
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTarget-triggered cascade assembly of a catalytic network as an artificial enzyme for highly efficient sensing†
Lei
Zhang‡
,
Fengjiao
Ma‡
,
Jianping
Lei
 *,
Jintong
Liu
and
Huangxian
Ju
*
*,
Jintong
Liu
and
Huangxian
Ju
*
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: jpl@nju.edu.cn; hxju@nju.edu.cn; Tel: +86 25 89681922
First published on 28th April 2017
Abstract
Determining the catalytic activity of artificial enzymes is an ongoing challenge. In this work, we design a porphyrin-based enzymatic network through the target-triggered cascade assembly of catalytic nanoparticles. The nanoparticles are synthesized via the covalent binding of hemin to amino-coated gold nanoparticles and then the axial coordination of the Fe center with a dual-functional imidazole or pyridine derivative. The network, which is specifically formed by coordination polymerization triggered by Hg2+ as the target, shows high catalytic activity due to the triple amplification of enzymatic activity during the cascade assembly. The catalytic dynamics are comparable to those of natural horseradish peroxidase. The catalytic characteristics can be ultrasensitively regulated by the target, leading to a selective methodology for the analysis of sub-attomolar Hg2+. It has also been used for “signal-on” imaging of reactive oxygen species in living cells. This work provides a new avenue for the design of enzyme mimics, and a powerful biocatalyst with signal switching for the development of biosensing protocols.
Introduction
In nature, enzymes often participate in self-assembly processes to form “soft” nanostructures, such as spheres and tubes, demonstrating their catalytic functions. Many synthetic catalysts that behave as enzymes can mimic the functions of natural systems.1–4 However, the performance of some artificial enzymes is very inferior to that of corresponding natural enzymes, possibly as a result of the aggregation of metal macrocycles, such as porphyrins and phthalocyanines, to form inactive dimers that are arranged in a disordered manner.5 To improve the catalytic efficiency, much effort has been devoted to the self-assembly of porphyrins into ever-larger structures involving porphyrinic dendrimers, nanocrystals, and square nanosheets,6–9 and the incorporation of porphyrin-containing hemin in a guanine quadruplex to dissociate the aggregate.10,11 The assembly of porphyrins on nanomaterials has been demonstrated to be a simple and efficient approach for achieving high catalytic activity, specific recognition and even signal transduction,12,13 and it was used to produce multifunctional nanoscale systems for artificial photosynthesis, catalysis, and biosensing.14–23 Porphyrinic metal–organic frameworks as crystalline molecular materials have shown unique enzyme-like activities.24–26 Inspired by the promising applications of porphyrin assemblies as artificial enzymes, this work designs a target-triggered cascade assembly mechanism for the formation of a porphyrin-based enzymatic network, which leads to an ultrasensitive signal switch for target-related bioanalysis. The assembly of porphyrins on nanomaterial surfaces is generally performed through chemisorption, π–π stacking interactions, covalent binding and axial ligation.27–31 Owing to the lack of functional groups on biocompatible gold nanoparticles (AuNPs), which are extensively used in biomedicine, it is difficult to immobilize porphyrinic catalysts on AuNPs. Thus, this work describes the preparation of amino group modified AuNPs (N-AuNPs) using a rationally designed ligand, pamidronic acid disodium salt (PADS), for effective chemical combination with metalloporphyrins (hemin) via an amide reaction (Fig. 1A). The Au-Hem conjugates then assemble with two dual-functional small molecules: 5-(1H-imidazol-4-ylmethylene)-pyrimidine-2,4,6-trione (MPT) via the model shown in Fig. 1B, and 5-(pyridin-4-ylmethylene)-pyrimidine-2,4,6-trione (PMPT) through axial coordination. In the presence of Hg2+ as a target, the coordination polymerization of functionalized AuNPs (Au-Hem-MPT or Au-Hem-PMPT) via N–HgII–N bond connectivities produces enzyme-active networks (Au-Hem-Net and Au-Hem-pNet). Both of the networks show catalytic activities that are much higher than that of free hemin due to the triple amplification of enzymatic activity during the cascade assembly (Fig. 1C). The ordered network provides catalytic dynamics that are comparable to those of the natural enzyme horseradish peroxidase (HRP). Moreover, the catalytic characteristics can be flexibly regulated by trace amounts of Hg2+, a highly toxic metal ion that damages the central nervous and endocrine systems.32,33 This work provides a powerful protocol for the ultrasensitive sensing of sub-attomolar Hg2+. More importantly, the network showed low cytotoxicity and it can be conveniently used for the sensitive fluorescence imaging of intracellular enzyme-related species, such as reactive oxygen species (ROS). Thus, the cascade synthesis of target-controllable catalytic systems provides a new concept for the design and application of enzyme mimics.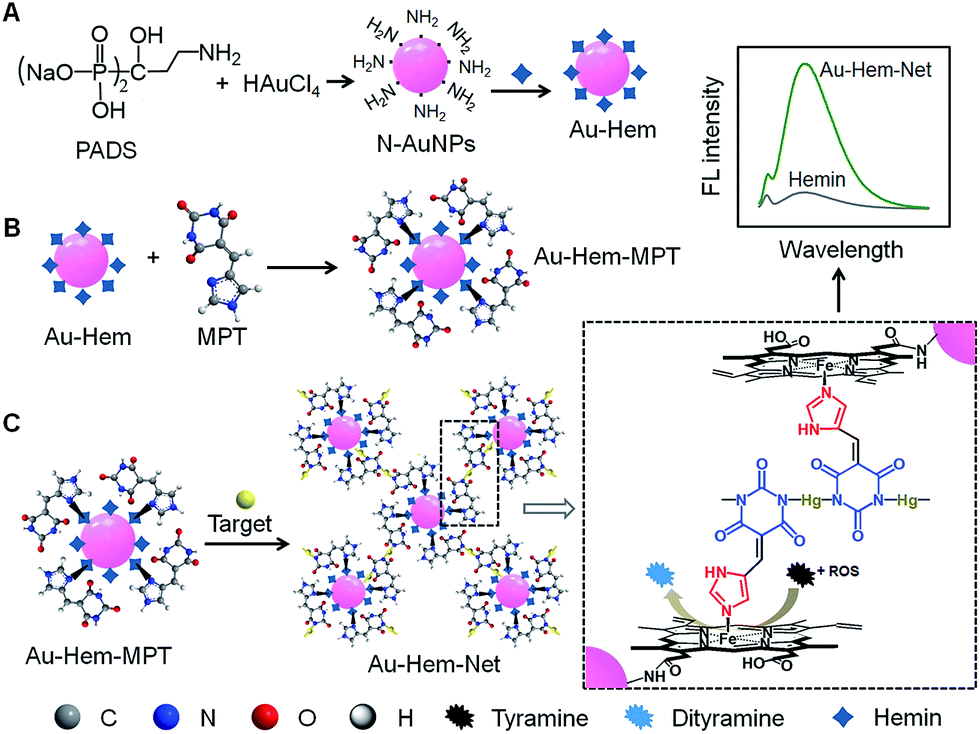 | ||
| Fig. 1 Schematic representation of the cascade assembly of high catalytic networks. (A) The synthesis of N-AuNPs using PADS as both a reducing agent and a protection ligand, and Au-Hem conjugates via an amide reaction. (B) The assembly of MPT on Au-Hem through axial interactions. (C) The preparation of an enzymatic network via Hg2+-triggered polymerization and the catalytic oxidation of tyramine by the network in the presence of ROS. | ||
Results and discussion
Construction and characterization of enzyme-active networks
The N-AuNPs were synthesized via the reaction of HAuCl4 with PADS as both the reducing reagent and the protection ligand in a boiling aqueous solution. At different mass ratios of PADS:HAuCl4, the obtained N-AuNPs showed different colours related to size and dispersion (Fig. 2A). The sharp absorption peak in their UV-vis spectra (Fig. 2B) and dynamic light scattering measurements (Table S1†) indicated a relatively narrow size distribution at ratios from 3.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 11.0:1. Transmission electron micrographs (TEMs) and zeta-potential analysis further demonstrated that the best dispersion of N-AuNPs was at a ratio of 5.0:1 (Fig. S1 and Table S1†), which showed the largest zeta potential, a diameter of around 17 nm, and the characteristic surface plasmon band centered at 525 nm.
:1 to 11.0:1. Transmission electron micrographs (TEMs) and zeta-potential analysis further demonstrated that the best dispersion of N-AuNPs was at a ratio of 5.0:1 (Fig. S1 and Table S1†), which showed the largest zeta potential, a diameter of around 17 nm, and the characteristic surface plasmon band centered at 525 nm.
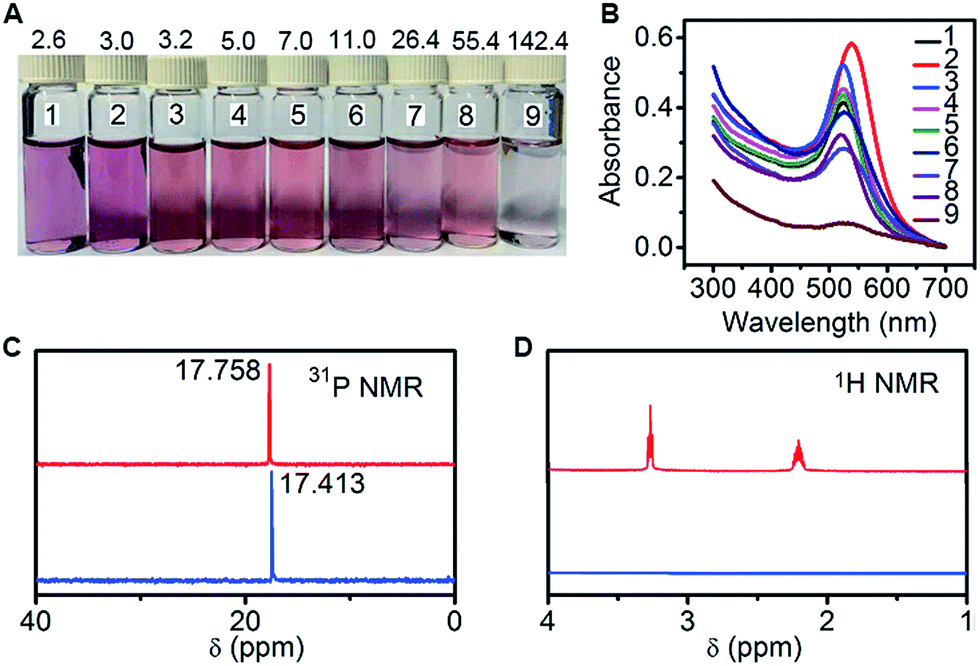 | ||
| Fig. 2 (A) Photograph and (B) UV-vis spectra of AuNPs prepared with PADS and HAuCl4 at mass ratios of 2.6, 3.0, 3.2, 5.0, 7.0, 11.0, 26.4, 55.4 and 142.4 from 1 to 9. (C) 31P NMR and (D) 1H NMR spectra of PADS (red) and the reaction mixture at a mass ratio of 5.0 (blue). | ||
The formation mechanism of N-AuNPs was studied using 1-hydroxyethane-1,1-diphosphonic acid (HEDP), which does not contain an amino group at the tertiary carbon, and 3-aminopropylphosphonic acid (APPA), which does not contain a hydroxyl group at the carbon adjacent to the phosphonic group, as controls. An obvious colour change occurred in the mixtures of HAuCl4 with PADS and HEDP, while no reaction and no plasmon absorption of the AuNPs were observed in the mixture of HAuCl4 with APPA (Fig. S2†), demonstrating that the hydroxyl group at the tertiary carbon was involved in the oxidation reaction of PADS or HEDP by HAuCl4.34,35 The oxidation reaction led to a shift in the 31P NMR peak to a higher magnetic field, accompanied by the signal disappearance of two multiple peaks that were attributed to two methylene groups in the 1H NMR spectrum (Fig. 2C and D), helping to identify the formation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond via the cleavage of another group from the carbon, as shown in the electrospray ionization mass spectra (Fig. S3†). The N-AuNPs prepared with PADS remained as a deep red suspension for several months and exhibited better stability and dispersion (Fig. S2†) due to the presence of an excess amount of PADS on their surfaces. The presence of excess PADS on the nanoparticles was supported by the characteristic vibration frequencies of primary amines at 3228.9 and 3406.1 cm−1 for N-AuNPs in the infrared absorption spectrum (Fig. 3A). More importantly, the adsorbed PADS not only protected the AuNPs against aggregation, but also provided the –NH2 group for covalent binding with hemin, which was characterized by the infrared absorption of the amide bond at 1698.4 and 1298.6 cm−1 (Fig. 3A).
O bond via the cleavage of another group from the carbon, as shown in the electrospray ionization mass spectra (Fig. S3†). The N-AuNPs prepared with PADS remained as a deep red suspension for several months and exhibited better stability and dispersion (Fig. S2†) due to the presence of an excess amount of PADS on their surfaces. The presence of excess PADS on the nanoparticles was supported by the characteristic vibration frequencies of primary amines at 3228.9 and 3406.1 cm−1 for N-AuNPs in the infrared absorption spectrum (Fig. 3A). More importantly, the adsorbed PADS not only protected the AuNPs against aggregation, but also provided the –NH2 group for covalent binding with hemin, which was characterized by the infrared absorption of the amide bond at 1698.4 and 1298.6 cm−1 (Fig. 3A).
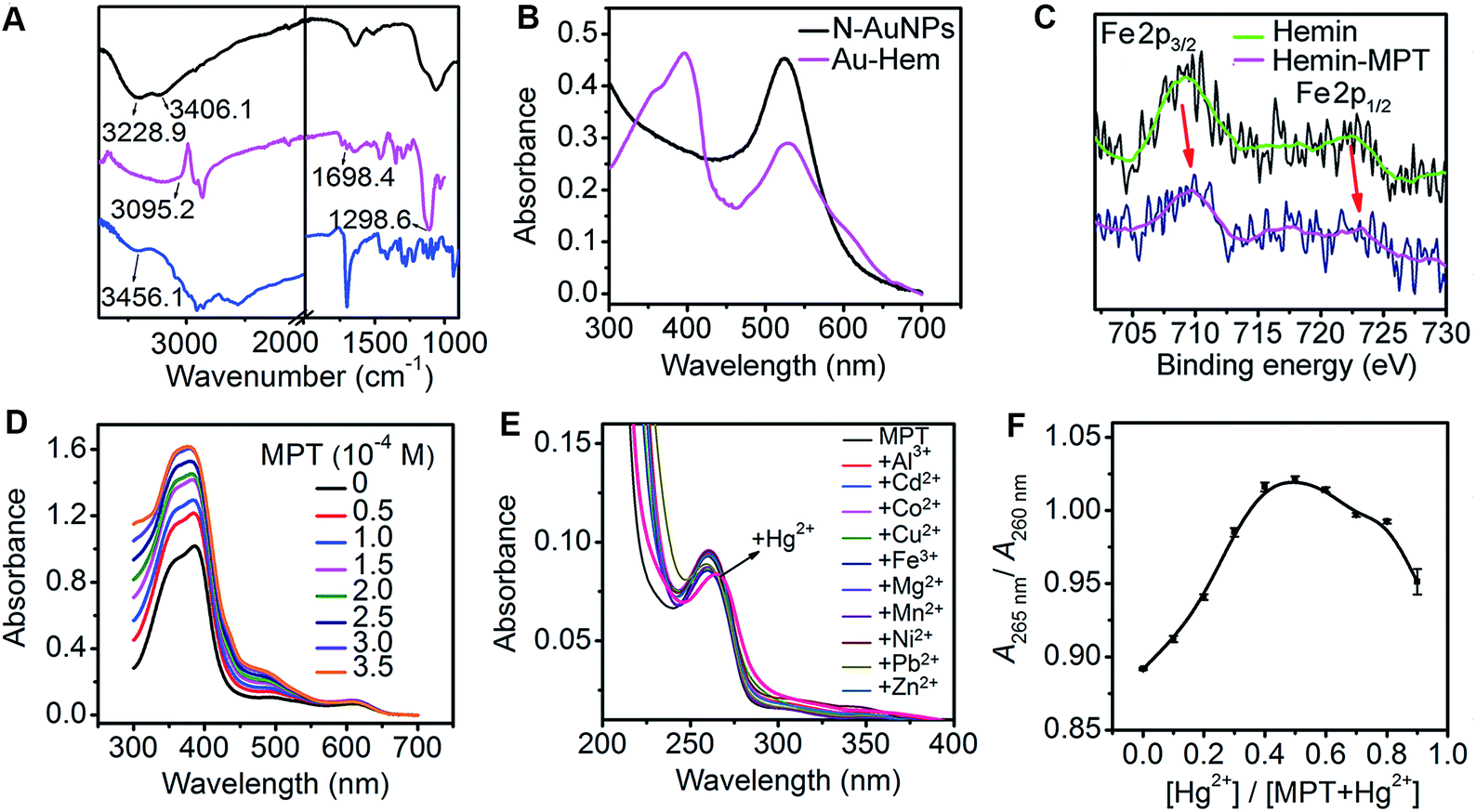 | ||
| Fig. 3 (A) The Fourier transform infrared spectra of N-AuNPs (black), hemin (blue) and Au-Hem (magenta). (B) The UV-vis spectra of N-AuNPs and Au-Hem (1 μM equivalent hemin) solutions. (C) The Fe 2p XPS spectra of hemin and the hemin–MPT complex. (D) The change in the UV-vis spectrum upon the titration of MPT to hemin (20 μM) in pH 7.4 Tris–HCl at 36 °C. (E) The UV-vis spectra of MPT (10 μM) in the presence of different metal ions (100 μM). (F) A Job’s plot for the binding of Hg2+ to MPT. The total concentration of MPT and Hg2+ was kept constant at 100 μM in pH 7.4 Tris–HCl. | ||
The formed Au-Hem showed a single Soret band at around 400 nm (Fig. 3B). The disappearance of the dimeric absorption of hemin at 385 nm and the presence of monomeric absorption at 400 nm and around 350 nm (Fig. S4A†) suggested that the hemin on the N-AuNPs was in its monomeric state, the preferred form for obtaining a high catalytic ability.36 The number of hemin complexes on each Au nanoparticle was calculated to be 73 from the Soret band intensity of hemin using UV-vis spectroscopy (see the ESI Experimental section and Fig. S4B†), indicating an acceptable capacity of the enzyme mimic system for amplifying the catalytic activity.
The Au-Hem was further modified with MPT or PMPTvia the axial coordination of their imidazole or pyridine group with the Fe center of hemin. Both MPT and PMPT were synthesized to contain a pyrimidinetrione moiety for the preparation of the enzyme-active network by coordination polymerization. The MPT and PMPT structures were firstly characterized using MS and NMR information (see the ESI Experimental section, and Fig. S5 and S6†). The Fe 2p X-ray photoelectron spectra (XPS) (Fig. 3C and S7A†) demonstrate that the axial coordination interaction of hemin with both MPT and PMPT increased the Fe 2p1/2 binding energy due to a change in the electron density on the Fe atom.27 The coordination numbers and the binding constants (n and K) were determined via the titration of the hemin solution with MPT and PMPT,37 which led to a slight red shift of the Soret band of hemin at 385 nm (Fig. 3D and S7B†), further suggesting the formation of an axial ligation to the Fe center of hemin.38 As expected, a six-coordinated structure of hemin with two MPT complexes at both axial sides could be formed. However, it occurred only at relatively low temperatures, and showed a much higher binding constant than the five-coordinated structure formed at relatively high temperatures due to the exothermic axial coordination reaction (Fig. S8†). Interestingly, only one PMPT complex could axially coordinate to the Fe center to form the five-coordinated hemin structure due to the weaker coordination ability of the pyridine group compared to that of the imidazole group, which led to relatively lower binding constants than those of the hemin–MPT complex (Table S2†). The ΔrH and ΔrS values for the formation of the hemin-PMPT complex were calculated to be −156.30 kJ mol−1 and −432.33 J mol−1 K−1, respectively. Considering the fact that the dual axial coordination blocked the access of substrate to the Fe sites for catalytic oxidation,39 the Au-Hem-MPT was synthesized at 37 °C, the physiological temperature, for ensuring effective enzymatic catalytic activity.
The interaction between the imide site of thymine and Hg2+ to form a “N–Hg(II)–N” structure is well known and is used for bioanalysis.40,41 The designed pyrimidinetrione moiety in both MPT and PMPT contained two imide sites. Using MPT as an example, the absorption peak of MPT was red-shifted from 260 nm to 265 nm upon the addition of Hg2+ in the MPT solution, while other ions did not change the absorption peak (Fig. 3E), demonstrating the specific recognition of the two imide sites of MPT to Hg2+ to form a “N–Hg(II)–N” like binding motif.42 Using the absorbance ratio of A265 nm to A260 nm in the UV-vis spectra, the Job’s plot analysis revealed a 1:1 binding stoichiometry of the coordination reaction (Fig. 3F). Thus the structure of the MPT–Hg2+ coordination polymer is represented in the inset of Fig. S9A.† The matrix-assisted laser desorption/ionization time-of-fight mass spectrum of the mixture of MPT and Hg2+ further verified the formation of the 1:1 MPT–Hg2+ coordination polymer (Fig. S9A†), in which the mass peaks at m/z 406.6, 612.6, 655.9 and 847.6 could be attributed to the characteristic fragments MPT + Hg-1H, 2MPT + Hg-2H, 2MPT + 2Hg-2 imidazole groups-3H and 2MPT + 2Hg + Cl-3H, respectively.
The 1:1 pyrimidinetrione-Hg2+ coordination reaction induced the formation of Au-Hem-Net and Au-Hem-pNet, as shown in Fig. 1C. After the Hg2+-triggered polymerization of Au-Hem-MPT, both the dynamic light scattering measurements and the transmission electron micrographs showed a greatly increased nanoparticle size from ∼30 to ∼130 nm (Fig. 4A and B). The presence of two clusters of MS peaks at around m/z 1258.6 and 1465.0 also supported the formation of Au-Hem-Net (Fig. S9B†). Here the characteristic peaks at m/z 652.0 and 859.4 corresponded to hemin and hemin–MPT, and the peaks at around m/z 1258.6 and 1465.0 could be attributed to hemin–MPT + 2Hg–2H and hemin–MPT + 2Hg + MPT–3H, respectively.
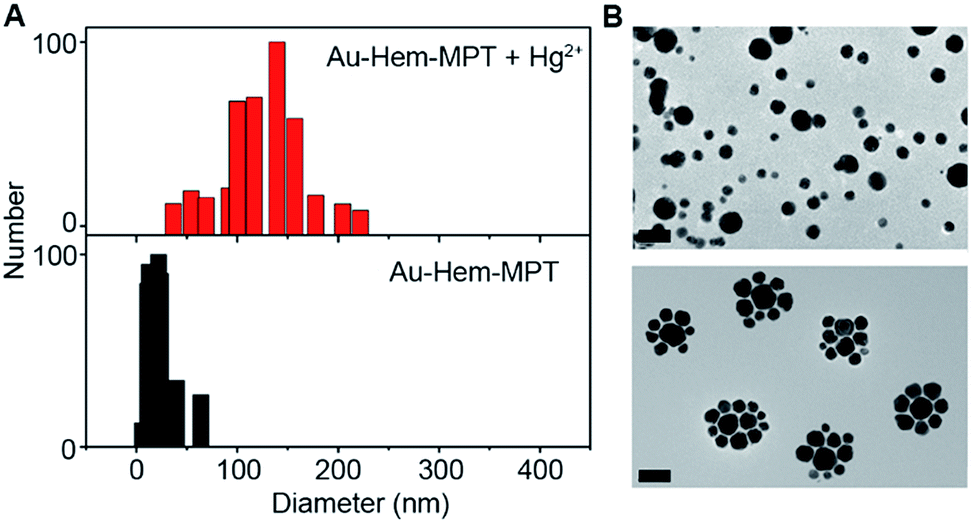 | ||
| Fig. 4 (A) DLS assays of Au-Hem-MPT (0.1 μM equivalent hemin) before and after the addition of Hg2+ (0.1 μM). (B) The TEM images of Au-Hem-MPT (0.1 μM equivalent hemin) in the absence (top) and presence (bottom) of Hg2+ (0.1 μM). The scale bars represent 50 nm. | ||
Catalytic performance
To evaluate the catalytic performance of the enzyme mimics proposed in this work, a fluorescence assay of the tyramine oxidation reaction was performed, in which the nonfluorescent tyramine (0.14 mM) was oxidized by hydrogen peroxide (0.4 mM) to give fluorescent dityramine,43 thus the fluorescent signal represented the catalytic activity of these enzymes as catalysts. The amplified catalytic activity was demonstrated by the strong fluorescent signal of the oxidation product of tyramine by H2O2 in the presence of Au-Hem, when compared to the N-AuNPs and free hemin in aqueous solution (Fig. 5A). | ||
| Fig. 5 (A) Fluorescence intensities of the oxidation product of tyramine (0.14 mM) catalyzed by N-AuNPs (1.0 nM), hemin, Au-Hem, Au-Hem-MPT and Au-Hem-Net (10 nM equivalent hemin) in the presence of H2O2 (0.4 mM) at 37 °C. (B) The fluorescence responses of the oxidation product of tyramine (0.14 mM) by H2O2 (0.4 mM), which is catalyzed by the adduct of Au-Hem (10 nM equivalent hemin) and MPT at marked concentrations in the absence (black) and presence (red) of Hg2+ (1.0 nM) at 37 °C. (C) The fluorescence response of the oxidation product of tyramine (0.14 mM) by 0.4 mM H2O2 for 15 min at 37 °C in the presence of an incubated mixture of Au-Hem (10 nM equivalent hemin) and different concentrations of Hg2+. (D) The fluorescence intensities of the oxidation product of tyramine (0.14 mM) by H2O2 (0.4 mM), which is catalyzed by Au-Hem-MPT (10 nM equivalent hemin) in the presence of marked metal ions at 37 °C. The concentration of Hg2+ is 1.0 nM, and the concentrations of the other metal ions are 10 nM. | ||
On increasing the MPT concentration in Au-Hem, the catalytic activity was slightly enhanced (Fig. 5B), suggesting that the axial coordination of MPT to the Fe center of hemin changed its electronic and geometric structure, which increased the rate of the oxidation–reduction reaction.27 However, the catalytic activity greatly increased upon the addition of MPT in the mixture of Au-Hem and Hg2+, and reached a maximum at a hemin/MPT concentration ratio of 1:1. At this concentration ratio, the formed mimic produced a 2.6, 3.1 and 14.9 times higher fluorescence intensity than that of Au-Hem-MPT, Au-Hem and hemin, respectively, (Fig. 5A), suggesting an improved peroxidase activity in each assembly step. As a control, different concentrations of Hg2+ were added in the Au-Hem solution, and the catalytic oxidation product did not show any fluorescence change (Fig. 5C). More interestingly, this method showed high selectivity. Other metal ions did not produce an obvious fluorescence change (Fig. 5D). Thus, high peroxidase activity resulted from the Hg2+-induced coordination polymerization of Au-Hem-MPT. That is, Hg2+-triggered nanoparticle aggregation results in ordered multiscale organization and the strong interparticle coupling of surface plasmons between neighboring nanoparticles.44,45 Thus, the plasmon enhancement effect accelerates the electronic transfer between H2O2 and FeIII hemin with the production of an initial intermediate (a ferrylporphyrin radical cation),46 leading to the improved catalytic performance of the Hg2+ triggered network. In addition, the networks could be formed by the different sized N-AuNPs with average diameters of 26.1–6.4 nm, which was deduced from the increased fluorescence intensity after the addition of Hg2+, among which small-sized particles had less of a tendency to form the network (Fig. S10†).
The Hg2+-regulated catalytic characteristics of Au-Hem-MPT could be applied to the sensitive detection of Hg2+ using the catalytic fluorescence signal. Using the optimized incubation (9 min) and catalytic reaction (15 min) time (Fig. 6A and B), a remarkable fluorescence increase was observed upon addition of Hg2+ in the mixture of Au-Hem-MPT, tyramine and H2O2 at 37 °C (Fig. 6C). The plot of fluorescence intensity vs. the logarithm of Hg2+ concentration showed good linearity in the range 1.0 aM to 10 pM with a correlation coefficient of 0.992 (Fig. 6D). It is worth noting that the detection limit at 3σ was 0.30 aM, which was 7 orders of magnitude lower than that of conventional analytical methods,47–50 and also the most sensitive method available for detection of Hg2+ (Table S3†). Since Hg2+ is very difficult to biodegrade and can accumulate in organisms to cause various human diseases,51 the coordination polymerization-amplified catalytic reaction is of great importance in monitoring Hg2+ at ultralow abundances and the slow accumulation of Hg2+ in organisms.
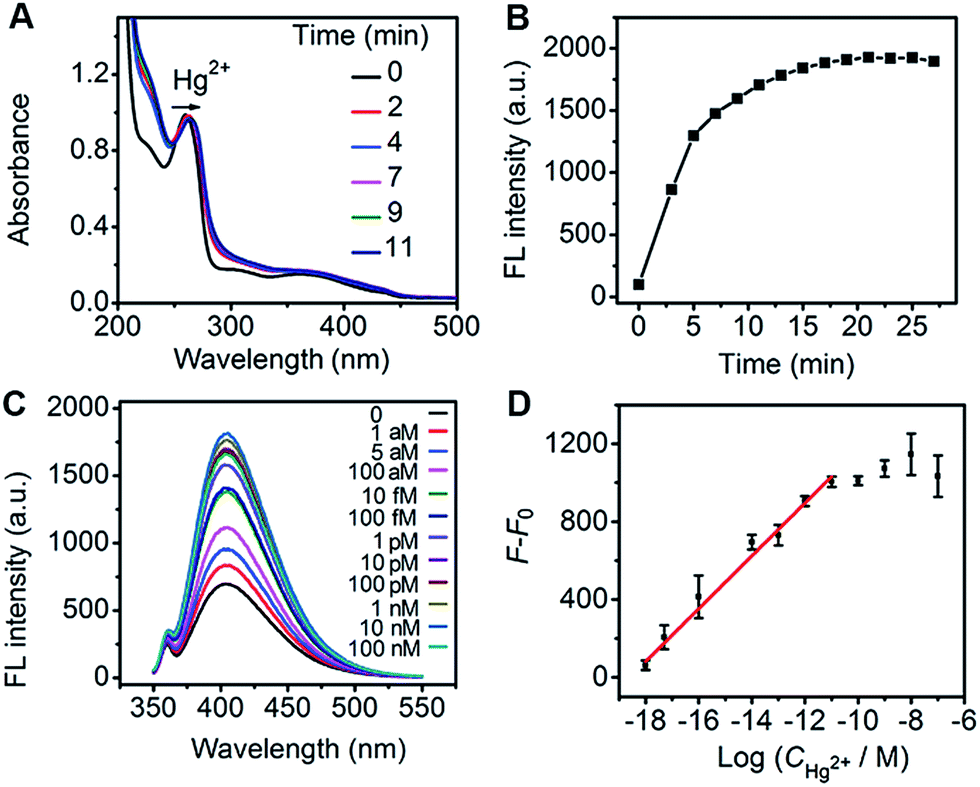 | ||
| Fig. 6 (A) Time-dependent UV-vis spectra of MPT (100 μM) in the presence of Hg2+ (100 μM). (B) The time-dependent fluorescence intensity of the oxidation product of tyramine (0.14 mM) catalyzed by Au-Hem-Net (10 nM equivalent hemin) in the presence of 0.4 mM H2O2 at 37 °C. (C) The fluorescence spectra of the oxidation product of tyramine (0.14 mM) by H2O2 (0.4 mM), which is catalyzed by Au-Hem-MPT (10 nM equivalent hemin) in the presence of Hg2+ at marked concentrations at 37 °C. (D) A plot of F − F0vs. the logarithmic value of Hg2+ concentration. F0 and F are the fluorescence intensity in the absence and presence of Hg2+, respectively. | ||
The Hg2+-triggered formation of Au-Hem-pNet could also be used for Hg2+ detection with the same detection system, and showed a detectable concentration range of 1.0 aM to 100 pM with a detection limit of 0.78 aM (Fig. S11A and B†). Similarly, the formed highly catalytic network could be applied to the detection of H2O2. Using Au-Hem-Net as an example, the mixture of Au-Hem-Net and tyramine showed linearly increasing fluorescence intensity with increasing H2O2 concentration in the range 4.4 to 100 μM (Fig. S11C and D†).
To evaluate the catalytic dynamics of the designed network, the oxidation product of pyrogallol by H2O2 was detected using UV-vis spectroscopy. At pyrogallol concentrations that were much higher than those of the enzyme mimics, three enzyme mimics, Au-Hem, Au-Hem-PMPT and Au-Hem-pNet, all showed a linear Lineweaver–Burk plot at the initial oxidation stage (Fig. 7), which led to two important kinetic parameters, Km (the Michaelis constant) and kcat (the catalytic kinetic constant), as listed in Table 1. The Km value of Au-Hem was a surprisingly low value of 0.40 mM, which was less than that of HRP (0.81 mM), indicating a good affinity of pyrogallol to the Au-Hem conjugate. Meanwhile, Au-Hem-PMPT and Au-Hem-pNet showed comparable affinities to HRP. The kcat values of Au-Hem (419.6 min−1), Au-Hem-PMPT (714.3 min−1) and Au-Hem-pNet (1054.5 min−1) were 2–3 orders of magnitude higher than that of free hemin (2.4 min−1) and higher than those of other carrier-supported hemin composites.20,21,52,53 Moreover, the kinetic constant of Au-Hem-pNet was comparable to that of natural HRP (1750 min−1), highlighting the excellent catalytic activity of the enzyme-active network, which was greatly facilitated by the substrate being close to the active site of the enzyme and cooperative binding.54
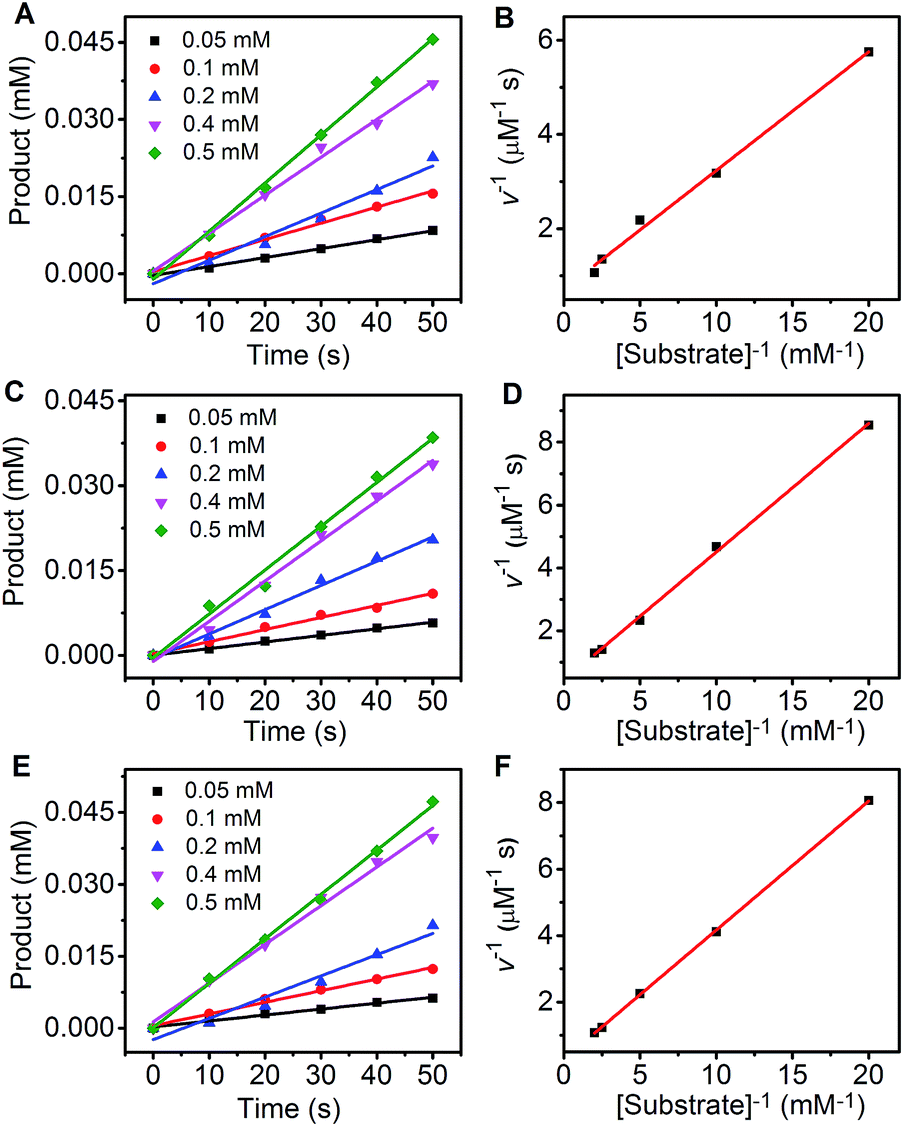 | ||
| Fig. 7 The fitting curve of the initial pyrogallol oxidation profile at marked pyrogallol concentrations (A, C and E) and a Lineweaver–Burk plot of the pyrogallol oxidation (B, D and F) catalyzed by the Au-Hem conjugate (A and B, 0.2 μM hemin equivalent), Au-Hem-PMPT (C and D, 0.2 μM hemin equivalent) and Au-Hem-pNet (E and F, 0.2 μM hemin equivalent) in the presence of 0.4 mM H2O2. | ||
| Mimics | K m (mM) | k cat (min−1) | Ref. |
|---|---|---|---|
| a FeTMPyP: tetramethylpyridylporphyrin iron. | |||
| Au-Hem | 0.40 | 419.6 | This work |
| Au-Hem-PMPT | 0.97 | 714.3 | This work |
| Au-Hem-pNet | 1.36 | 1054.5 | This work |
| Hemin | 87.1 | 2.4 | 52 |
| Hemin–graphene | 1.22 | 246 | 21 |
| Hemin–hydrogel | — | 19.9 | 20 |
| Hemin–polymer | — | 7.6 | 20 |
| FeTMPyPa | — | 83 | 53 |
| FeTMPyP–graphene | 0.96 | 545 | 21 |
| FeTMPyP–antibody | 8.6 | 680 | 53 |
| HRP | 0.81 | 1750 | 53 |
Fluorescence imaging of intracellular reactive oxygen species
In view of the high catalytic activity towards the substrate oxidation by different ROS (Fig. 8A), the cascade-assembled network, Au-Hem-Net, was used for the fluorescence imaging of ROS in living cells with o-phenylenediamine as the substrate. The cytotoxicity of Au-Hem-Net along with the substrate was examined using MTT assay (a colorimetric assay for assessing cell metabolic activity, see the ESI materials and reagents, Experimental section†) by incubating HeLa cells with the mixture, which maintained about 90% of the cell viability after 5 h of incubation (Fig. 8B). The confocal fluorescence imaging of intracellular ROS was performed by incubating the HeLa cells with Au-Hem-Net and o-phenylenediamine at 37 °C for different times. The fluorescence from the HeLa cells was noticeable after 2 h and increased with increasing incubation time, reaching a maximum fluorescence intensity at 4 h (Fig. 8C), suggesting the formation of a fluorescent product from the Au-Hem-Net catalyzed oxidation of o-phenylenediamine by intracellular ROS. In contrast, individual substrate or Au-Hem-Net treated HeLa cells did not show appreciable changes in the fluorescence images (Fig. 8D and E). Therefore, the cascade-assembled Au-Hem-Net possesses promising potential for application in the sensitive fluorescence imaging of intracellular ROS and/or other catalytic reaction-related biomolecules.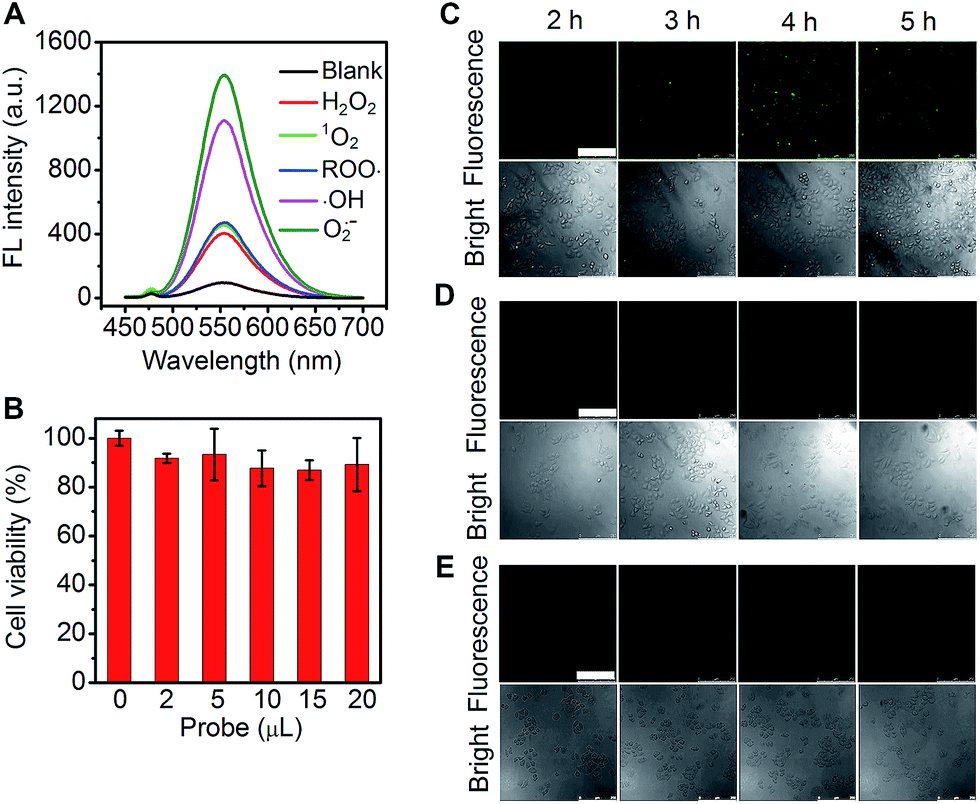 | ||
| Fig. 8 (A) Fluorescence responses of o-phenylenediamine (0.14 mM) oxidized by different ROS for 15 min in the presence of Au-Hem-Net (10 nM equivalent hemin) at an excitation wavelength of 410 nm. (B) The viability of HeLa cells that were detected with MTT after treatment with 100 μL of culture medium containing o-phenylenediamine (0.14 mM) and different volumes of Au-Hem-Net (100 nM equivalent hemin) for 5 h at 37 °C. (C–E) Confocal fluorescence images of HeLa cells after incubation with o-phenylenediamine and Au-Hem-Net (C), o-phenylenediamine (D) and Au-Hem-Net (E) for different times at 37 °C. o-Phenylenediamine: 0.14 mM; Au-Hem-Net: 10 nM equivalent hemin. The scale bars represent 250 μm. | ||
Conclusions
We present a highly catalytic network using axial coordination and coordination polymerization to achieve target-triggered cascade assembly based on the specific recognition of pyrimidinetrione moieties to the target. Two dual-functional pyrimidinetrione-containing derivates of imidazole (MPT) and pyridine (PMPT) are designed to link the target Hg2+ and the Fe center of hemin that is covalently assembled on amino group modified AuNPs. The high catalytic activity of the network results from the triple amplification of the enzymatic activity during the cascade assembly: monomeric hemin loading on the N-AuNPs, axial coordination of MPT or PMPT to the Fe center of hemin to form a five-coordinated hemin structure, and coordination polymerization via a 1:1 binding stoichiometry to form a “N–Hg(II)–N” like binding motif for the ordered aggregation of the catalytic monomers. The catalytic activity of the network can be flexibly controlled by Hg2+, which leads to an extremely sensitive strategy for Hg2+ detection. The sub-attomolar detection limit is 7 orders of magnitude lower than those of conventional analytical methods. This catalytic system shows low cytotoxicity and has been successfully utilized in sensitive fluorescence imaging of intracellular ROS. The proposed catalytic network showed excellent affinity and high catalytic kinetics that are comparable to those of natural HRP. The cascade synthesis of the target-controllable catalytic system opens up new perspectives for not only the assembly of metalloporphyrin in the discovery and design of enzyme mimics, but also the development of biosensing protocols.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (21375060, 21635005, 21605082, 21675084).Notes and references
- J. P. Collman, N. K. Devaraj, R. A. Decréau, Y. Yang, Y. L. Yan, W. Ebina, T. A. Eberspacher and C. E. Chidsey, Science, 2007, 315, 1565–1568 CrossRef CAS PubMed.
- A. M. Beauchemin, Nat. Chem., 2013, 5, 731–732 CrossRef CAS PubMed.
- M. Fischlechner, Y. Schaerli, M. F. Mohamed, S. Patil, C. Abell and F. Hollfelder, Nat. Chem., 2014, 6, 791–796 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- T. C. Bruice, Acc. Chem. Res., 1991, 24, 243–249 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- S. J. Lee, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, Adv. Mater., 2008, 20, 3543–3549 CrossRef CAS.
- Z. C. Wang, Z. Y. Li, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2007, 129, 2440–2441 CrossRef CAS PubMed.
- M. Shema-Mizrachi, G. M. Pavan, E. Levin, A. Danani and N. G. Lemcoff, J. Am. Chem. Soc., 2011, 133, 14359–14367 CrossRef CAS PubMed.
- P. Travascio, Y. F. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef CAS PubMed.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153–1165 RSC.
- F. Bedioui, Coord. Chem. Rev., 1995, 144, 39–68 CrossRef CAS.
- L. G. Marinescu and M. Bols, Angew. Chem., Int. Ed., 2006, 45, 4590–4593 CrossRef CAS PubMed.
- T. Hasobe, H. Imahori, P. V. Kamat and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 14962–14963 CrossRef CAS PubMed.
- W. Hieringer, J. Am. Chem. Soc., 2011, 133, 6206–6222 CrossRef CAS PubMed.
- Y. M. Wang, X. F. Wang, S. K. Ghosh and H. P. Lu, J. Am. Chem. Soc., 2009, 1, 1479–1487 CrossRef PubMed.
- L. Zhang, J. P. Lei, F. J. Ma, P. H. Ling, J. T. Liu and H. X. Ju, Chem. Commun., 2015, 51, 10831–10834 RSC.
- Y. Zang, J. P. Lei, P. H. Ling and H. X. Ju, Anal. Chem., 2015, 87, 5430–5436 CrossRef CAS PubMed.
- L. Chen, Y. Yang and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- Q. G. Wang, Z. M. Yang, X. Q. Zhang, X. D. Xiao, C. K. Chang and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 4285–4289 CrossRef CAS PubMed.
- T. Xue, S. Jiang, Y. Q. Qu, Q. Su, R. Cheng, S. Dubin, C. Y. Chiu, R. Kaner, Y. Huang and X. F. Duan, Angew. Chem., Int. Ed., 2012, 51, 3822–3825 CrossRef CAS PubMed.
- S. Jiang, R. Cheng, X. Wang, T. Xue, Y. Liu, A. Nel, Y. Huang and X. F. Duan, Nat. Commun., 2013, 4, 2225 Search PubMed.
- T. Xue, B. Peng, M. Xue, X. Zhong, C. Y. Chiu, S. Yang, Y. Q. Qu, L. Y. Ruan, S. Jiang, S. Dubin, R. B. Kaner, J. I. Zink, M. E. Meyerhoff, X. F. Duan and Y. Huang, Nat. Commun., 2014, 5, 3200 Search PubMed.
- O. K. Farha, A. M. Shultz, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 5652–5655 CrossRef CAS PubMed.
- D. W. Feng, Z. Y. Gu, J. R. Li and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- R. G. Cao, R. Thapa, H. Kim, X. D. Xu, M. G. Kim, Q. Li, N. Park, M. L. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 Search PubMed.
- Y. H. Lin, L. Wu, Y. Y. Huang, J. S. Ren and X. G. Qu, Chem. Sci., 2015, 6, 1272–1276 RSC.
- H. Li, Y. Y. Luk and M. Mrksich, Langmuir, 1999, 15, 4957–4959 CrossRef CAS.
- T. Belser, M. Stçhr and A. Pfaltz, J. Am. Chem. Soc., 2005, 127, 8720–8731 CrossRef CAS PubMed.
- S. Debobrata, H. Partha and K. P. Tapan, Angew. Chem., Int. Ed., 2013, 52, 13314–13318 CrossRef PubMed.
- E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed.
- F. Zahir, S. J. Rizwi, S. K. Haq and R. H. Khan, Environ. Toxicol. Pharmacol., 2005, 20, 351–360 CrossRef CAS PubMed.
- C. H. Su, P. L. Wu and C. S. Yeh, J. Phys. Chem. B, 2003, 107, 14240–14243 CrossRef CAS.
- K. K. Caswell, C. M. Bender and C. J. Murphy, Nano Lett., 2003, 3, 667–669 CrossRef CAS.
- E. S. Ryabova, J. Biol. Inorg Chem., 2004, 9, 385–395 CrossRef CAS PubMed.
- Z. C. Chen, L. H. Wang and J. P. M. Schelvis, Biochemistry, 2003, 42, 2542–2551 CrossRef CAS PubMed.
- F. A. Walker, M. W. Lo and M. T. Ree, J. Am. Chem. Soc., 1976, 98, 5552–5560 CrossRef CAS PubMed.
- J. P. Collman, T. Kodadek, S. A. Raybuck and B. Meunier, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 7039–7041 CrossRef CAS.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172–2173 CrossRef CAS PubMed.
- X. J. Xue, F. Wang and X. G. Liu, J. Am. Chem. Soc., 2008, 130, 3244–3245 CrossRef CAS PubMed.
- J. N. Wang, L. Zhang, Q. Qi, S. H. Li and Y. B. Jiang, Anal. Methods, 2013, 5, 608–611 RSC.
- Q. B. Wang, N. Xu, Z. Gui, J. P. Lei, H. X. Ju and F. Yan, Chem. Commun., 2014, 50, 15362–15365 RSC.
- N. L. Pacioni, M. González-Béjar, E. Alarcón, K. L. McGilvray and J. C. Scaiano, J. Am. Chem. Soc., 2010, 132, 6298–6299 CrossRef CAS PubMed.
- W. Yang, K. H. Liu, D. Song, Q. Du, R. N. Wang and H. M. Su, J. Phys. Chem. C, 2013, 117, 27088–27095 CAS.
- X. J. Yang, C. L. Fang, H. C. Mei, T. J. Chang, Z. H. Cao and D. H. Shangguan, Chem.–Eur. J., 2011, 17, 14475–14484 CrossRef CAS PubMed.
- J. S. Lee, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 4093–4096 CrossRef CAS PubMed.
- Y. K. Yang, K. J. Yook and J. S. Tae, J. Am. Chem. Soc., 2005, 127, 16760–16761 CrossRef CAS PubMed.
- S. Ando and K. Koide, J. Am. Chem. Soc., 2011, 133, 2556–2566 CrossRef CAS PubMed.
- R. Freeman, X. Q. Liu and I. Willner, J. Am. Chem. Soc., 2011, 133, 11597–11604 CrossRef CAS PubMed.
- P. W. Davidson, G. J. Myers, C. Cox, C. Axtell, C. Shamlaye, J. Sloane-Reeves, E. Cernichiari, L. Needham, A. Choi, Y. Wang, M. Berlin and T. W. Clarkson, J. Am. Med. Assoc., 1998, 280, 701–707 CrossRef CAS PubMed.
- Q. G. Wang, Z. M. Yang, M. L. Ma, C. K. Chang and B. Xu, Chem.–Eur. J., 2008, 14, 5073–5078 CrossRef CAS PubMed.
- H. Yamaguchi, K. Tsubouchi, K. Kawaguchi, E. Horita and A. Harada, Chem.–Eur. J., 2004, 10, 6179–6186 CrossRef CAS PubMed.
- Q. Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, and supporting figures and tables. See DOI: 10.1039/c7sc01453h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |