
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective palladium-catalyzed diboration of 1,1-disubstituted allenes†
Jiawang
Liu‡
 a,
Ming
Nie‡
a,
Qinghai
Zhou
bd,
Shen
Gao
a,
Wenhao
Jiang
a,
Lung Wa
Chung
*b,
Wenjun
Tang
*a and
Kuiling
Ding
*acd
a,
Ming
Nie‡
a,
Qinghai
Zhou
bd,
Shen
Gao
a,
Wenhao
Jiang
a,
Lung Wa
Chung
*b,
Wenjun
Tang
*a and
Kuiling
Ding
*acd
aState Key Laboratory of Organometallic Chemistry, State Key Laboratory of Bio-Organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Ling Ling Rd, Shanghai 200032, China. E-mail: tangwenjun@sioc.ac.cn; kding@sioc.ac.cn
bDepartment of Chemistry, South University of Science and Technology of China, Shenzhen 518055, China. E-mail: oscarchung@sustc.edu.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dCollaborative Innovation Center of Chemical Science and Engineering, Tianjin 300071, China
First published on 16th May 2017
Abstract
A practical and enantioselective palladium-catalyzed diboration of 1,1-disubstituted allenes is developed by employing a P-chiral monophosphorus ligand, BI-DIME. A series of diboronic esters containing a chiral tertiary boronic ester moiety are formed in excellent yields and ee’s with the palladium loading as low as 0.2 mol%. DFT calculations revealed a concerted mechanism of oxidative addition of bis(pinacolato)diboron and allene insertion, as well as a critical dispersion effect on the origins of the enantioselectivity. The method is successfully applied to the concise and enantioselective synthesis of brassinazole.
Chiral boronic esters have become versatile building blocks in synthetic organic chemistry.1 The synthesis of chiral tertiary boronic esters has attracted considerable interest recently and some notable methods have been developed (Fig. 1), including the Aggarwal–Matteson lithiation–borylation methodology from chiral secondary alcohols,2 asymmetric hydroboration of 1,1-disubstituted alkenes,3 and asymmetric borylation of allylic carbonates4 and Michael acceptors,5 as well as tertiary halides.6 The asymmetric diboration of 1,1-disubstituted alkenes or allenes7 would not only provide a chiral tertiary boronic ester moiety, but also form an additional boronic ester component for further transformations. Work by Morken on asymmetric diboration8,9 with various transition metal catalysts (Rh, Pt, or Pd) has provided significant progress in forming secondary boronic ester products with excellent ee’s. However, the asymmetric diboration of 1,1-disubstituted alkenes or allenes to form chiral tertiary boronic esters remains challenging, with either low ee’s or low yields.9b Herein we communicate our results on palladium-catalyzed asymmetric diboration of 1,1-disubstituted allenes that have led to a series of diboronic esters containing a chiral tertiary boronic ester moiety in excellent enantioselectivities and yields with the employment of a P-chiral monophosphorus ligand BI-DIME. The chiral diboronic ester products are useful chiral building blocks, and have led to the first enantioselective synthesis of a specific brassinosteroid biosynthetic inhibitor—brassinazole.19
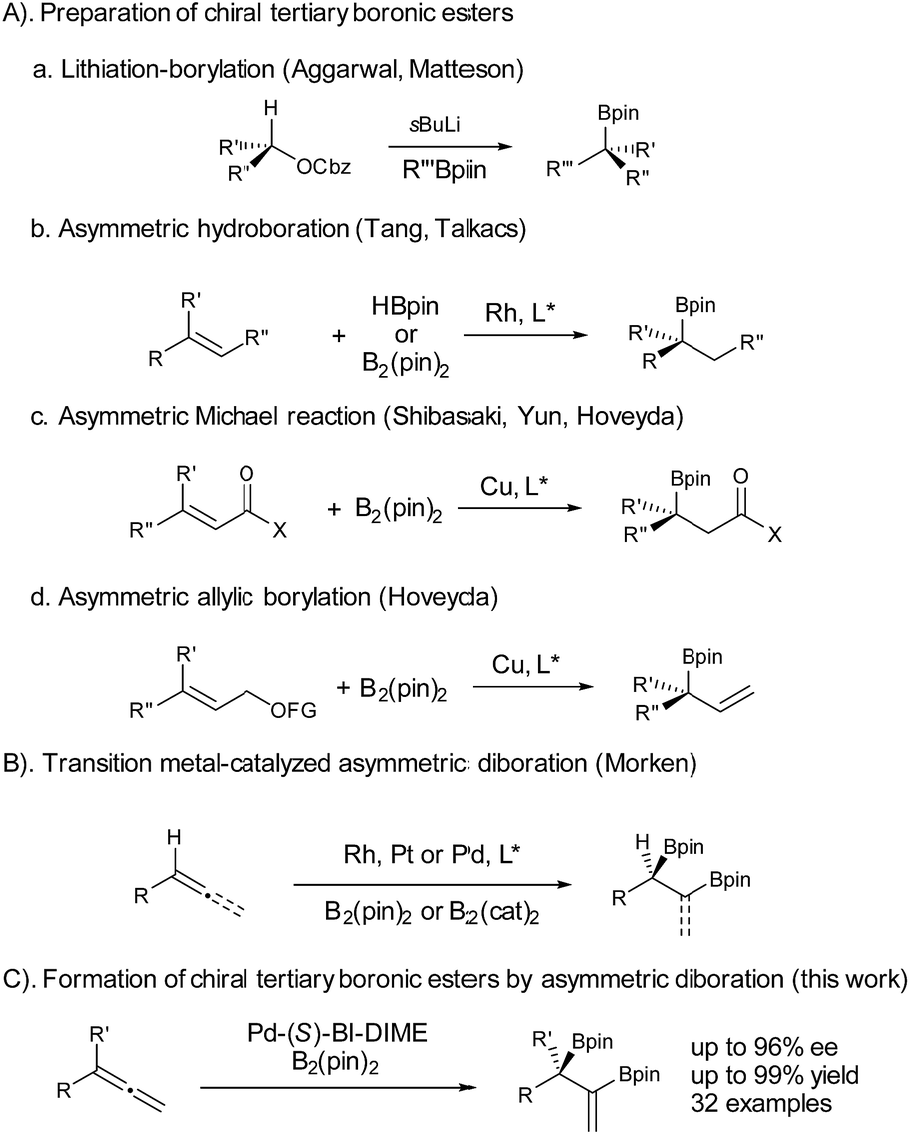 | ||
| Fig. 1 Formation of chiral tertiary boronic esters by asymmetric diboration. | ||
We chose to use buta-2,3-dien-2-ylbenzene (1a) as the substrate to investigate the palladium-catalyzed asymmetric diboration of 1,1-disubstituted allenes (Table 1). The reactions were carried out at rt in cyclohexane for 24 h with bis(pinacolato)diboron as the reagent in the presence of Pd2(dba)3 (1 mol%) and a chiral phosphorus ligand (2.5 mol%) (Table 1). It was found that chelating phosphorus ligands such as BINAP and SDP did not provide any reactivity (entries 1–2).
|
|
||||
|---|---|---|---|---|
| Entrya | L* | Solvent | Yieldb (%) | eec (%) |
| a Unless otherwise specified, the reactions were performed under nitrogen at rt for 24 h with 1a (0.20 mmol), 2 (0.24 mmol), L* (2.5 mol%) and Pd2(dba)3 (1.0 mol%) in the specified solvent. Product 3a was the only detectable product. The R absolute configuration of 3a was determined by comparing its optical rotation with reported data.14 b Isolated yields. c Determined by HPLC on a chiral IC-3 column. d Pd(OAc)2 instead of Pd2(dba)3 was employed as the precursor. e 1a (36.0 mmol), Pd2(dba)3 (0.1 mmol%), L8 (0.25 mmol%), 72 h. | ||||
| 1 | L1 | CyH | 0 | — |
| 2 | L2 | CyH | 0 | — |
| 3 | L3 | CyH | 15 | 52 |
| 4 | L4 | CyH | 95 | 59 |
| 5 | L5 | CyH | 76 | 79 |
| 6 | L6 | CyH | 45 | 12 |
| 7 | L7 | CyH | 60 | 91 |
| 8 | L8 | CyH | 98 | 94 |
| 9 | L8 | Toluene | 82 | 92 |
| 10 | L8 | THF | 51 | 91 |
| 11 | L8 | Dioxane | 27 | 92 |
| 12 | L8 | DCM | 0 | — |
| 13d | L8 | CyH | 33 | 92 |
| 14e | L8 | CyH | 97 | 94 |
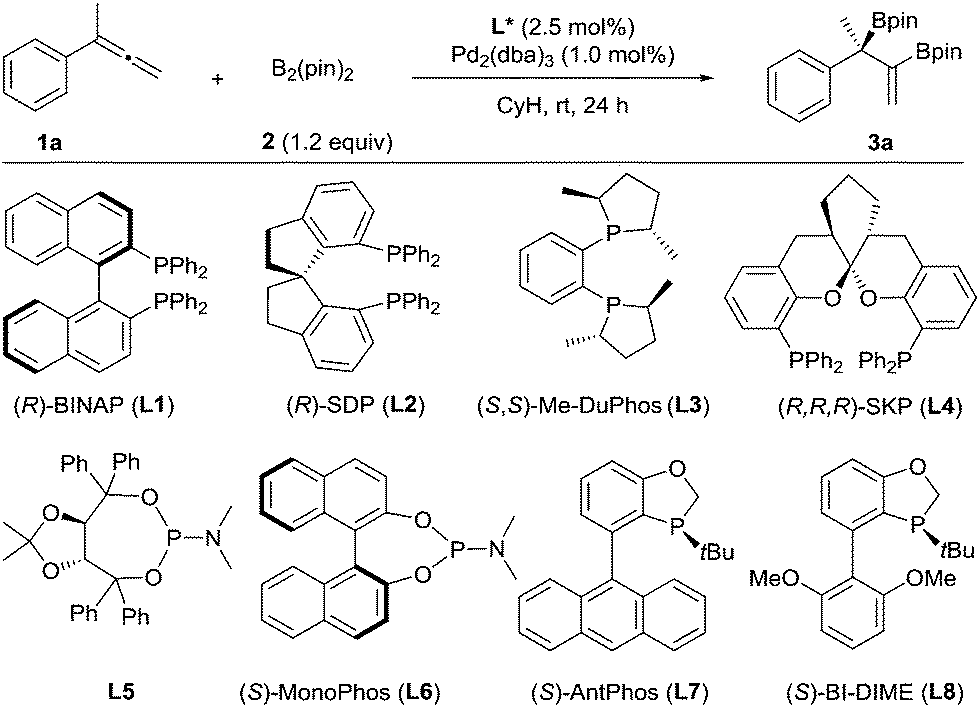
A low yield (15%) and ee (52% ee) were observed when Me-DuPhos was employed as the ligand (entry 3). It should be noted that the diboration occurred exclusively on the substituted double bond of the allene, providing product 3a, which contains both a tertiary boronic ester moiety and an alkenyl boronic ester moiety. Interestingly, the SKP ligand10 (L4) with a large bite angle led to an excellent yield (95%) and a moderate ee (59% ee) (entry 4). We thus predicted that the reaction could be better promoted with a monophosphorus ligand, as observed by Morken in the diboration of monosubstituted allenes.9 Thus, a TADDOL-derived monophosphoramidite ligand, L5, led to the formation of 3a in 76% yield and 79% ee (entry 5). Another monophosphoramidite ligand, L6, derived from a chiral BINOL backbone, proved to be less effective, indicating the importance of the ligand scaffold for both the reactivity and enantioselectivity of the reaction. Encouragingly, the P-chiral monophosphorus ligand AntPhos11 (L7) provided a moderate yield (60%) and an excellent ee (91% ee) (entry 7). Further study of the P-chiral phosphorus ligands developed in our laboratory showed that BI-DIME12 (L8) provided an almost quantitative yield and the highest ee (94%) (entry 8). Screening of the solvent showed that the reaction was facilitated with a nonpolar and non-coordinating solvent, as a diminished yield was observed in toluene, THF or dioxane. No reaction was observed when dichloromethane was employed (entries 8–12). A Pd(0) precursor appeared to be advantageous for the reaction since a diminished yield was observed when Pd(OAc)2 was applied (entry 13). Finally, the diboration was studied at a low catalytic loading (0.2 mol% Pd, 0.25 mol% L8) and at a gram scale (36 mmol 1a, 4.8 g). The product, 3a (13.6 g) was obtained in 97% yield and in 94% ee (entry 14), demonstrating the practicality of this asymmetric transformation.
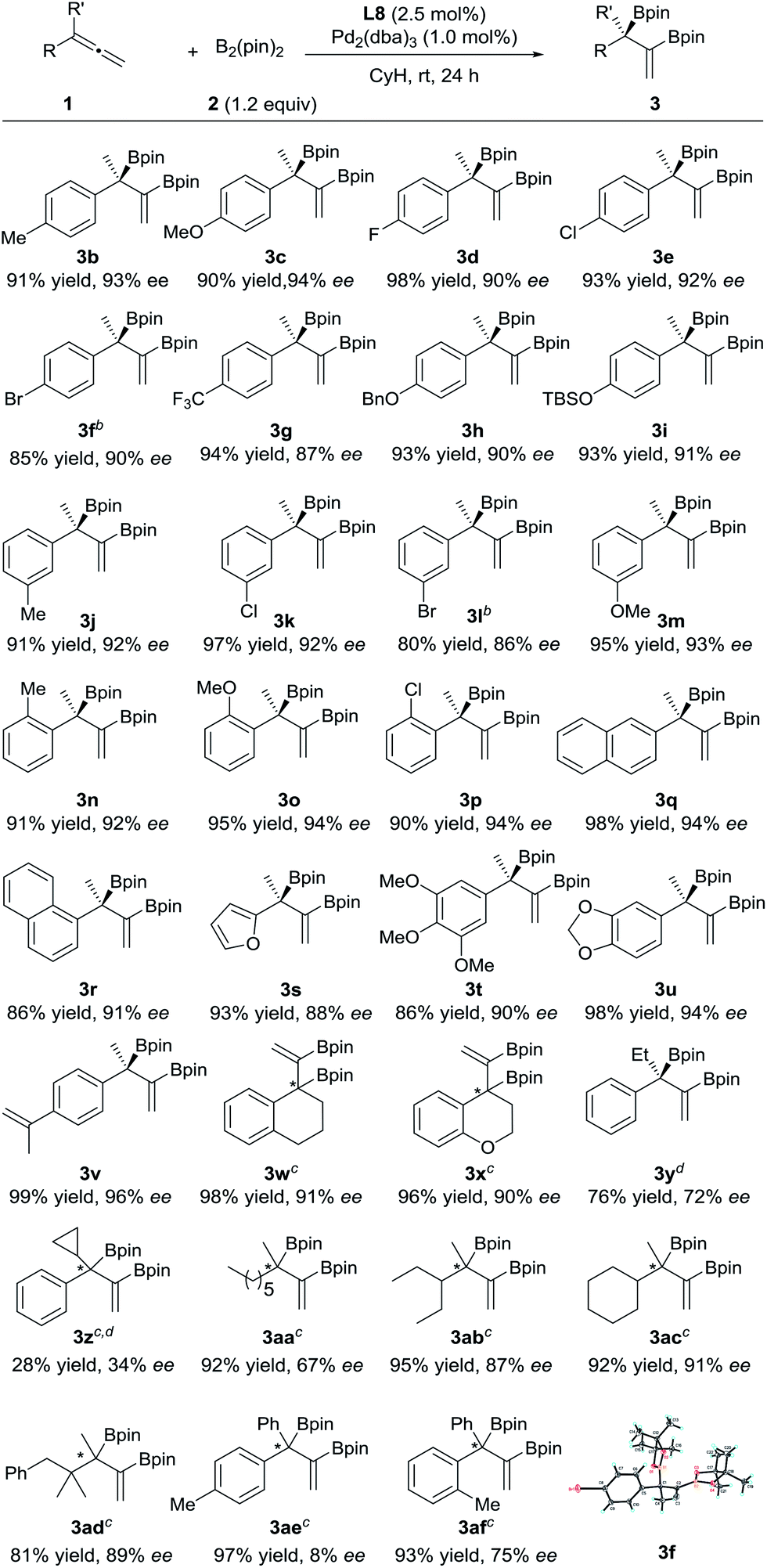
The substrate scope of this asymmetric diboration was then investigated. As depicted in Table 2, a series of diboronic esters with various electronic properties and substitution patterns on the benzene ring (3b–p) were smoothly formed at rt in excellent ee’s and yields with Pd-L8 as the catalyst. The enantioselectivities obtained were slightly higher with substrates having electron-donating substituents, but substituents such as fluoro groups (3d) and trifluoromethyl groups (3g) were well applicable. Substrates with an ortho substituent (3n–p) were also tolerable. The reactions of both 1- and 2-naphthyl substrates provided excellent yields and ee’s (3q–r). A chiral furyl product, 3s, was also synthesized successfully. Substrates with multiple substituents on the benzene ring were equally effective for the transformation (3t–u). In order to test the chemoselectivity between an allene and an olefin, a substrate containing both moieties was subjected to diboration and only the allene moiety was reactive under the current conditions, forming product 3v in 99% yield and 96% ee. Both cyclic and heterocyclic substrates were applicable to smoothly afford 3w and 3x, respectively, in excellent yields and ee’s. Switching the methyl substituent on the allene with an ethyl group resulted in an inferior ee (72% ee, 3y). The introduction of a cyclopropyl group instead of the methyl substituent was less effective (3z). 1,1-Dialkylallenes were also applicable. While a moderate ee (67%) was obtained for product 3aa, bearing two primary alkyl substituents on the quaternary stereocenter, high ee’s (87% and 91%) were obtained for products 3ab and 3ac, respectively, which contain both a methyl and a secondary alkyl group at the chiral center. A good ee was also achieved for 3ad, bearing a tertiary alkyl group. Finally, the diboration of 1,1-diarylallenes was studied. While a low ee was obtained for product 3ae, indicating little difference between the phenyl substituent and the para-tolyl group, a moderate ee (75%) was achieved for product 3af, bearing both a phenyl and an ortho-tolyl substituent at the quaternary stereocenter.
| a Unless otherwise specified, the reactions were performed under nitrogen at rt for 24 h with 1 (0.20 mmol), 2 (0.24 mmol), L8 (2.5 mol%) and Pd2(dba)3 (1.0 mol%) in cyclohexane (2.0 mL). The yields of the isolated products are shown here. The ee values were determined by HPLC on a chiral column. The R absolute configuration of 3f was determined by X-ray crystallographic analysis;13 the others were assigned by analogy. b Pd2(dba)3 (2.0 mol%) and L8 (5.0 mol%) were employed. c The absolute configurations were not determined. d Incomplete conversions. |
|---|
|
To understand the mechanism of this catalytic asymmetric reaction, the diboration of 1a was investigated with a scalemic mixture of ligand L8. A perfect linear relationship between the ee of L8 and the ee of product 3a was observed, indicating that the reaction was catalyzed by a palladium catalyst composed of a single monophosphorus ligand, L8. Variation of the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L8 ratio from 1:1, 1:2, and 2:1 did not lead to a significant change in either the yield or the enantioselectivity, further demonstrating the presence of a single L8 in the active palladium catalyst. To understand the perfect regioselectivity and the stereochemical model of this asymmetric diboration, the energetics of the catalytic transformation were calculated at the B3LYP-D3/6-31G(d)+SDD level. As shown in Fig. 2a, the reaction initiates from the palladium species RC, which undergoes oxidative addition of bis(pinacolato)diboron concerted with allene insertion, where the boryl group migrates to the middle carbon of the allene to give an η3 Pd(II)-allyl intermediate, INTSi or INTRe. Notably, this initial oxidative boryl migration16 is in good agreement with the perfect regioselectivity of diboration on the internal double bond of the allene, and this process is an irreversible and stereo-determining step of the transformation.15TS1Re is computed to be lower in free energy than TS1Si by 1.4 kcal mol−1, which is in qualitative agreement with the observed enantioselectivity (ΔGexp: 2.1 kcal mol−1).
:L8 ratio from 1:1, 1:2, and 2:1 did not lead to a significant change in either the yield or the enantioselectivity, further demonstrating the presence of a single L8 in the active palladium catalyst. To understand the perfect regioselectivity and the stereochemical model of this asymmetric diboration, the energetics of the catalytic transformation were calculated at the B3LYP-D3/6-31G(d)+SDD level. As shown in Fig. 2a, the reaction initiates from the palladium species RC, which undergoes oxidative addition of bis(pinacolato)diboron concerted with allene insertion, where the boryl group migrates to the middle carbon of the allene to give an η3 Pd(II)-allyl intermediate, INTSi or INTRe. Notably, this initial oxidative boryl migration16 is in good agreement with the perfect regioselectivity of diboration on the internal double bond of the allene, and this process is an irreversible and stereo-determining step of the transformation.15TS1Re is computed to be lower in free energy than TS1Si by 1.4 kcal mol−1, which is in qualitative agreement with the observed enantioselectivity (ΔGexp: 2.1 kcal mol−1).
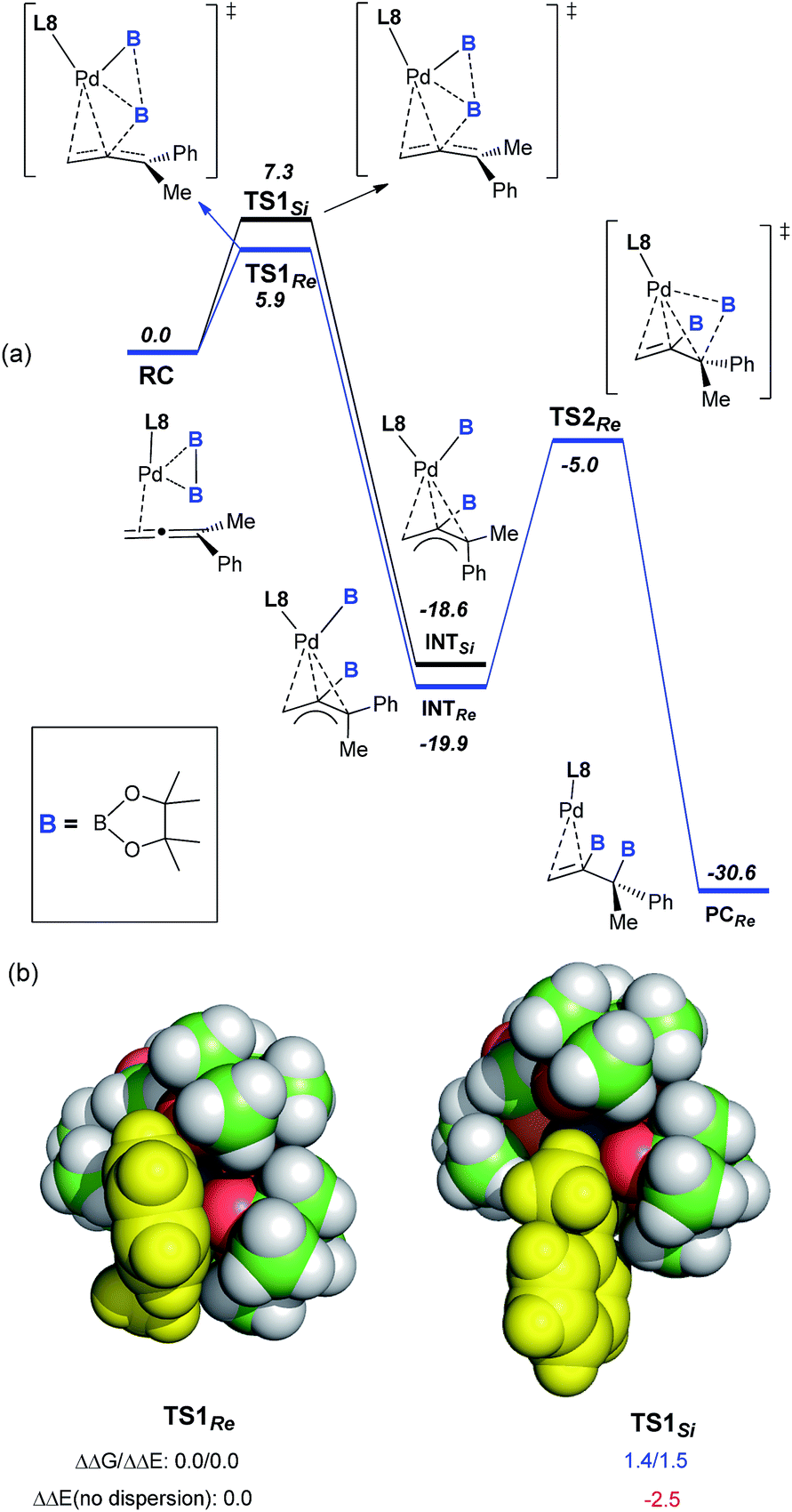 | ||
| Fig. 2 (a) Free-energy profile (in kcal mol−1) for the Pd-catalyzed asymmetric diboration of buta-2,3-dien-2-ylbenzene (1a) with (S)-BI-DME (L8) as the ligand at the B3LYP-D3/6-31G(d)+SDD level.15 (b) VdW representation of the optimized TS1Re and TS1Si, with relative free and electronic energies in kcal mol−1. | ||
Distortion/interaction analysis17 reveals that there is a greater dispersion interaction between the phenyl group17b on the allene and a boryl group in TS1Re than in TS1Si, which is considered as the key factor for the enantioselectivity (Fig. 2b).15 When excluding the dispersion contribution, the enantioselectivity is computed to be reversed. Finally, the reductive elimination viaTS2Re proceeds from INTRe to produce PCRe, with a barrier of about 14.9 kcal mol−1. In contrast to Morken’s proposal that the oxidative addition of diboron to Pd proceeded prior to migratory insertion and that the oxidative addition of diboron to Pd was computed as the rate-determining step,9l our calculations on the diboration of 1a revealed a concerted mechanism of oxidative addition of bis(pinacolato)diboron and allene insertion, provided the first computational insight into the origins of the enantioselectivity (molecular details and a critical dispersion effect), and disclosed the final reductive elimination step as the rate-determining step.
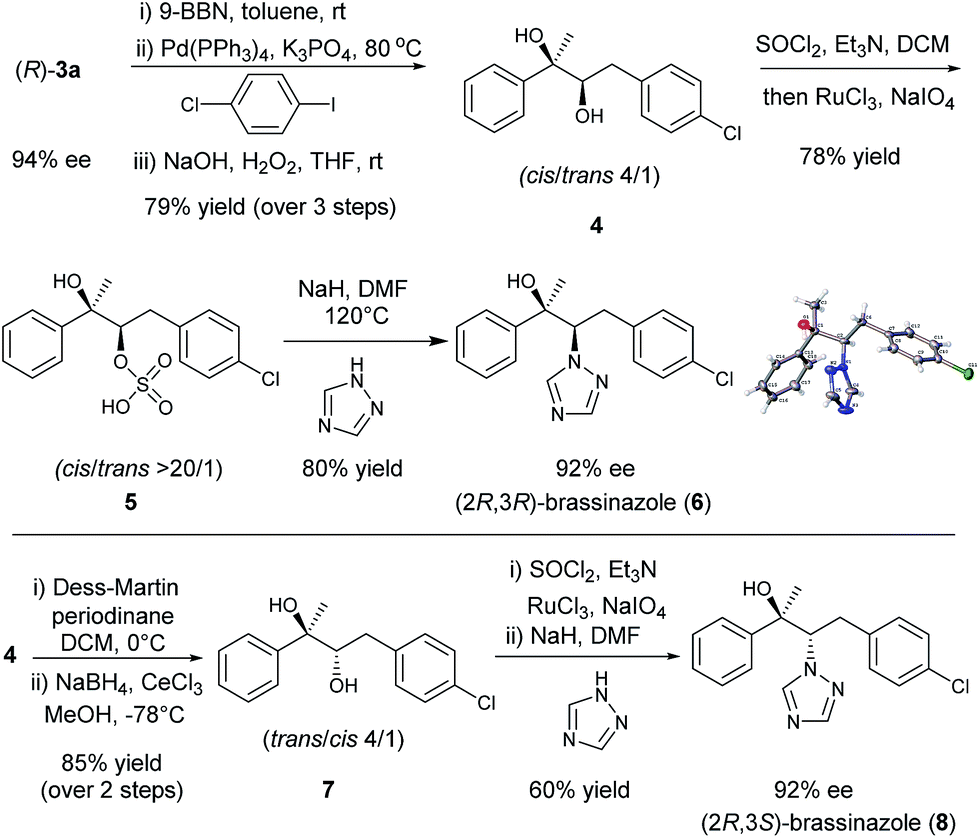
Chiral diboronic ester products are versatile building blocks in organic synthesis. For example, Aggarwal reported a stereospecific allylation between the diboronic ester (S)-3a and benzaldehyde to form a tetrasubstituted alkene after a Suzuki–Miyaura cross-coupling.14,18,9k To further explore the synthetic applications of such chiral diboronic esters, an enantioselective synthesis of brassinazole,19 a specific inhibitor of brassinosteroid biosynthesis, was studied with (R)-3a as the starting material (Scheme 1). Surprisingly, despite its significant biological properties, its enantioselective synthesis had not been reported to our knowledge. We envisioned that the diboration product (R)-3a would provide rapid and efficient access to brassinazole through simple transformations. Thus, (R)-3a was subjected to a one-pot, three step sequence: (a) hydroboration by treatment with 9-BBN;20 (b) Suzuki–Miyaura coupling with 1-chloro-4-iodobenzene; and (c) oxidation under conditions of H2O2/NaOH. The chiral diol 4 was formed smoothly with a cis/trans ratio of 4:1 in 79% overall yield. Under Ley’s conditions,21 diol 4 was readily oxidized to form the corresponding sulfate, 5, which was isolated as a pure cis product in 78% yield. Finally, treatment of sulfate 5 with 1,2,4-triazole under conditions of NaH/DMF yielded (2R,3R)-brassinazole (6) in 80% yield. The absolute configuration was confirmed by X-ray crystallography.13 It should be noted that sulfate 5 proceeded first through an intramolecular SN2 reaction to form an epoxide, which was subsequently attacked by 1,2,4-triazole to undergo a 2nd SN2 reaction, yielding product 6 with net retention of stereochemistry. Compound 4 was also transformed to its diastereomer 7 through an oxidation–reduction procedure, which ultimately led to the formation of (2R,3S)-brassinazole (8) via sulfate formation and nucleophilic substitution. Thus, we accomplished a concise and first enantioselective synthesis of brassinazole.
 | ||
| Scheme 1 Asymmetric synthesis of brassinazole. | ||
Conclusions
In summary, we have developed the practical and enantioselective palladium-catalyzed diboration of 1,1-disubstituted allenes. This has led to the synthesis of a series of diboronic esters containing a chiral tertiary boronic ester with excellent yields and enantioselectivities, with the palladium loading as low as 0.2 mol%. The reaction enjoys a broad substrate scope and good functional group compatibility. The chiral ligand BI-DIME has proven to be crucial for the success of the reaction. DFT calculations identified a concerted mechanism of oxidative addition of bis(pinacolato)diboron and allene insertion, and revealed a critical dispersion effect on the origins of the enantioselectivity. Finally, the application of the chiral diboronic ester to a concise and first enantioselective synthesis of brassinazole has been successfully demonstrated.Acknowledgements
This work is supported by the Strategic Priority Research Program of the Chinese Academy of Sciences XDB20000000, NSFC-21432007, 21272254, 21572246, 21232009, 20421091, CAS (QYZDY-SSW-SLH012, SLH029).Notes and references
- For reviews, see: (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS PubMed; (b) D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2015, 54, 1082 CrossRef CAS PubMed; (c) H. K. Scott and V. K. Aggarwal, Chem.–Eur. J., 2011, 17, 13124 CrossRef CAS PubMed.
- (a) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed; (b) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS PubMed; (c) V. Bagutski, R. M. French and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 5142 CrossRef CAS PubMed; (d) A. P. Pulis, D. J. Blair, E. Torres and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 16054 CrossRef CAS PubMed.
- (a) N. Hu, G. Zhao, Y. Zhang, X. Liu, G. Li and W. Tang, J. Am. Chem. Soc., 2015, 137, 6746 CrossRef CAS PubMed; (b) V. M. Shoba, N. C. Thacker, A. J. Bochat and J. M. Takacs, Angew. Chem., Int. Ed., 2016, 55, 1465 CrossRef CAS PubMed.
- A. Guzman-Martinez and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10634 CrossRef CAS PubMed.
- (a) I.-H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664 CrossRef CAS PubMed; (b) J. M. O’Brien, K.-S. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10630 CrossRef PubMed; (c) X. Feng and J. Yun, Chem.–Eur. J., 2010, 16, 13609 CrossRef CAS PubMed; (d) S. Rodomkit and A. H. Hoveyda, Angew. Chem., Int. Ed., 2014, 53, 3387 CrossRef PubMed.
- A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693 CrossRef CAS PubMed.
- For reviews of allenes, see: (a) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989 CrossRef CAS PubMed; (b) P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524 CrossRef CAS PubMed; (c) E. Soriano and I. Fernández, Chem. Soc. Rev., 2014, 43, 3041 RSC; (d) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed; (e) S. Ma, Acc. Chem. Res., 2009, 42, 1679 CrossRef CAS PubMed; (f) M. Jeganmohan and C.-H. Cheng, Chem. Commun., 2008, 3101 RSC; (g) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed; (h) S. Ma, Acc. Chem. Res., 2003, 36, 701 CrossRef CAS PubMed; (i) R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067 CrossRef CAS PubMed; (j) Y. Yamamoto and U. Radhakrishnan, Chem. Soc. Rev., 1999, 28, 199 RSC.
- For reviews of diboration, see: (a) J. Takaya and N. Iwasawa, ACS Catal., 2012, 9, 1993 CrossRef; (b) L. Dang, Z. Lin and T. B. Marder, Chem. Commun., 2009, 3987 RSC; (c) J. Ramírez, C. Lillo, A. M. Segarra and E. C. R. Fernández, C. R. Chim., 2007, 10, 138 CrossRef; (d) H. E. Burks and J. P. Morken, Chem. Commun., 2007, 4717 RSC; (e) I. B. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed; (f) T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 3, 271 CrossRef CAS PubMed; (g) T. B. Marder and N. C. Norman, Top. Catal., 1998, 5, 63 CrossRef CAS; (h) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed.
- For asymmetric rhodium-catalyzed diborations, see: (a) J. B. Morgan, S. P. Miller and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 8702 CrossRef CAS PubMed; (b) S. Trudeau, J. B. Morgan, M. Shrestha and J. P. Morken, J. Org. Chem., 2005, 70, 9538 CrossRef CAS PubMed; (c) S. P. Miller, J. B. Morgan, F. J. Nepveux and J. P. Morken, Org. Lett., 2004, 6, 131 CrossRef CAS PubMed; (d) K. Toribatake and H. Nishiyama, Angew. Chem., Int. Ed., 2013, 52, 11011 CrossRef CAS PubMed. For platinum-catalyzed diboration: (e) L. T. Kliman, S. N. Mlynarski and J. P. Morken, J. Am. Chem. Soc., 2009, 131, 13210 CrossRef CAS PubMed; (f) L. T. Kliman, S. N. Mlynarski, G. E. Ferris and J. P. Morken, Angew. Chem., Int. Ed., 2012, 51, 521 CrossRef CAS PubMed; (g) J. R. Coombs, D. Haefner, L. T. Kliman and J. P. Morken, J. Am. Chem. Soc., 2013, 135, 11222 CrossRef CAS PubMed; (h) S. N. Mlynarski, C. H. Schuster and J. P. Morken, Nature, 2014, 505, 386 CrossRef CAS PubMed. For asymmetric palladium-catalyzed diboration: (i) X. Guo, A. K. Nelson, C. Slebodnick and W. L. Santos, ACS Catal., 2015, 5, 2172 CrossRef CAS; (j) N. F. Pelz, A. R. Woodward, H. E. Burks, J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2004, 126, 16328 CrossRef CAS PubMed; (k) J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2006, 128, 74 CrossRef CAS PubMed; (l) H. E. Burks, S. Liu and J. P. Morken, J. Am. Chem. Soc., 2007, 129, 8766 CrossRef CAS PubMed.
- (a) Z.-Y. Cao, X. M. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197 CrossRef CAS PubMed; (b) X. M. Wang, P. Guo, Z. Han, X. B. Wang, Z. Wang and K. Ding, J. Am. Chem. Soc., 2014, 136, 405 CrossRef CAS PubMed; (c) X. M. Wang, F. Meng, Y. Wang, Z. Han, Y. Chen, L. Liu, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 9276 CrossRef CAS PubMed; (d) X. M. Wang, Z. Han, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 936 CrossRef CAS PubMed; (e) J. Liu, Z. Han, X. Wang, Z. Wang and K. Ding, J. Am. Chem. Soc., 2015, 137, 15346 CrossRef CAS PubMed.
- (a) W. Fu, M. Nie, A. Wang, Z. Cao and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 2520 CrossRef CAS PubMed; (b) N. Hu, K. Li, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2016, 55, 5044 CrossRef CAS PubMed; (c) G. Xu, Q. Zhao and W. Tang, Chin. J. Org. Chem., 2014, 34, 1919 CrossRef CAS; (d) C. Li, D. Chen and W. Tang, Synlett, 2016, 27, 2183 CrossRef CAS.
- (a) W. Tang, N. D. Patel, G. Xu, X. Xu, J. Savoie, S. Ma, M.-H. Hao, S. Keshipeddy, A. G. Capacci, X. Wei, Y. Zhang, J. J. Gao, W. Li, S. Rodriguez, B. Z. Lu, N. K. Yee and C. H. Senanayake, Org. Lett., 2012, 14, 2258 CrossRef CAS PubMed; (b) G. Xu, W. Fu, G. Liu, C. H. Senanayanke and W. Tang, J. Am. Chem. Soc., 2014, 136, 570 CrossRef CAS PubMed; (c) K. Du, P. Guo, Y. Chen, Z. Cao, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3033 CrossRef CAS PubMed; (d) M. Nie, W. Fu, Z. Cao and W. Tang, Org. Chem. Front., 2015, 2, 1322 RSC.
- CCDC 1517472 and 1517578† contain the supplementary crystallographic data for this paper.
- M. J. Hesse, S. Essafi, C. G. Watson, J. N. Harvey, D. Hirst, C. L. Willis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 6145 CrossRef CAS PubMed.
- See ESI† for computational details, results and discussion.
- X. Zhang, L. W. Chung and Y.-D. Wu, Acc. Chem. Res., 2016, 49, 1302 CrossRef CAS PubMed.
- (a) C. Y. Legault, Y. Garcia, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 12664 CrossRef CAS PubMed; (b) A dispersion effect could also be applied to 1,1-dialkyl substrates 3ab, 3ac, and 3ad for the observed high ee’s.
- A. R. Woodward, H. E. Burks, L. M. Chan and J. P. Morken, Org. Lett., 2005, 7, 5505 CrossRef CAS PubMed.
- (a) T. Asami, Y. K. Min, N. Nagata, K. Yamagishi, S. Takatsuto, S. Fujioka, N. Murofushi, I. Yamaguchi and S. Yoshida, Plant Physiol., 2000, 123, 93 CrossRef CAS PubMed; (b) S. Yoshida, A. Fujioka and T. Asami, Jpn. Kokai Tokkyo Koho, JP2000053657 (A), 2000; (c) Y. K. Min, T. Asami, S. Fujioka, N. Murofushi, E. Yamaguchi and S. Yoshida, Bioorg. Med. Chem. Lett., 1999, 9, 425 CrossRef CAS PubMed.
- N. F. Pelz and J. P. Morken, Org. Lett., 2006, 8, 4557 CrossRef CAS PubMed.
- (a) Y. Gao and K. B. Sharpless, J. Am. Chem. Soc., 1988, 110, 7538 CrossRef CAS; (b) J. E. Baldwin, R. M. Adlington and D. W. Goilins, Tetrahedron, 1995, 51, 5169 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1517472 and 1517578. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01254c |
| ‡ J. Liu and M. Nie contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |