
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective total synthesis of (−)-colchicine, (+)-demecolcinone and metacolchicine: determination of the absolute configurations of the latter two alkaloids†
Bo
Chen‡
a,
Xin
Liu‡
ac,
Ya-Jian
Hu
a,
Dong-Mei
Zhang
b,
Lijuan
Deng
b,
Jieyu
Lu
a,
Long
Min
a,
Wen-Cai
Ye
 b and
Chuang-Chuang
Li
*a
b and
Chuang-Chuang
Li
*a
aDepartment of Chemistry, South University of Science and Technology of China, Shenzhen 518055, China. E-mail: ccli@sustc.edu.cn
bCollege of Pharmacy, Jinan University, Guangzhou 510632, China
cInstitute of Chinese Medical Sciences, University of Macau, Macao, China
First published on 5th May 2017
Abstract
Here, we describe a concise, enantioselective, and scalable synthesis of (−)-colchicine (9.2% overall yield, >99% ee). Moreover, we have also achieved the first syntheses of (+)-demecolcinone and metacolchicine, and determined their absolute configurations. The challenging tricyclic 6-7-7 core of colchicinoids was efficiently introduced using an intramolecular oxidopyrylium-mediated [5 + 2] cycloaddition reaction. Notably, the synthesized colchicinoid 23 exhibited potent inhibitory activity toward the cell growth of human cancer cell lines (IC50 = ∼3.0 nM), and greater inhibitory activity towards microtubule assembly than colchicine, making it a promising lead in the search for novel anticancer agents.
Introduction
Colchicine (1), an alkaloid natural product, was the first tubulin-destabilizing agent to be reported in the literature (Fig. 1a). The effects of this compound against a variety of indications have been investigated owing to its remarkable antimitotic activity. Furthermore, colchicine (1) has been used to treat several diseases, including acute gout, familial Mediterranean fever, and chronic myelocytic leukemia.1 Colchicine (1) has also been used as a neurotoxin in animal models of Alzheimer's disease and epilepsy.2 The development of novel colchicinoids with enhanced antitumor properties continues to pose a significant challenge to drug discovery scientists.3 From a structural perspective,4 the unusual 6-7-7-membered ring system of colchicinoids 1–4, as well as the stereocenter at C-7 and the aR-configured stereogenic axis defined by the pivot bond joining the A and C rings, represents a formidable synthetic challenge. The regioselective construction of the highly oxidized tropolone C ring and the stereoselective installation of the C-7-acetamido group undoubtedly represent two of the more challenging features of any prospective colchicinoid synthesis.5 It is noteworthy that (+)-demecolcinone (3), which was the first naturally occurring dextrorotatory colchicinoid to be reported, contains a constrained azabicyclo[3.2.1]octane (tropane) framework that is unprecedented in nature.6 Nevertheless, the relative configuration of naturally occurring (+)-demecolcinone (3) was proposed only based on the energy-minimized representation of the molecule and the postulated biosynthetic pathway, owing to the lack of direct and conclusive evidence. The absolute configuration of (+)-demecolcinone (3) has not been determined through X-ray crystallographic analysis or the exciton chirality circular dichroism method. Furthermore, metacolchicine (4) is the first example of a naturally occurring colchicinoid bearing an additional carbon unit at its C-10 position.7 However, the absolute configuration of the unique stereogenic axis defined by the pivot bond joining the A and C rings of metacolchicine (4) has not previously been addressed, and there have been no reports on the biological activity of this new colchicinoid.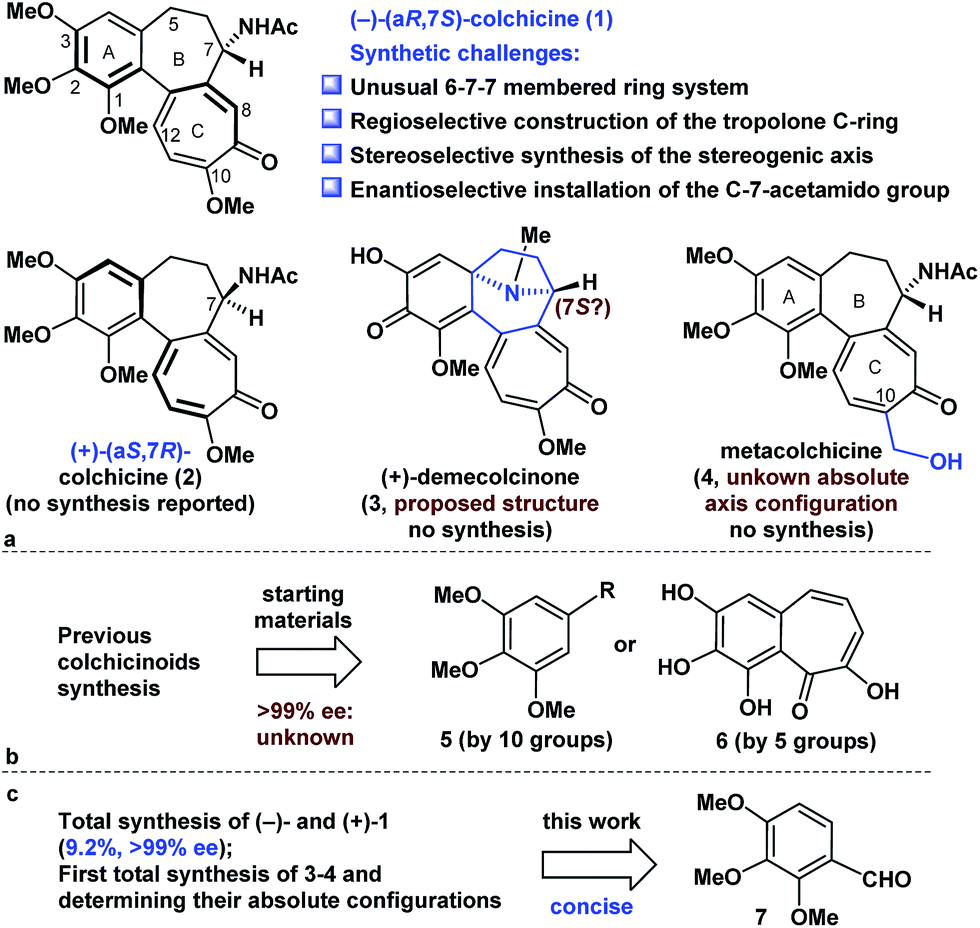 | ||
| Fig. 1 Selected colchicinoids and a summary of their syntheses. | ||
The fascinating structural motifs and promising pharmacological properties of the colchicinoids have attracted considerable interest from the synthetic chemistry community over the past 60 years, and this has culminated in an large number of interesting reports.5a,8,9 However, most of the existing routes to this class of compounds are heavily reliant on the use of symmetric C-5-substituted pyrogallol trimethyl ethers (5) and purpurogallin (6) as starting materials (Fig. 1b).5a All previous asymmetric installations of the C-7-amino group are limited to the following procedure: asymmetric reduction of the ketone to the alcohol, subsequent conversion of the alcohol to the azide, and reduction of the azide to the amino group.8b,8c,8d The highly enantioselective (≥99% ee) synthesis of colchicine (1) also remains a great challenge because of the partial racemization of relative compounds at the C-7 proton.8b Notably, there have been no reports in the literature to date pertaining to the total synthesis of compounds 2–4. Herein, we describe a highly concise and enantioselective synthesis of (−)-colchicine and (+)-colchicine (>99% ee). We also describe the first reported syntheses of (+)-demecolcinone (3) and metacolchicine (4), including determination of their absolute configurations, using 2,3,4-trimethoxybenzaldehyde (7) as a new starting material.
Results and discussion
Retrosynthetic analysis of colchicinoids
Retrosynthetically (Fig. 2), demecolcinone (3) could be prepared from colchicine (1) in a biomimetic manner through an intramolecular oxidative dearomatization–azaspiroannulation reaction.10 Furthermore, metacolchicine (4) could be derived from colchicine (1) by regioselective hydroxymethylation at its C-10 position. It was envisioned that colchicine (1) could be generated from 8 through a chemoselective oxidation, followed by regioselective formation of the tropolone C-ring. Inspired by previous work,11 tricyclic 8 could itself be synthesized from 9 in an intramolecular oxidopyrylium-mediated [5 + 2] cycloaddition reaction.12,13 Compound 9 can be derived from 10 through the Achmatowicz reaction,14 and compound 10 could be synthesized enantioselectively by reaction of amide 12 with furfuryl alcohol (13) followed by the diastereoselective reductive amination of the resulting ketone with Ellman auxiliary 11.15 In this sense, it was envisioned that 11 could be used as a chiral directing group to provide the required amino group as a single enantiomer. Amide 12 could be synthesized by reaction of commercially available and inexpensive compound 7 with N-methoxy-N-methylacrylamide (14) via a transition-metal-catalysed C–H functionalization reaction16 using the aldehyde as a directing group, followed by a Seyferth–Gilbert homologation.17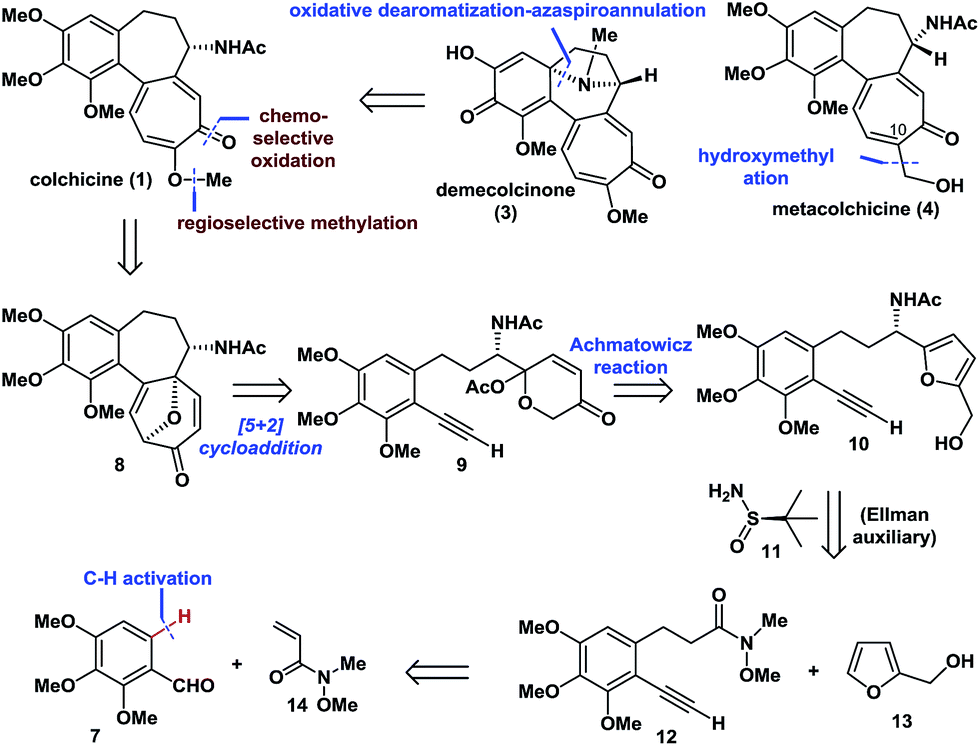 | ||
| Fig. 2 Retrosynthetic analysis of colchicinoids 1, 3 and 4. | ||
Enantioselective total synthesis of (−)- and (+)-colchicine
The synthesis began with the transition-metal-catalyzed C–H bond functionalization of 7 with 14 (Scheme 1). Inspired by Li's seminal work,18 we applied the strategy to compound 7. Pleasingly, after optimization, we successfully generated the N-sulfonyl imine in situ by reaction of 7 with TsNH2 (15) in the presence of anhydrous CuSO4 in THF. Furthermore, subsequent treatment of this imine with [RhCp*Cl2]2 (1 mol%), AgSbF6 (4 mol%), NaOAc (2.0 equiv.), and 14 (2.0 equiv.) at 80 °C afforded ortho-olefinated benzaldehyde 16 in good yield (90% on a 0.5 g scale; 70% on a 5.0 g scale). This modified catalytic C–H bond activation involved a transient directing group.19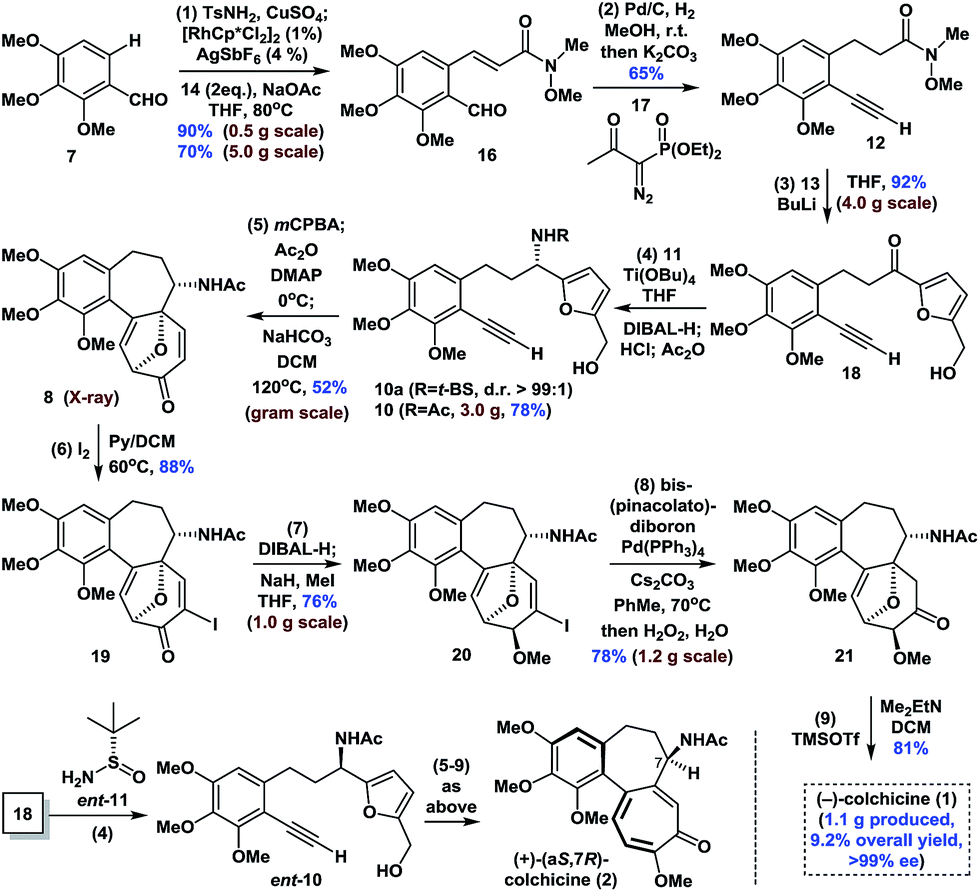 | ||
| Scheme 1 Enantioselective synthesis of (−)-colchicine and (+)-colchicine. | ||
Moving forward, the subsequent selective hydrogenation of 16 (Pd/C, H2), followed by the Seyferth–Gilbert homologation of the aldehyde moiety in the presence of 17 gave 12 in one pot in 65% yield. 1,2-Addition of the organolithium reagent prepared by treatment of furfuryl alcohol (13) with BuLi to 12 provided ketone 18 in 92% yield (4.0 g scale). Subsequent condensation of ketone 18 with Ellman auxiliary 11 using Ti(OBu)4 (2.0 equiv.) in THF, followed by reduction of the resulting tert-butanesulfinyl (t-BS) ketimine with DIBAL-H afforded 10a (R = t-BS) with very high diastereoselectivity. Treatment of 10a with HCl and methanol in the same pot resulted in clean cleavage of the sulfinyl group to give the corresponding amine, which was acetylated in situ in the presence of NaHCO3 to give 10 (R = Ac, >99% ee) in 78% yield (one-pot, 3.0 g scale, see the ESI for details†). Oxidative rearrangement of 10 using 3-chloroperoxybenzoic acid (mCPBA), followed by acetylation of the anomeric hydroxyl and intramolecular [5 + 2] cycloaddition of acetoxypyranone 9 under our optimized one-pot reaction conditions, gave tricyclic 6-7-7 core-containing 8 in 52% yield (1.0 g scale), which was confirmed by X-ray crystallography.
Next, we continued with our proposed total synthesis of colchicine (1) from 8. After extensive experimentation, we found that treatment of 8 with iodine in a mixture of pyridine and DCM, afforded α-iodoenone 19 in 88% yield. Reduction of the ketone group in 19 followed by in situ chemoselective methylation of the resulting alcohol afforded 20 in 76% yield (1.0 g scale). A palladium-catalyzed cross-coupling reaction of bis(pinacolato)diboron with 20 followed by oxidation under mild conditions (H2O2/H2O) gave ketone 21 with 78% yield (1.2 g scale). Finally, double elimination of the oxa-bridge in 21 using a slightly modified version of Cha's procedure8c in the presence of TMSOTf and Me2EtN proceeded smoothly to complete our total synthesis of (−)-1 in >99% ee. We also achieved the first synthesis of (+)-(aS,7R)-colchicine (2, ent-1), using Ellman auxiliary ent-11 for condensation with ketone 18, according to a similar sequence to that shown in Scheme 1. Notably, this route provided facile access to a total of 1.1 g of (−)-1, thereby highlighting the robust nature of this chemistry.
Enantioselective synthesis of (+)-demecolcinone (3) and metacolchicine (4)
With (−)-colchicine (1) in hand, we proceeded to investigate our proposed syntheses of the remaining colchicinoids (Scheme 2). Demecolcine (22, R = Me) and 23 (R = CHO) were rapidly prepared from colchicine (1) through a series of slightly modified procedures from the literature (see ESI†). The application of a modified version of Brossi's procedure20 (H2SO4 in DCM at 45 °C) to 22 gave 2-demethyldemecolcine (24) in 72% yield. It is worth noting that compound 22 exhibited much better chemoselectivity towards O-demethylation than did colchicine (1) and 23 under the same conditions.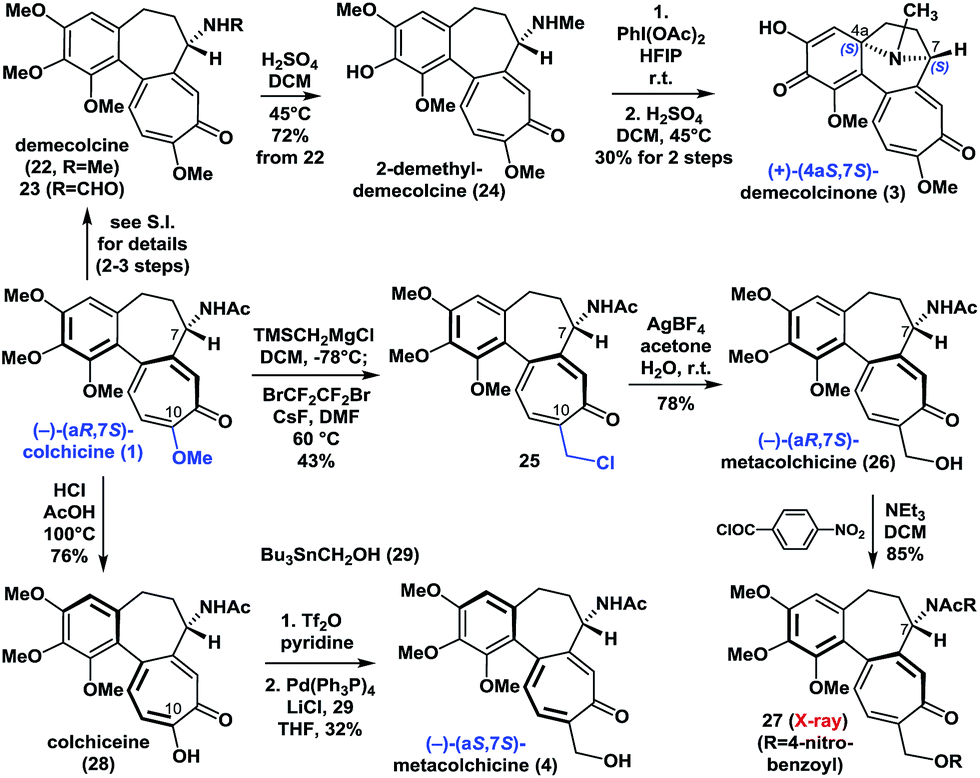 | ||
| Scheme 2 Enantioselective synthesis of (+)-demecolcinone (3) and metacolchicine (4), and determination of their absolute configurations. | ||
Our initial efforts to construct the 8-azabicyclo[3.2.1]octane structure in demecolcinone (3) involved the intramolecular oxidative dearomatization–azaspiroannulation reaction of 22 using a variety of hypervalent iodine sources21 in several different solvents. Unfortunately, none of these reactions afforded any of the desired products. However, after extensive experimentation, we found that treatment of 24 with PhI(OAc)2 in the highly polar solvent hexafluoroisopropanol at 25 °C followed by demethylation completed our synthesis of (+)-demecolcinone (3). The 1H and 13C NMR spectra of synthetic (+)-demecolcinone (3), as well as its optical rotation, were identical to those of the natural product. Thus, the absolute configuration of naturally occurring (+)-3, which was the first naturally occurring dextrorotatory colchicinoid to be reported in the literature,6 was unambiguously established to be 4aS,7S based on our total synthesis. It is noteworthy that this study represents the first reported application of an oxidative dearomatization–azaspiroannulation strategy to the synthesis of a medicinally significant tropane structure.22
We then continued to investigate the synthesis of metacolchicine (4), although we believed it would be challenging to regioselectively install the hydroxymethyl group at the C-10 position of colchicine (1). After a long period of exploration, we pleasingly found that treatment of colchicine (1) with (trimethylsilyl)methylmagnesium chloride, followed by halogenation in the presence of CsF and BrCF2CF2Br in the same pot, gave 25. This novel process involved a series of sequential reactions, including the regioselective 1,8-conjugate addition of (trimethylsilyl)methylmagnesium chloride to colchicine (1) at C10, elimination of the C10-methoxyl group, and desilylation, bromination, and chlorination (see ESI†). It is noteworthy that the chlorine atom in 25 is derived from the (trimethylsilyl)methylmagnesium chloride. Later, chloride 25 was smoothly hydrolyzed with AgBF4 in acetone and H2O to afford (−)-(aR,7S)-metacolchicine (26) in 78% yield. The structure of 26 was determined by 2D-NMR spectroscopy and confirmed by X-ray crystallographic analysis of its derivative 27. Surprisingly, however, the 1H and 13C NMR spectra of 26 differed from those of the natural product of metacolchicine (4).7 The absolute configuration of naturally occurring metacolchicine (4) has only two possibilities: aR,7S or aS,7S. Thus, the absolute configuration of naturally occurring metacolchicine (4) was determined to be aS,7S according to our synthesis. To the best of our knowledge, naturally occurring (−)-(aS,7S)-metacolchicine (4) is the first compound to be identified that has a different absolute configuration from that of other colchicine-type natural products.
Finally, the C10-methoxyl group of colchicine (1) was demethylated with high regioselectivity to give colchiceine (28) in good yield. Triflation of 28, followed by a Stille–Migita coupling reaction (Pd(PPh3)4, Bu3SnCH2OH (29)) furnished metacolchicine (4) in 32% overall yield. The 1H and 13C NMR spectra and the optical rotation (synthetic: [α]D20 = −167 (c = 1.0, CHCl3); natural: [α]D21 = −160 (c = 0.39, CHCl3)) of newly synthesized metacolchicine (4) were identical to those of the natural product. In this process (from 1 to 4) clean inversion of axial stereochemistry was found, indicating that the inversion probably occurred during the conversion of 1 to 28 at 100 °C in the presence of acid. This approach could also be applied to the syntheses of structurally diverse analogues of colchicinoids bearing different groups at their C-10 positions and with different stereogenic axes. This would make it possible to carry out structure–activity relationship (SAR) studies on these compounds.
Cell growth inhibitory activities of colchicinoids and tubule-targeting activities of 23
The cell growth inhibitory activities of compounds 1–4 and 22–28 were determined in three human cancer cell lines, namely lung adenocarcinoma (A549), breast carcinoma (MDA-MB-231), and colon adenocarcinoma (LoVo) cells (Table 1, see ESI for details†). Of the compounds tested in this study, compound 23 displayed the most potent inhibitory effects, with IC50 values of 2.8, 3.2, and 3.5 nM against A549, MDA-MB-231, and LoVo cells, respectively.| Compounds | IC50a (×±SD) μM | ||
|---|---|---|---|
| A549 | MDA-MB-231 | LoVo | |
| a IC50 values are expressed as the mean values ± S.D. from three independent experiments. | |||
| 1 | 0.0710 ± 0.0110 | 0.0332 ± 0.0310 | 0.0087 ± 0.0023 |
| 2 | >50 | >50 | >50 |
| 3 | >50 | >50 | >50 |
| 4 | 0.4481 ± 0.1190 | 0.5752 ± 0.4881 | 0.4030 ± 0.0647 |
| 22 | 0.0276 ± 0.0106 | 0.0310 ± 0.0047 | 0.0346 ± 0.0032 |
| 23 | 0.0028 ± 0.0009 | 0.0032 ± 0.0003 | 0.0035 ± 0.0006 |
Compound 23 was selected as a representative example to determine whether the synthesized colchicinoids acted as microtubule-targeting agents. As shown in Fig. 3a (see ESI†), 23 inhibited the polymerization of tubulin in a dose-dependent manner, thereby exhibiting similar behavior to that of colchicine (1). Microscale thermophoresis (MST) was used with purified recombinant tubulin to further confirm that 23 directly interfered with the assembly of tubulin monomers into microtubules. Colchicine (1) was also analyzed by MST as a positive control. The resulting MST measurements gave dissociation constants (Kd) of 0.5 ± 0.3 and 1.3 ± 0.4 μM for 23 and colchicine (1), respectively (Fig. 3b). Furthermore, pre-treatment of recombinant tubulin with 23 did not lead to any discernible difference in the binding affinity of colchicine (1) (1.9 ± 1.2 μM) for tubulin, which indicates that 23 binds to a different binding site on tubulin than does colchicine (1). It is noteworthy that the inhibitory activity of 23 towards the polymerization of tubulin and the binding affinity of 23 to tubulin were both more potent than those of colchicine (1) in vitro.
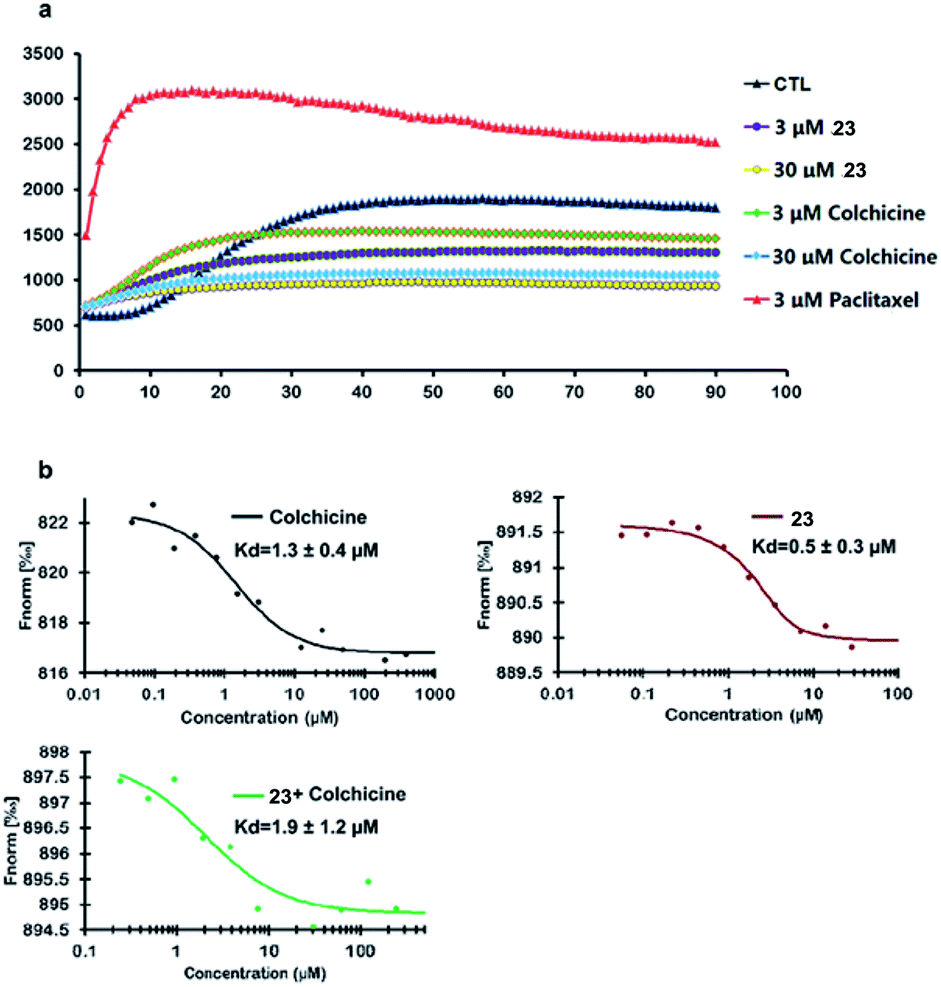 | ||
| Fig. 3 Inhibition of the polymerization of tubulin by 23 and direct binding of 23 to tubulin in vitro. | ||
Microtubule-targeting drugs damage the microtubule structure of cells and induce cell cycle arrest in the G2/M phase by disrupting the formation of the mitotic spindle required for mitosis.1d We observed morphological changes in the microtubule structure of MDA-MB-231 cells after they had been exposed to 23 or colchicine (1) for 12 h. Treatment of MDA-MB-231 cells with 23 resulted in considerable disruption to their microtubule network with short microtubules. Similar effects were also observed for colchicine (1) (Fig. 4a). As shown in Fig. 4b, treatment of MDA-MB-231 cells with 23 led to a considerable dose-dependent increase in the number of cells in the G2/M phase of the cell cycle, with values increasing from 25.19 ± 1.97% (CTL) to 46.03 ± 4.42% (30 nM), 78.85 ± 0.94% (60 nM) and 92.32 ± 2.08% (120 nM). These results therefore indicate that the activity of 23 stems from its effect on the depolymerization of the microtubules.
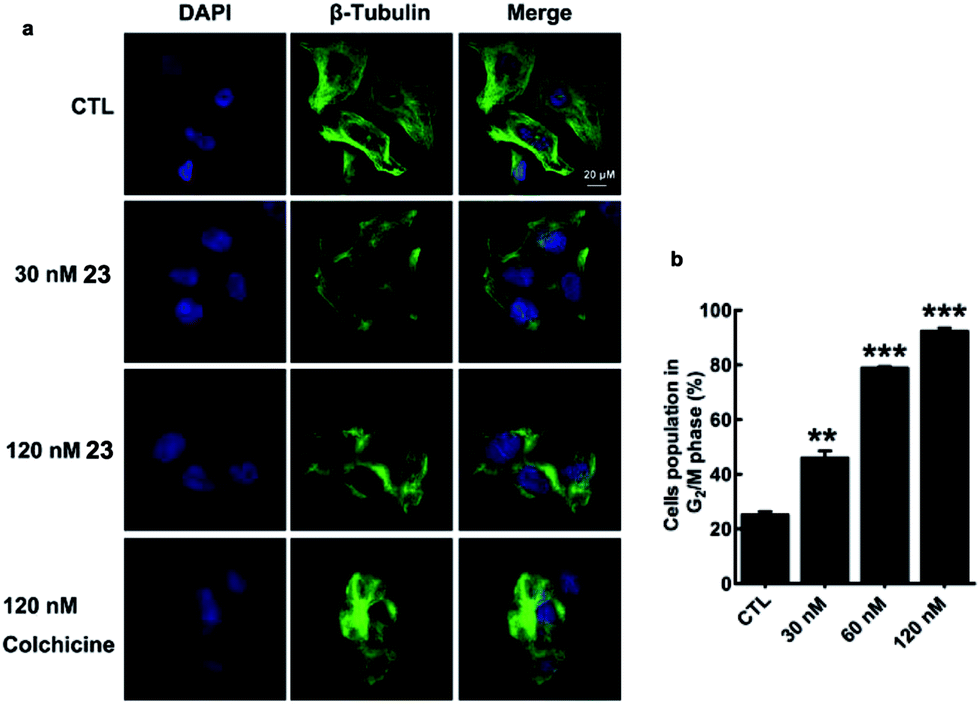 | ||
| Fig. 4 Disruption of the microtubule structure and induction of G2/M cell cycle arrest in MDA-MB-231 cells by 23. | ||
Conclusions
In summary, we have developed a concise and highly enantioselective synthesis of colchicine (1) (1.1 g, >99% ee) in nine steps23 and 9.2% overall yield, without the need for protecting groups.24 An intramolecular oxidopyrylium-mediated [5 + 2] cycloaddition reaction was used as a key step in this synthesis for efficient formation of the challenging tricyclic 6-7-7 core. In addition to achieving the most enantioselective total synthesis of colchicine (1) reported to date, we have also succeeded in describing the first reported asymmetric syntheses of (+)-demecolcinone (3) and metacolchicine (4), including the determination of their absolute configurations. Notably, demecolcinone (3), consisting of a novel and challenging azabicyclo[3.2.1]octane structure, was synthesized via an unusual oxidative dearomatization–azaspiroannulation reaction. It is noteworthy that an Ellman auxiliary was successfully used in the current study as a chiral directing group for the stereo- and enantioselective installation of the C7-acetamido group in a single step. Furthermore, the in vitro biological evaluation of compound 23 revealed that this material displayed the most potent inhibitory effects of all the compounds tested toward A549, MDA-MB-231, and LoVo cells (IC50 = ∼3.0 nM), as well as more potent inhibition of tubulin assembly than colchicine (1). These results therefore suggest that colchicinoid 23 could be used as a promising lead for the development of novel anticancer agents with improved properties.Acknowledgements
This paper is dedicated to Professor Yun-Dong Wu on the occasion of his 60th birthday. This work was supported by the Natural Science Foundation of China (Grant no. 21402083, 21502087 and 21522204), Guangdong Science and Technology Department (2016A050503011), the Shenzhen Science and Technology Innovation Committee (JSGG20160301103446375 and KQTD2015071710315717).Notes and references
- (a) O. Boye and A. Brossi, in The Alkaloids, ed. A. Brossi and G. A. Cordell, Academic Press, San Diego, 1992, vol. 41, p. 125 Search PubMed; (b) C. Le Hello, in The Alkaloids, G. A. Cordell, Academic Press, San Diego, 2000, vol. 53, p. 287 Search PubMed; (c) B. Bhattacharyya, D. Panda, S. Gupta and M. Banerjee, Med. Res. Rev., 2008, 28, 155 CrossRef CAS PubMed; (d) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253 CrossRef CAS PubMed; (e) R. B. G. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198 CrossRef CAS PubMed.
- J. L. Weiner, A. V. Buhler, V. J. Whatley, R. A. Harris and T. V. J. Dunwiddie, J. Pharmacol. Exp. Ther., 1998, 284, 95 CAS.
- Y. Finkelstein, S. E. Aks, J. R. Hutson, D. N. Juurlink, P. Nguyen, G. Dubnov-Raz, U. Pollak, G. Koren and Y. Bentur, Clin. Toxicol., 2010, 48, 407 CrossRef CAS PubMed.
- (a) M. J. S. Dewar, Nature, 1945, 155, 141 CrossRef CAS; (b) M. V. King, J. L. De Vries and R. Pepinsky, Acta Crystallogr., 1952, 5, 437 CrossRef CAS.
- (a) T. Graening and H. G. Schmalz, Angew. Chem., Int. Ed., 2004, 43, 3230 CrossRef CAS PubMed; (b) N. Liu, W. Z. Song, C. M. Schienebeck, M. Zhang and W. P. Tang, Tetrahedron, 2014, 70, 9281 CrossRef CAS PubMed.
- F. Q. Alali, T. El-Elimat, C. Li, A. Qandil, A. Alkofahi, K. Tawaha, J. P. Burgess, Y. Nakanishi, D. J. Kroll, H. A. Navarro, J. O. Falkinham, M. C. Wani and N. H. Oberlies, J. Nat. Prod., 2005, 68, 173 CrossRef CAS PubMed.
- M. Kitajima, A. Tanaka, N. Kogure and H. Takayama, Heterocycles, 2011, 83, 1903 CrossRef CAS.
- For first total synthesis of colchicine, see: (a) J. Schreiber, W. Leimgruber, M. Pesaro, P. Schudel and A. Eschenmoser, Angew. Chem., 1959, 71, 637 CrossRef CAS; For asymmetric total synthesis, see: (b) M. G. Banwell, Pure Appl. Chem., 1996, 68, 539 CrossRef CAS; (c) J. C. Lee, S. J. Jin and J. K. Cha, J. Org. Chem., 1998, 63, 2804 CrossRef CAS; (d) T. Graening, V. Bette, J. Neudorfl, J. Lex and H. G. Schmalz, Org. Lett., 2005, 7, 4317 CrossRef CAS PubMed.
- For selected synthesis, see: (a) E. E. van Tamelen, T. A. Spencer Jr, D. S. Allen Jr and R. L. Ovis, J. Am. Chem. Soc., 1959, 81, 6341 CrossRef CAS; (b) R. B. Woodward, Harvey Lectures Series. 1963, vol. 59, p. 31 Search PubMed; (c) D. A. Evans, S. P. Tanis and D. J. Hart, J. Am. Chem. Soc., 1981, 103, 5813 CrossRef CAS; (d) D. L. Boger and C. E. Brotherton, J. Am. Chem. Soc., 1986, 108, 6713 CrossRef CAS; (e) T. Graening, W. Friedrichsen, J. Lex and H. G. Schmalz, Angew. Chem., Int. Ed., 2002, 41, 1524 CrossRef CAS; (f) F. D. Boyer and I. Hanna, Org. Lett., 2007, 9, 2293 CrossRef CAS PubMed; (g) For a summary of colchicinoid synthesis, see ref. 5a and Table S4 in ESI† for details.
- (a) C. X. Zhuo, W. Zhang and S. L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (b) G. Scheffler, H. Seike and E. J. Sorensen, Angew. Chem., Int. Ed., 2000, 39, 4593 CrossRef CAS.
- Previous attempts to employ a similar oxidopyrylium-mediated [5+2] cycloaddition strategy for the synthesis of colchicine have been unsuccessful because of the failure of the cycloaddition products to react further, see: (a) P. G. Sammes, Gazz. Chim. Ital., 1986, 116, 109 CAS; (b) A. Lupi, M. Patamia and F. Arcamone, Gazz. Chim. Ital., 1990, 120, 277 CAS; (c) B. J. Mcbride and M. E. Garst, Tetrahedron, 1993, 49, 2839 CrossRef CAS.
- For recent reviews, see: (a) H. Pellissier, Adv. Synth. Catal., 2011, 353, 189 CrossRef CAS; (b) K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244 CrossRef CAS PubMed.
- For related studies of of [5+2] cycloaddition reaction from our group, see: (a) G. J. Mei, X. Liu, C. Qiao, W. Chen and C. C. Li, Angew. Chem., Int. Ed., 2015, 54, 1754 CrossRef CAS PubMed; (b) G. J. Mei, H. Yuan, Y. Q. Gu, W. Chen, L. W. Chung and C. C. Li, Angew. Chem., Int. Ed., 2014, 53, 11051 CrossRef CAS PubMed.
- O. Achmatowicz Jr, P. Bukowski, B. Szechner, Z. Zwierzchowska and A. Zamojski, Tetrahedron, 1971, 27, 1973 CrossRef.
- J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984 CrossRef CAS PubMed.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- S. Muller, B. Liepold, G. J. Roth and H. J. Bestmann, Synlett, 1996, 521 CrossRef.
- T. Zhang, L. M. Wu and X. W. Li, Org. Lett., 2013, 15, 6294 CrossRef CAS PubMed.
- (a) F. L. Zhang, K. Hong, T. J. Li, H. Park and J. Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed; (b) F. Mo and G. Dong, Science, 2014, 345, 68 CrossRef CAS PubMed.
- M. Rosner, F. L. Hsu and A. Brossi, J. Org. Chem., 1981, 46, 3686 CrossRef.
- P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS PubMed.
- G. P. Pollini, S. Benetti, C. De Risi and V. Zanirato, Chem. Rev., 2006, 106, 2434 CrossRef CAS PubMed.
- For the way to count steps, see: (a) L. Jørgensen, S. J. McKerrall, C. A. Kuttruff, F. Ungeheuer, J. Felding and P. S. Baran, Science, 2013, 341, 878 CrossRef PubMed; (b) H. Chu, J. M. Smith, J. Felding and P. S. Baran, ACS Cent. Sci., 2017, 3, 47 CrossRef CAS PubMed.
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1547674 and 1524362. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01341h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |