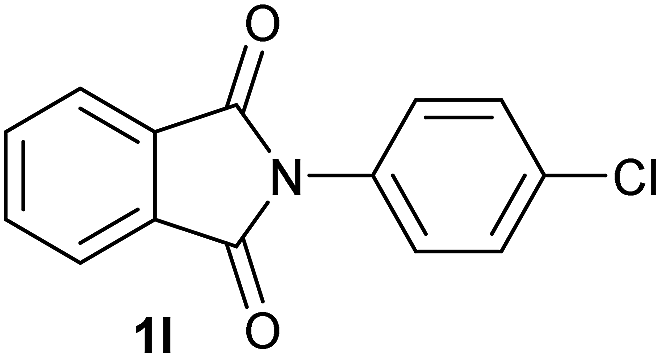
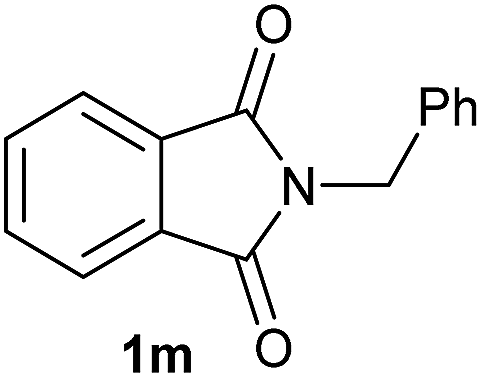
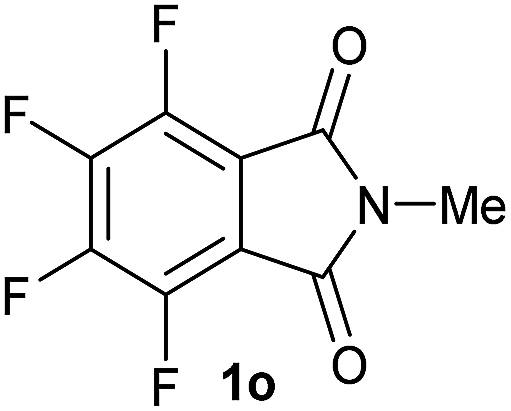
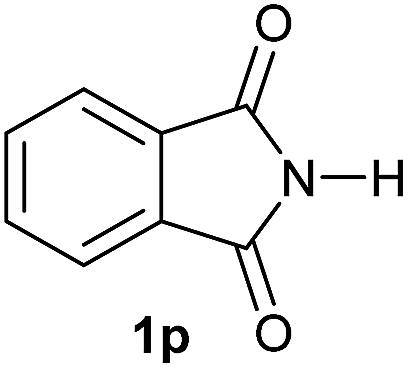
Gratifyingly, excellent selectivity for the monoalkoxylation product was observed for all the studied substrates. N-Alkyl substituted phthalimides (1a–1e) afforded 3-methoxylated isoindolinones 2a–2e in very good isolated yields (89–97%, Table 2, entries 1–5). To study the influence on the catalytic activity of the electronic character of the N-substituent, different N-aryl or N-benzyl substituted phthalimides (1f–n) were tested with the cobalt system. Phthalimides containing fluoro-, trifluoromethyl-, methoxy-, methylthio-, hydroxy- and chloride-substituted aryl rings in ortho-, meta- and para-position were successfully converted affording the corresponding methoxylated products 2f–n in high isolated yields (86–99%, Table 2, entries 6–14). Notably, this unusual reductive transformation worked well for the fluorinated phthalimide derivative 1o obtaining the methoxy compound 2o in high isolated yield (86%, Table 2, entry 15). For the first time, the [Co/triphos] allowed for this reductive transformation of NH-phthalimides. In fact, the system showed an excellent activity for the methoxylation of 1p affording the methoxy product 2p in excellent yield (89%, Table 2, entry 16). Finally, different N-substituted succinimides (1q–u) were tested, too. N-Methyl, N-phenyl, and N-benzyl succinimides, were smoothly methoxylated giving the desired 3-alkoxy-pyrrolidin-2-one derivatives 2q–u in good to very good isolated yields (80–94%, Table 2, entries 17–21). Unfortunately, when N-benzyl-2,3-pyridinedicarboximide and N-anisoyl-2-pyrrolidinone, as examples of heterocyclic and linear imides respectively, were subjected to the optimized reaction conditions no desired product could be obtained.
Once we had shown the generality of our protocol, we became interested to study the selective reduction of non-symmetrical phthalimides (Scheme 2). These are more challenging substrates as the reactivity of the two carbonyl functions might be similar. Up to date, there is only one ruthenium catalyst described,14 which showed moderate to good regioselectivities in such transformations (see Fig. 2). Therefore, the development of new non-precious metal-based strategies to selectively functionalize one of the carbonyl groups still remains a challenging task.
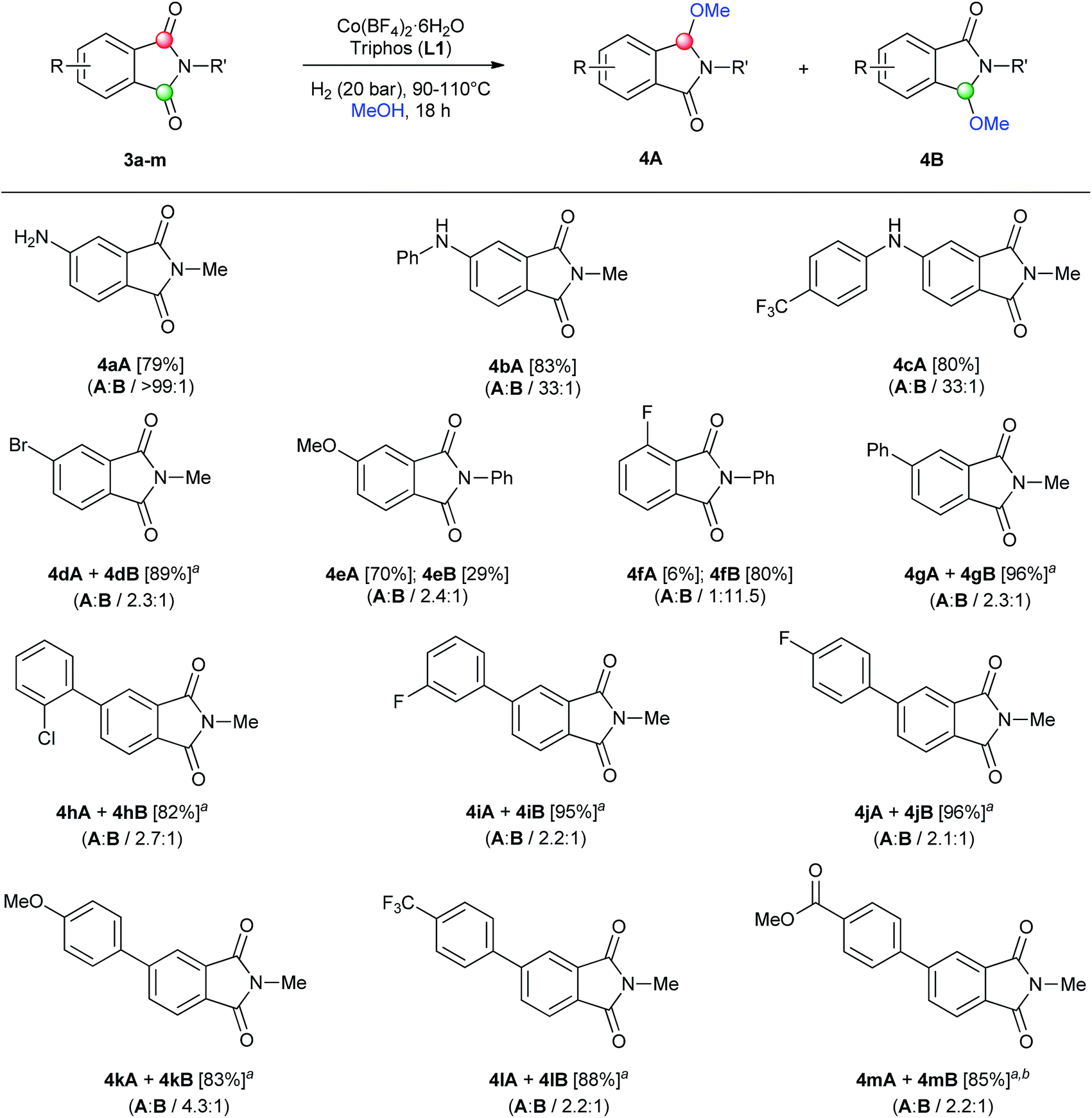 | ||
| Scheme 2 Regioselective [Co/triphos (L1)]-catalysed reductive methoxylation of several asymmetrical ring-substituted phthalimides. Standard reaction conditions: phthalimide (0.5 mmol), Co(BF4)2·6H2O (4–6 mol%), triphos L1 (6–9 mol%, 1.5 eq. to Co), H2 (20 bar), MeOH (2 mL), 90–110 °C and 18 h. Specific reaction conditions for cyclic imides 3a, 3e and 3g: Co(BF4)2·6H2O (4 mol%) at 110 °C, for cyclic imides 3b–d and 3f: Co(BF4)2·6H2O (4 mol%) at 90 °C and for cyclic imides 3h–m: Co(BF4)2·6H2O (6 mol%) at 110 °C. Isolated yields of the products are given between brackets. In parentheses is shown the relative selectivity for each regioisomer A and B, calculated by GC-MS and 1H-NMR analysis (see ESI†). aThe isolated yield corresponds to the unseparable mixture of regioisomers A and B. bSmall amounts (<5%) of products A and B containing ester group hydrogenated to alcohol were detected. | ||
As shown in Scheme 2 the regioselective monomethoxylation of unsymmetrical aryl ring-substituted phthalimides using the cobalt/triphos system proceeded selectively. Most of the phthalimides used for this study are not commercially available and had to be synthetized (see ESI for Experimental details†). To our delight, C4-substituted phthalimides with nitrogen-based electron-donating groups (3a–c), exhibited excellent regioselectivities (>33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for the monofunctionalization on the C2 carbonyl group (isomer A), affording the corresponding isoindolinones in good yields. On the other hand, C4-substituted phthalimides with an electron-withdrawing group such as Br (3d), or an oxygen-based electron-donating group like methoxy (3e), gave lower regioselectivities (2.3:1 and 2.4:1) to the same carbonyl group than nitrogen-based substituents. Furthermore, excellent isolated yields were achieved for the mixture of regioisomer products (4dA + 4dB) (89%) as well as for the regioisomers 4eA and 4eB (70 and 29% yield, respectively). The difference between the observed regioselectivities for an amino and a methoxy C4-substituted phthalimide can be explained by the more important coordinating character of the nitrogen, that can direct the cobalt complex to functionalize the C2 carbonyl.11a Interestingly, a C3-fluorine substituted N-phenyl phthalimide (3f) afforded a good regioselectivity for the functionalization in the carbonyl group but at position C7, hence giving isomer B. This switch in the regioselectivity could be exploited as a synthetic tool. Both regioisomers (4fA and 4fB) were isolated separately in 6 and 80% yield, respectively. Next, a small family of C4-aryl substituted N-methyl phthalimides 3g–m was synthetized (see Scheme S2†) and their reductive alkoxylation was studied. Different substituents such as o-Cl (4h), m- and p-F (4i–j), p-OMe (4k), p-CF3 (4l) and p-C(O)OMe (4m) aryl groups afforded moderate to good regioselectivities (>2.2:1) to the carbonyl A position, with no influence of their electronic character. For all of these examples, alkoxylated products 4g–m were successfully isolated in up to 96% yield as a mixture of regioisomers (A and B). Functional groups like halogen, ether, trifluoromethyl and ester groups were tolerated in the presence of this cobalt-based system.
:1) for the monofunctionalization on the C2 carbonyl group (isomer A), affording the corresponding isoindolinones in good yields. On the other hand, C4-substituted phthalimides with an electron-withdrawing group such as Br (3d), or an oxygen-based electron-donating group like methoxy (3e), gave lower regioselectivities (2.3:1 and 2.4:1) to the same carbonyl group than nitrogen-based substituents. Furthermore, excellent isolated yields were achieved for the mixture of regioisomer products (4dA + 4dB) (89%) as well as for the regioisomers 4eA and 4eB (70 and 29% yield, respectively). The difference between the observed regioselectivities for an amino and a methoxy C4-substituted phthalimide can be explained by the more important coordinating character of the nitrogen, that can direct the cobalt complex to functionalize the C2 carbonyl.11a Interestingly, a C3-fluorine substituted N-phenyl phthalimide (3f) afforded a good regioselectivity for the functionalization in the carbonyl group but at position C7, hence giving isomer B. This switch in the regioselectivity could be exploited as a synthetic tool. Both regioisomers (4fA and 4fB) were isolated separately in 6 and 80% yield, respectively. Next, a small family of C4-aryl substituted N-methyl phthalimides 3g–m was synthetized (see Scheme S2†) and their reductive alkoxylation was studied. Different substituents such as o-Cl (4h), m- and p-F (4i–j), p-OMe (4k), p-CF3 (4l) and p-C(O)OMe (4m) aryl groups afforded moderate to good regioselectivities (>2.2:1) to the carbonyl A position, with no influence of their electronic character. For all of these examples, alkoxylated products 4g–m were successfully isolated in up to 96% yield as a mixture of regioisomers (A and B). Functional groups like halogen, ether, trifluoromethyl and ester groups were tolerated in the presence of this cobalt-based system.
Furthermore, we envisaged the possibility to perform selective intramolecular reductive alkoxylations. This route gives straightforward access to interesting building blocks for the synthesis of alkaloids and intermediates for the production of a stereogenic carbon on the α-positon to the nitrogen lactam.3c,d,g Using several N-(3-hydroxypropyl)phthalimides (5a–e), it was possible to efficiently synthesize these tricyclic compounds22 in one-step (Scheme 3). In order to achieve full conversions, the reactions were conducted under 20 bar of hydrogen in methanol at 90 or 110 °C in the presence of 2.5–6 mol% catalyst. N-(3-Hydroxypropyl)phthalimide 5a with no substitution in the aromatic ring afforded cyclic compound 6a in an excellent isolated yield (94%). Encouraged by this result, different C4-substituted N-(3-hydroxypropyl)phthalimides were studied in order to explore the regioselectivity of the process. Substrates with an electron-donating group in C4 position such as NH-Ph (5b) or OMe (5c), showed good to excellent regioselectivities (8:1 and 3.5:1, respectively) to the attack of carbonyl group A. The better regioselectivity for the amino substituted phthalimide 5b in comparison with the methoxy one 5c can be also explained by the directing effect of the nitrogen.11a Regioselectivity was moderate towards isomer A in the case of a phthalimide substituted with an electron-withdrawing group such as F (5d), giving products 6dA and 6dB in 86% yield. Finally, no regioselectivity was detected in the intramolecular alkoxylation of the phthalimide 5e, containing an alkyl substituent in the aromatic ring. Two regioisomeric positions A and B were reacted with the same selectivity, affording the mixture of regioisomers 6eA and 6eB in 87% isolated yield.
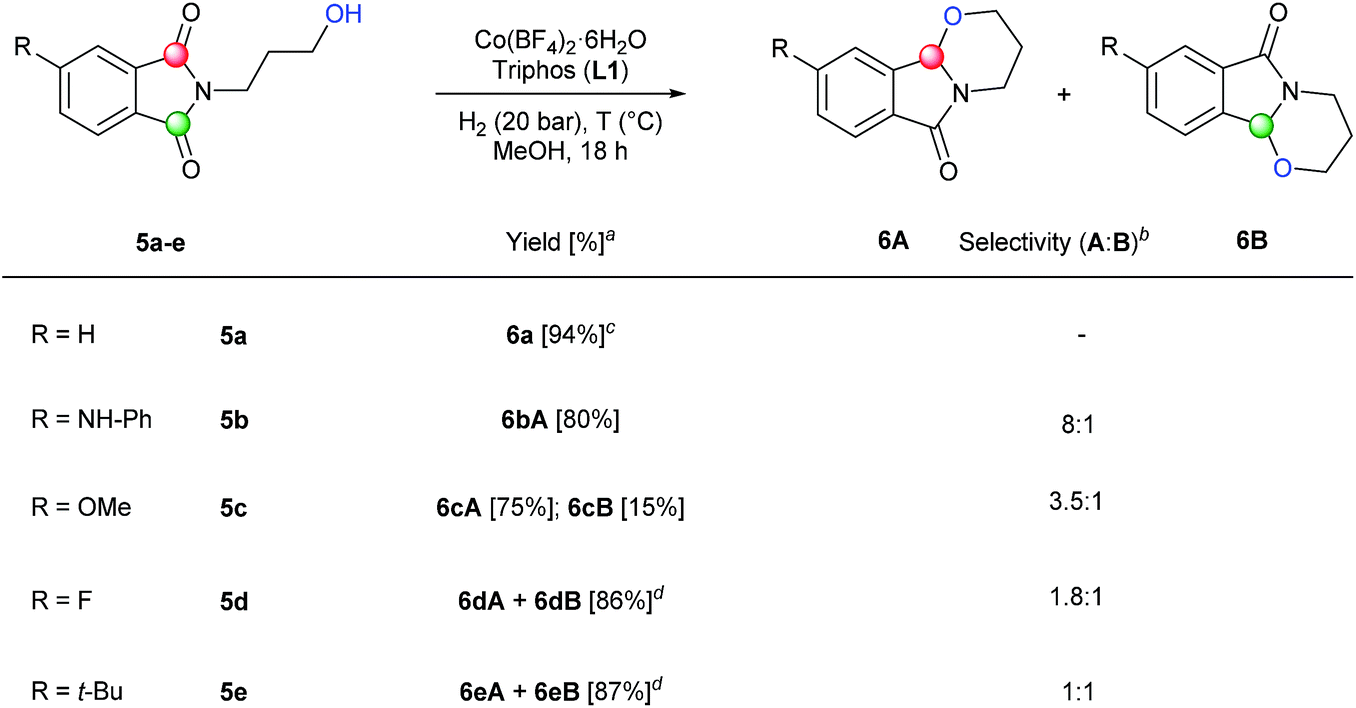 | ||
| Scheme 3 Synthesis of tricyclic compounds by one-step regioselective [Co/triphos (L1)]-catalysed intramolecular reductive cyclization of different C4-substituted N-hydroxypropyl phthalimides. Standard reaction conditions: phthalimide (0.5 mmol), Co(BF4)2·6H2O (2.5–6 mol%), triphos L1 (5–9 mol%, 1.5–2 eq. to Co), H2 (20 bar), MeOH (2 mL), 90–110 °C and 18 h. When the reaction was carried out using 6 mol% of cobalt precatalyst, 1.5 eq. of ligand L1 respect to the metal was added. Specific reaction conditions for cyclic imide 5a: Co(BF4)2·6H2O (2.5 mol%) at 90 °C, for cyclic imides 5b and 5d: Co(BF4)2·6H2O (6 mol%) at 90 °C and for cyclic imides 5c and 5e: Co(BF4)2·6H2O (6 mol%) at 110 °C. aIsolated yield of the products are given. bThe relative selectivity for each cyclic regioisomer A and B was calculated by GC-MS and 1H-NMR analysis (see ESI†). cWhen (R = H) compounds A and B are the same. dIsolated yield of the unseparable mixture of products A and B is given. | ||
Finally, we decided to investigate the general applicability of different alcohols in this reductive functionalization, reacting phthalimide 1a with a wide range of alcohols under neat conditions (Scheme 4). All the reactions were conducted under the previously optimized conditions for methanol (2.5 mol% Co, 5 mol% L1, 20 bar of hydrogen, 90 °C, 18 h) and in specific cases, higher catalyst loadings were required to obtain full conversions of 1a. Both aliphatic primary alcohols (ethanol, 2-methoxyethanol, pentanol, cyclopentanemethanol) and secondary ones (isopropanol, 3-pentanol) afforded the corresponding 3-alkoxylated isoindolinones 7a–f with excellent isolated yields (83–95%). In addition, benzyl and phenethyl alcohols also reacted successfully to give the corresponding C3 functionalized isoindolinones in very good yields (89 and 91%, respectively). In conclusion, this cobalt-catalysed transformation allows the straightforward synthesis of a variety of functionalized isoindolinone derivatives.
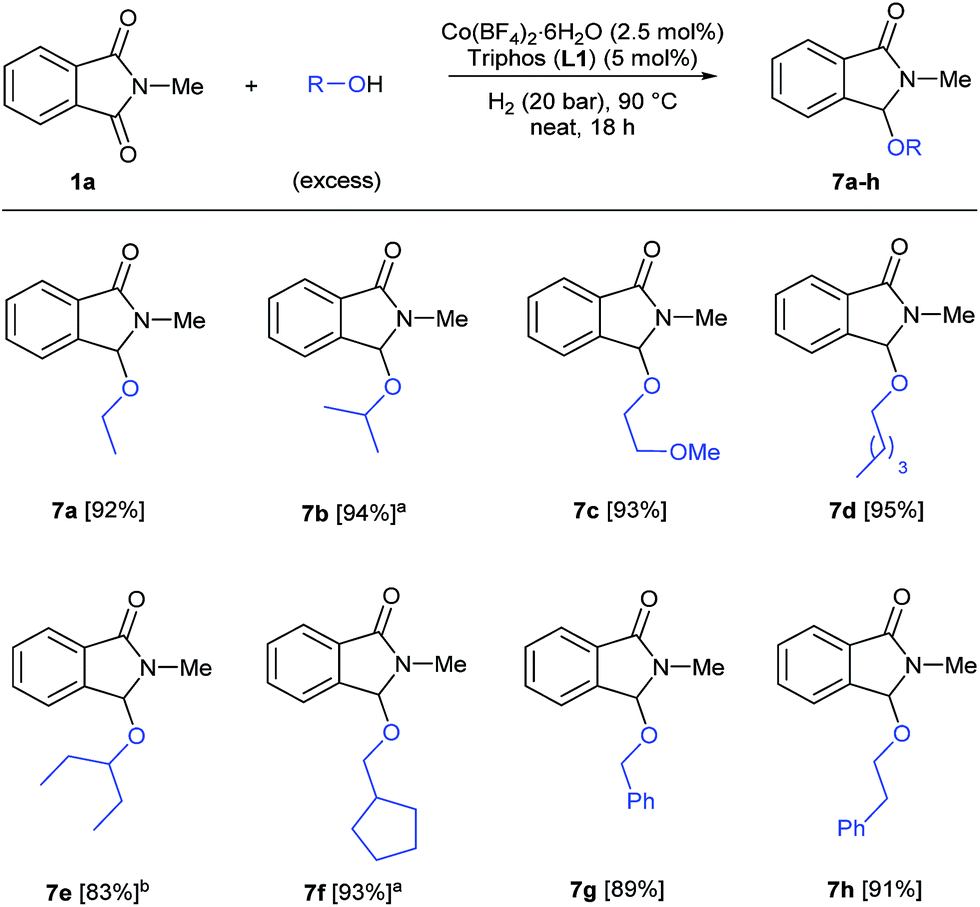 | ||
| Scheme 4 [Co/triphos]-catalysed reductive alkoxylation of N-methylphthalimide 1a with different alcohols. Standard reaction conditions: N-methylphthalimide 1a (82.2 mg, 0.5 mmol), Co(BF4)2·6H2O (4.25 mg, 0.0125 mmol, 2.5 mol%), triphos L1 (16.7 mg, 0.025 mmol, 5 mol%, 2 eq. to Co), H2 (20 bar), alcohol (2 mL), 90 °C and 18 h. Isolated yields of the products are given. [a] Run with Co(BF4)2·6H2O (4 mol%) and triphos L1 (6 mol%). [b] Run with Co(BF4)2·6H2O (6 mol%) and triphos L1 (9 mol%). | ||
To gain insight in the mechanism of the cobalt-catalysed reaction, some kinetic studies (see Fig. 3 and S1–S4†) were performed. Fig. 3 (up) shows the yield/time kinetic profiles for the formation of 3-methoxy-2-methylisoindolin-1-one 2a in the reductive methoxylation of 1a at different hydrogen pressures: (A) 30 bar, (B) 20 bar and (C) 10 bar. No induction period was detected in any experiment, indicating that the catalytically active species can be formed easily. The comparison of the different kinetic profiles reveals that the initial rates (r0), expressed as [yield (%) of 2a × t (min)−1], decrease notably from 30 to 10 bar of hydrogen (0.7325, 0.4379 and 0.149, respectively). Therefore, the reaction exhibits a strong dependence on the hydrogen pressure indicating that the initial hydrogenation of the phthalimide 1a to the intermediate hemiaminal 1aI is the rate limiting step of the overall process. Thus, the subsequent methoxylation of 1aI is expected to be the fast step. In order to confirm these assumptions, additional kinetic experiments using the hemiaminal 1aI as substrate were performed. Fig. 3 (bottom) shows the yield/time kinetic profile of the reductive methoxylation of 1aI under 20 bar of hydrogen (see also Fig. S4†). The initial rate for the formation of 2a in this case (r0 = 2) is almost five times larger than the one obtained using phthalimide 1a as starting material (r0 = 0.4379).
 | ||
| Fig. 3 (Up) Yield/time kinetic profile for the formation of product 2a in the reductive methoxylation of N-methylphthalimide 1a using methanol at 90 °C under different pressures of molecular hydrogen: (A) 30 bar, (B) 20 bar and (C) 10 bar. (Bottom) Yield/time kinetic profile for the formation of product 2a from the intermediate hemiaminal 1aI using methanol and molecular hydrogen (20 bar) at 90 °C. Insets correspond to the initial rate plots where (r0) is the slope of the linear equation: [yield (%) = r0 × time (min)] defined at initial reaction times and expressed as [yield (%) of product × t (min)−1]. Standard reaction conditions: substrate 1a or 1aI (3.0 mmol), Co(BF4)2·6H2O (25.5 mg, 0.075 mmol, 2.5 mol%), triphos L1 (100.5 mg, 0.15 mmol, 5 mol%, 2 eq. to Co), MeOH (12.0 mL) and H2 (10, 20 or 30 bar) at 90 °C. Yields of product 2a were calculated by GC using hexadecane as internal standard. Vertical error bar (5%) for all data points is shown. | ||
This observation supports the methoxylation of 1aI to 2a as the fast step, and the hemiaminal 1a as a real intermediate of this transformation.23 Indeed, additional control experiments starting from 1aI corroborate this observation (see Scheme S5†). The reaction of the hemiaminal 1aI in the presence of lower catalyst loadings (0.5 mol% Co) afforded good yields of the methoxylated product 2a. Moreover, 2a can be produced in quantitative yields (98%) from 1aI with ligand-free [Co(BF4)2·6H2O] as catalyst.4m Apparently, this simple cobalt salt is able to catalyze the alkoxylation process. Interestingly, when the same reaction is performed adding ligand L1 and in the absence of hydrogen, N-methylphthalimide (1a) was detected in 19% yield as a by-product coming from the de-hydrogenation reaction of 1aI mediated by [Co/L1].
In addition, poisoning studies with TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl), a radical inhibitor, and TMTU (tetramethylurea), a binding poison, were performed (Table S2†).18f In the case of TEMPO the reductive methoxylation proceeded successfully, indicating that no radicals are involved in the mechanism. In contrast, TMTU caused a complete inhibition of the reaction with only half equivalent with respect to the catalyst. This effect, previously observed for the carboxylic acids hydrogenation with the same catalyst,18f can be explained by either a dinuclear cobalt species or TMTU acting as a bridging poison.
With the aim of understanding in more detail the catalytic system, high-resolution electrospray ionization mass spectrometry (HR ESI-MS) experiments were performed (Fig. S5–S9†). At short reaction times of 0.5 and 2 hours a signal at m/z 728.156 was detected, consistent with [Co(L1)(COO−)]+. The formation of this species is justified by the use of MeOH/0.1% HCOOH mixture as solvent for the ESI Experiment. In fact, in samples containing a simple mixture of Co(BF4)2·6H2O/L1 and Co(BF4)2·6H2O/L1/2a, the same peak was detected. Any of these tests showed a cobalt species coordinated to N-methylphthalimide 1a. Interestingly, a species with m/z 1309.404 corresponding to [Co(L1)2(H)2]+ could be detected as an important peak in samples at short reaction times. However, in a sample taken at the end of the reaction, this species becomes less important and, using acetonitrile as ESI-MS solvent, a new peak at m/z 901.245 appears. The latter signal is consistent with [Co(L1)(CH3CN)(2a)]+, a possible resting state containing the methoxylated product 2a.
Based on all these observations, a plausible mechanism for the [Co/triphos (L1)]-catalysed reductive alkoxylation of cyclic imides is depicted in Fig. 4. The [Co(L1)2(H)2]+ species, detected by ESI-MS, is proposed as the active catalyst for the hydrogenation of 1a to the hemiaminal intermediate 1aI. At the end of the reaction, [Co(L1)(CH3CN)(2a)]+ is detected as the resting state.
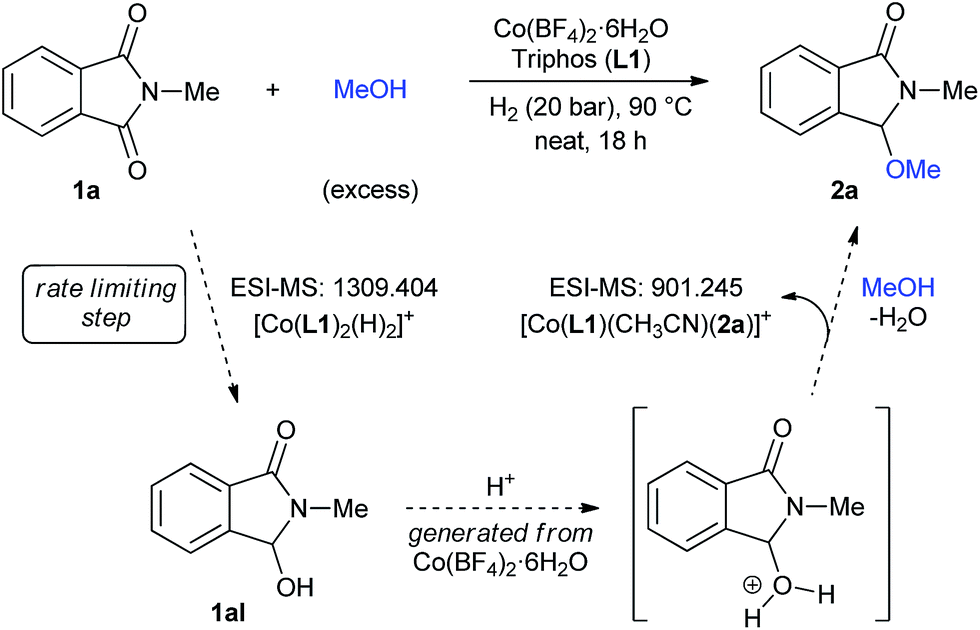 | ||
| Fig. 4 Possible reaction mechanism for the [Co/L1]-catalysed reductive methoxylation of cyclic imides. | ||
Conclusions
In conclusion, a general and efficient cobalt-catalysed reductive alkoxylation of cyclic imides was presented for the first time. This green protocol avoids the use of stoichiometric amounts of silanes or metal hydrides. Hydrogenation of the aromatic ring of the phthalimide core does not take place and excellent chemoselectivity to the mono-alkoxylation products is obtained. A wide range of phthalimides/succinimides are selectively functionalized under mild conditions. Notably, the [Co/triphos] system is active without the need of any acid additive to give 3-alkoxy-2,3-dihydro-1H-isoindolin-1-one and 3-alkoxy-pyrrolidin-2-one derivatives in high isolated yields. Furthermore, this cobalt based catalyst allows the selective functionalization of one of the carbonyl groups in non-symmetrical aryl ring-substituted phthalimides. Additionally, the reaction can be performed in an intramolecular fashion, giving N,O-acetal tricyclic compounds in one-step with high yields. Kinetic investigations revealed that the initial hydrogenation of the phthalimide to the hemiaminal intermediate is the rate limiting step of the overall process. This novel base metal protocol opens a door to the development of environmentally-benign processes for the selective synthesis of functionalized N-heterocyclic compounds.Experimental details
General procedure for the reductive methoxylation of N-methylphthalimide (1a)
A 4 mL glass vial containing a stirring bar was sequentially charged with N-methylphthalimide 1a (82.2 mg, 0.5 mmol), Co(BF4)2·6H2O (4.25 mg, 0.0125 mmol, 2.5 mol%), triphos L1 (16.75 mg, 0.025 mmol, 5 mol%, 2.5 eq. to Co), n-hexadecane (50.0 mg) as an internal standard and MeOH (2.0 mL) as solvent. Afterwards, the reaction vial was capped with a septum equipped with a syringe and set in the alloy plate, which was then placed into a 300 mL autoclave. Once sealed, the autoclave was purged three times with 30 bar of hydrogen, then pressurized to 20 bar and placed into an aluminium block, which was preheated at 90 °C. After 18 h, the autoclave was cooled in an ice bath, and the remaining gas was carefully released. Finally, the reaction mixture was diluted with ethyl acetate and analysed by GC.General procedure for the reductive alkoxylation of cyclic imides
A 4 mL glass vial containing a stirring bar was sequentially charged with cyclic imide (0.5 mmol), Co(BF4)2·6H2O (2.5–6 mol%), triphos L1 (5–9 mol%, 1.5–2 eq. to Co) and alcohol (2.0 mL) as solvent. Afterwards, the reaction vial was capped with a septum equipped with a syringe and set in the alloy plate, which was then placed into a 300 mL autoclave. Once sealed, the autoclave was purged three times with 30 bar of hydrogen, then pressurized to 20 bar and placed into an aluminium block, which was preheated at 90–130 °C. After 18 h, the autoclave was cooled in an ice bath, and the remaining gas was carefully released. Finally, the reaction mixture was diluted with ethyl acetate and purified by silica gel column chromatography (n-heptane/ethyl acetate mixtures) obtaining the desired alkoxylated derivatives.General procedure for the kinetic studies
A 100 mL glass inlet containing a stirring bar was sequentially charged with the corresponding substrate 1a or 1aI (3.0 mmol), Co(BF4)2·6H2O (25.5 mg, 0.075 mmol, 2.5 mol%), triphos L1 (100.5 mg, 0.15 mmol, 5 mol%, 2.5 eq. to Co), n-hexadecane (250.0 mg) as an internal standard and MeOH (12.0 mL) as solvent. Afterwards, the reaction inlet was then placed into a 100 mL autoclave. Once sealed, the autoclave was purged three times with 30 bar of hydrogen, then pressurized to 10, 20 or 30 bar and placed into an aluminium block, which was preheated at 90 °C. Periodically, aliquots of 200 μL were taken at different times of reaction, diluted with ethyl acetate and analysed by GC.Acknowledgements
This work was supported by the state of Mecklenburg-Vorpommern and the BMBF. J. R. C.-A. and R. A. thanks Ramon Areces Foundation for a postdoctoral fellowship.Notes and references
- (a) A. Kleemann, J. Engel, B. Kutschner and D. Reichert, Pharmaceutical Substances: Syntheses, Patents, Applications, Thieme, Stuttgart, New York, 5th edn, 2009 Search PubMed; (b) P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS; (c) H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, Industrial Organic Chemicals, Wiley-Interscience, New York, 3rd edn, 2013 Search PubMed; (d) D. Dodds and D. J. Cole-Hamilton, in Sustainable Catalysis: Challenges and practices for the Pharmaceutical and Fine Chemical Industries, ed. P. J. Dunn, K. K. Hii, M. J. Krische and M. T. Williams, John Wiley and Sons, Hoboken, NJ, 2013, pp. 1–36 Search PubMed; (e) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS; (f) A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed; (g) G. E. Arnott, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier B.V., 2nd edn, 2014, vol. 8, pp. 368–409 Search PubMed.
- For selected examples, see: (a) H. Miyachi, A. Azuma, A. Ogasawara, E. Uchimura, N. Watanabe, Y. Kobayashi, F. Kato, M. Kato and Y. Hashimoto, J. Med. Chem., 1997, 40, 2858–2865 CrossRef CAS PubMed; (b) K.-E. Kil, P. Poutiainen, Z. Zhang, A. Zhu, J.-K. Choi, K. Jokivarsi and A.-L. Brownell, J. Med. Chem., 2014, 57, 9130–9138 CrossRef CAS PubMed; (c) P. M. U. Ung, J. B. Dunbar Jr, J. E. Gestwicki and H. A. Carlson, J. Med. Chem., 2014, 57, 6468–6478 CrossRef CAS PubMed; (d) M. A. Bhat, M. A. Al-Omar, M. A. Ansari, K. M. A. Zoheir, F. Imam, S. M. Attia, S. A. Bakheet, A. Nadeem, H. M. Korashy, A. Voronkov, V. Berishvili and S. F. Ahmad, J. Med. Chem., 2015, 58, 8850–8867 CrossRef CAS PubMed.
- For selected examples, see: (a) V. G. Stygles, J. F. Newton and J. B. Hook, Res. Commun. Chem. Pathol. Pharmacol., 1977, 18, 329–340 CAS; (b) E. J. Corey and M. M. Mehrotra, Tetrahedron Lett., 1988, 29, 57–60 CrossRef CAS; (c) D. Romo and A. I. Meyers, Tetrahedron, 1991, 47, 9503–9569 CrossRef CAS; (d) A. A. Bahajaj, P. D. Bailey, M. H. Moore, K. M. Morgan and J. M. Vernon, J. Chem. Soc., Chem. Commun., 1994, 2511–2512 RSC; (e) J. T. Link, S. Raghavan and S. J. Danishefsky, J. Am. Chem. Soc., 1995, 117, 552–553 CrossRef CAS; (f) M. H. Norman, D. J. Minick and G. C. Rigdon, J. Med. Chem., 1996, 39, 149–157 CrossRef CAS PubMed; (g) J. M. Andrés, I. Herráiz-Sierra, R. Pedrosa and A. Pérez-Encabo, Eur. J. Org. Chem., 2000, 1719–1726 CrossRef; (h) U. C. Yoon, Y. X. Jin, S. W. Oh, C. H. Park, J. H. Park, C. F. Campana, X. Cai, E. N. Duesler and P. S. Mariano, J. Am. Chem. Soc., 2003, 125, 10664–10671 CrossRef CAS PubMed; (i) B. E. Maryanoff, H.-C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431–1628 CrossRef CAS PubMed; (j) J. Royer, M. Bonin and L. Micouin, Chem. Rev., 2004, 104, 2311–2352 CrossRef CAS PubMed; (k) I. R. Hardcastle, S. U. Ahmed, H. Atkins, A. H. Calvert, N. J. Curtin, G. Farnie, B. T. Golding, R. J. Griffin, S. Guyenne, C. Hutton, P. Kaellblad, S. J. Kemp, M. S. Kitching, D. R. Newell, S. Norbedo, J. S. Northen, R. J. Reid, K. Saravanan, H. M. G. Willems and J. Lunec, Bioorg. Med. Chem. Lett., 2005, 15, 1515–1520 CrossRef CAS PubMed; (l) I. R. Hardcastle, S. U. Ahmed, H. Atkins, G. Farnie, B. T. Golding, R. J. Griffin, S. Guyenne, C. Hutton, P. Kaellblad, S. J. Kemp, M. S. Kitching, D. R. Newell, S. Norbedo, J. S. Northen, R. J. Reid, K. Saravanan, H. M. G. Willems and J. Lunec, J. Med. Chem., 2006, 49, 6209–6221 CrossRef CAS PubMed; (m) S. Lee, C. Shinji, K. Ogura, M. Shimizu, S. Maeda, M. Sato, M. Yoshida, Y. Hashimoto and H. Miyachi, Bioorg. Med. Chem. Lett., 2007, 17, 4895–4900 CrossRef CAS PubMed; (n) H. J. Lee, S. J. Lim, S. J. Oh, D. H. Moon, D. J. Kim, J. Tae and K. H. Yoo, Bioorg. Med. Chem. Lett., 2008, 18, 1628–1631 CrossRef CAS PubMed; (o) J. H. Lee, S. R. Byeon, Y. Kim, S. J. Lim, S. J. Oh, D. H. Moon, K. H. Yoo, B. Y. Chung and D. J. Kim, Bioorg. Med. Chem. Lett., 2008, 18, 5701–5704 CrossRef CAS PubMed; (p) S. Ito, Y. Hirata, Y. Nagatomi, A. Satoh, G. Suzuki, T. Kimura, A. Satow, S. Maehara, H. Hikichi, M. Hata, H. Ohta and H. Kawamoto, Bioorg. Med. Chem. Lett., 2009, 19, 5310–5313 CrossRef CAS PubMed; (q) R. Ben Othman, R. Affani, M.-J. Tranchant, S. Antoniotti, V. Dalla and E. Dunach, Angew. Chem., Int. Ed., 2010, 49, 776–780 CrossRef CAS PubMed; (r) A. F. Watson, J. Liu, K. Bennaceur, C. J. Drummond, J. A. Endicott, B. T. Golding, R. J. Griffin, K. Haggerty, X. Lu, J. M. McDonnell, D. R. Newell, M. E. M. Noble, C. H. Revill, C. Riedinger, Q. Xu, Y. Zhao, J. Lunec and I. R. Hardcastle, Bioorg. Med. Chem. Lett., 2011, 21, 5916–5919 CAS; (s) C. Riedinger, M. E. Noble, D. J. Wright, F. Mulks, I. R. Hardcastle, J. A. Endicott and J. M. McDonnell, Chem. Biol. Drug Des., 2011, 77, 301–308 CrossRef CAS PubMed; (t) K. Xu, S. Zhang, Y. Hu, Z. Zha and Z. Wang, Chem.–Eur. J., 2013, 19, 3573–3578 CrossRef CAS PubMed; (u) M. Hamon, N. Dickinson, A. Devineau, D. Bolien, M.-J. Tranchant, C. Taillier, I. Jabin, D. C. Harrowven, R. J. Whitby, A. Ganesan and V. Dalla, J. Org. Chem., 2014, 79, 1900–1912 CrossRef CAS PubMed; (v) A. Daïch, A. Ghinet and B. Rigo, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier B.V., 2nd edn, 2014, vol. 2, p. 682 Search PubMed.
- For selected examples, see: (a) N. J. Lawrence, J. Liddle, S. M. Bushell and D. A. Jackson, J. Org. Chem., 2002, 67, 457–464 CrossRef CAS PubMed; (b) G. Stajer and F. Csende, Curr. Org. Chem., 2005, 9, 1277–1286 CrossRef CAS; (c) D. L. Comins, S. Schilling and Y. Zhang, Org. Lett., 2005, 7, 95–98 CrossRef CAS PubMed; (d) J. Guillaumel, S. Leonce, A. Pierre, P. Renard, B. Pfeiffer, P. B. Arimondo and C. Monneret, Eur. J. Med. Chem., 2006, 41, 379–386 CrossRef CAS PubMed; (e) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202–209 CrossRef CAS PubMed; (f) D. Hamprecht, F. Micheli, G. Tedesco, A. Checchia, D. Donati, M. Petrone, S. Terreni and M. Wood, Bioorg. Med. Chem. Lett., 2007, 17, 428–433 CrossRef CAS PubMed; (g) J. G. Pierce, D. L. Waller and P. Wipf, J. Organomet. Chem., 2007, 692, 4618–4629 CrossRef CAS PubMed; (h) X. Huang and J. Xu, J. Org. Chem., 2009, 74, 8859–8861 CrossRef CAS PubMed; (i) C. Li, X. Li and R. Hong, Org. Lett., 2009, 11, 4036–4039 CrossRef CAS PubMed; (j) A. A. Kilikli, C. Dengiz, S. Ozcan and M. Balci, Synthesis, 2011, 3697–3705 CAS; (k) A. Arizpe, F. J. Sayago, A. I. Jiménez, M. Ordóñez and C. Cativiela, Eur. J. Org. Chem., 2011, 2011, 6732–6738 CrossRef CAS; (l) L. Shi, L. Hu, J. Wang, X. Cao and H. Gu, Org. Lett., 2012, 14, 1876–1879 CrossRef CAS PubMed; (m) N. Lu, L. Wang, Z. Li and W. Zhang, Beilstein J. Org. Chem., 2012, 8, 192–200 CrossRef CAS PubMed; (n) S. K. Mamidyala and M. A. Cooper, Chem. Commun., 2013, 49, 8407–8409 RSC; (o) R. W. Foster, C. J. Tame, H. C. Hailes and T. D. Sheppard, Adv. Synth. Catal., 2013, 355, 2353–2360 CrossRef CAS PubMed; (p) R. Mancuso, I. Ziccarelli, D. Armentano, N. Marino, S. V. Giofre and B. Gabriele, J. Org. Chem., 2014, 79, 3506–3518 CrossRef CAS PubMed; (q) A. Verma, S. Patel, Meenakshi, A. Kumar, A. Yadav, S. Kumar, S. Jana, S. Sharma, C. D. Prasad and S. Kumar, Chem. Commun., 2015, 51, 1371–1374 RSC; (r) K. Kobayashi, Y. Chikazawa and K. Ezaki, Helv. Chim. Acta, 2015, 98, 604–610 CrossRef CAS; (s) K. Kobayashi and Y. Chikazawa, Tetrahedron, 2016, 72, 5100–5105 CrossRef CAS; (t) D. Niedek, S. M. M. Schuler, C. Eschmann, R. C. Wende, A. Seitz, F. Keul and P. R. Schreiner, Synthesis, 2017, 49, 371–382 CAS.
- D. E. Patton and R. S. Drago, J. Chem. Soc., Perkin Trans. 1, 1993, 1611–1615 RSC.
- R. Aoun, J.-L. Renaud, P. H. Dixneuf and C. Bruneau, Angew. Chem., Int. Ed., 2005, 44, 2021–2023 CrossRef CAS PubMed.
- (a) M. Ito, A. Sakaguchi, C. Kobayashi and T. Ikariya, J. Am. Chem. Soc., 2007, 129, 290–291 CrossRef CAS PubMed; (b) M. Ito, C. Kobayashi, A. Himizu and T. Ikariya, J. Am. Chem. Soc., 2010, 132, 11414–11415 CrossRef CAS PubMed.
- S. Takebayashi, J. M. John and S. H. Bergens, J. Am. Chem. Soc., 2010, 132, 12832–12834 CrossRef CAS PubMed.
- A. Arévalo, S. Ovando-Segovia, M. Flores-Alamo and J. J. García, Organometallics, 2013, 32, 2939–2943 CrossRef.
- A. M. Maj, I. Suisse, N. Pinault, N. Robert and F. Agbossou-Niedercorn, ChemCatChem, 2014, 6, 2621–2625 CrossRef CAS.
- (a) G. Ding, B. Lu, Y. Li, J. Wan, Z. Zhang and X. Xie, Adv. Synth. Catal., 2015, 357, 1013–1021 CrossRef CAS; (b) G. Ding, C. Li, Y. Shen, B. Lu, Z. Zhang and X. Xie, Adv. Synth. Catal., 2016, 358, 1241–1250 CrossRef CAS.
- L. Shi, X. Tan, J. Long, X. Xiong, S. Yang, P. Xue, H. Lv and X. Zhang, Chem.–Eur. J., 2017, 23, 546–548 CrossRef CAS PubMed.
- S. Das, D. Addis, L. R. Knoepke, U. Bentrup, K. Junge, A. Brueckner and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 9180–9184 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, I. Sorribes, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 387–391 CrossRef CAS PubMed.
- (a) H. T. Teunissen and C. J. Elsevier, Chem. Commun., 1997, 667–668 RSC; (b) M. C. van Engelen, H. T. Teunissen, J. G. de Vries and C. J. Elsevier, J. Mol. Catal. A: Chem., 2003, 206, 185–192 CrossRef CAS; (c) A. A. Núñez Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154–3156 RSC; (d) F. M. A. Geilen, B. Engendahl, M. Hölscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2011, 133, 14349–14358 CrossRef CAS PubMed; (e) D. L. Dodds, J. Coetzee, J. Klankermayer, S. Brosinski, W. Leitner and D. J. Cole-Hamilton, Chem. Commun., 2012, 48, 12249–12262 RSC; (f) J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinski, W. Leitner, A. M. Z. Slawin and D. J. Cole-Hamilton, Chem.–Eur. J., 2013, 19, 11039–11050 CrossRef CAS PubMed; (g) K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed; (h) Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed; (i) Y. Li, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 10476–10480 CrossRef CAS PubMed; (j) S. Savourey, G. Lefevre, J.-C. Berthet and T. Cantat, Chem. Commun., 2014, 50, 14033–14036 RSC; (k) T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hoelscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed; (l) X. Cui, Y. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed; (m) Y. Li, C. Topf, X. Cui, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 5196–5200 CrossRef CAS PubMed; (n) I. Sorribes, J. R. Cabrero-Antonino, C. Vicent, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 13580–13587 CrossRef CAS PubMed; (o) S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. v. Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC; (p) J. R. Cabrero-Antonino, E. Alberico, K. Junge, H. Junge and M. Beller, Chem. Sci., 2016, 7, 3432–3442 RSC; (q) J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Catal. Sci. Technol., 2016, 6, 7956–7966 RSC; (r) R. Adam, J. R. Cabrero-Antonino, K. Junge, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 11049–11053 CrossRef CAS PubMed.
- (a) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed; (b) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS PubMed; (c) L. C. Misal Castro, H. Li, J.-B. Sortais and C. Darcel, Green Chem., 2015, 17, 2283–2303 RSC; (d) D. S. Mérel, M. L. T. Do, S. Gaillard, P. Dupau and J.-L. Renaud, Coord. Chem. Rev., 2015, 288, 50–68 CrossRef; (e) Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS PubMed; (f) S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed; (g) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed; (h) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed; (i) J.-L. Renaud and S. Gaillard, Synthesis, 2016, 48, 3659–3683 CrossRef CAS; (j) S. W. M. Crossley, C. Obradors, R. M. Martínez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed; (k) A. Quintard and J. Rodriguez, ChemSusChem, 2016, 9, 28–30 CrossRef CAS PubMed; (l) A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- (a) D. A. Valyaev, G. Lavigne and N. Lugan, Coord. Chem. Rev., 2016, 308, 191–235 CrossRef CAS; (b) A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben David, N. A. E. Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed; (c) W. P. Liu, S. C. Richter, Y. J. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7747–7750 CrossRef CAS PubMed; (d) F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 CrossRef CAS PubMed; (e) J. R. Carney, B. R. Dillon and S. P. Thomas, Eur. J. Org. Chem., 2016, 2016, 3912–3929 CrossRef CAS; (f) S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed; (g) S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed; (h) S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 15364–15368 CrossRef CAS PubMed; (i) N. A. Espinosa-Jalapa, A. Nerush, L. J. W. Shimon, G. Leitus, L. Avram, Y. Ben-David and D. Milstein, Chem.–Eur. J., 2017, 23, 5934–5938 CrossRef CAS PubMed; (j) M. Pérez, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, ChemSusChem, 2017, 10, 83–86 CrossRef PubMed.
- (a) C. Federsel, C. Ziebart, R. Jackstell, W. Baumann and M. Beller, Chem.–Eur. J., 2012, 18, 72–75 CrossRef CAS PubMed; (b) G. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed; (c) D. Gärtner, A. Welther, B. R. Rad, R. Wolf and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2014, 53, 3722–3726 CrossRef PubMed; (d) D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed; (e) S. Rösler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef PubMed; (f) T. J. Korstanje, J. Ivar van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298 CrossRef CAS PubMed; (g) D. Zhang, E.-Z. Zhu, Z.-W. Lin, Z.-B. Wei, Y.-Y. Li and J.-X. Gao, Asian J. Org. Chem., 2016, 5, 1323–1326 CrossRef CAS; (h) A. Z. Spentzos, C. L. Barnes and W. H. Bernskoetter, Inorg. Chem., 2016, 55, 8225–8233 CrossRef CAS PubMed; (i) J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 1890–1893 CrossRef CAS PubMed.
- (a) M. Amezquita-Valencia and A. Cabrera, J. Mol. Catal. A: Chem., 2013, 366, 17–21 CrossRef CAS; (b) A. Mukherjee, D. Srimani, S. Chakraborty, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2015, 137, 8888–8891 CrossRef CAS PubMed; (c) Z. Shao, S. Fu, M. Wei, S. Zhou and Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 14653–14657 CrossRef CAS PubMed; (d) R. Adam, C. B. Bheeter, J. R. Cabrero-Antonino, K. Junge, R. Jackstell and M. Beller, ChemSusChem, 2017, 10, 842–846 CrossRef CAS PubMed.
- (a) S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561–4564 CrossRef CAS PubMed; (b) M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076–1080 CrossRef CAS PubMed; (c) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS PubMed; (d) S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588–8594 CrossRef CAS PubMed.
- (a) R. Xu, S. Chakraborty, H. Yuan and W. D. Jones, ACS Catal., 2015, 5, 6350–6354 CrossRef CAS; (b) R. Adam, J. R. Cabrero-Antonino, A. Spannenberg, K. Junge, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 3216–3220 CrossRef CAS PubMed.
- For selected examples, see: (a) C. J. Wharton and R. Wrigglesworth, J. Chem. Soc., Perkin Trans. 1, 1985, 809–813 RSC; (b) A. I. Meyers, B. A. Lefker, T. J. Sowin and L. J. Westrum, J. Org. Chem., 1989, 54, 4243–4246 CrossRef CAS; (c) D. Romo, J. L. Romine, W. Midura and A. I. Meyers, Tetrahedron, 1990, 46, 4951–4994 CrossRef CAS; (d) A. Mertens, H. Zilch, B. Koenig, W. Schaefer, T. Poll, W. Kampe, H. Seidel, U. Leser and H. Leinert, J. Med. Chem., 1993, 36, 2526–2535 CrossRef CAS PubMed; (e) S. M. Allin, C. J. Northfield, M. I. Page and A. M. Z. Slawin, Tetrahedron Lett., 1997, 38, 3627–3630 CrossRef CAS; (f) S. M. Allin, C. J. Northfield, M. I. Page and A. M. Z. Slawin, Tetrahedron Lett., 1998, 39, 4905–4908 CrossRef CAS; (g) S. M. Allin, C. J. Northfield, M. I. Page and A. M. Z. Slawin, Tetrahedron Lett., 1999, 40, 141–142 CrossRef CAS; (h) S. M. Allin, S. L. James, W. P. Martin and T. A. D. Smith, Tetrahedron Lett., 2001, 42, 3943–3946 CrossRef CAS.
- D. Cantillo, Eur. J. Inorg. Chem., 2011, 3008–3013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General information concerning experimental procedures, additional tables, figures, schemes, characterization data and NMR spectra of the isolated compounds are available. See DOI: 10.1039/c7sc01175j |
| This journal is © The Royal Society of Chemistry 2017 |