
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation and ligand-based reductive chemistry of bridged bis-alkylidene scandium(III) complexes†
Wangyang
Ma
 a,
Chao
Yu
a,
Yue
Chi
a,
Tianyang
Chen
a,
Lianjun
Wang
ac,
Jianhao
Yin
a,
Baosheng
Wei
a,
Ling
Xu
a,
Wen-Xiong
Zhang
*ab and
Zhenfeng
Xi
a
a,
Chao
Yu
a,
Yue
Chi
a,
Tianyang
Chen
a,
Lianjun
Wang
ac,
Jianhao
Yin
a,
Baosheng
Wei
a,
Ling
Xu
a,
Wen-Xiong
Zhang
*ab and
Zhenfeng
Xi
a
aBeijing National Laboratory for Molecular Sciences, MOE Key Laboratory of Bioorganic Chemistry and Molecular Engineering, College of Chemistry, Peking University, Beijing 100871, China. E-mail: wx_zhang@pku.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, China
cSchool of Chemistry and Chemical Engineering, Hunan Institute of Engineering, Xiangtan, 411104, China
First published on 31st July 2017
Abstract
The chemistry of rare-earth carbene and alkylidene complexes including their synthesis, structure and reaction is a challenging issue because of their high reactivity (or instability) and the lack of synthetic methods. In this work, we report the first synthesis of the bridged bis-alkylidene complexes which feature a 2-butene-1,1,4,4-tetraanion and four Sc–C(sp3) bonds by the reaction of 1,4-dilithio-1,3-butadienes with ScCl3. This reaction proceeds via two key intermediates: an isolable scandacyclopentadiene and a proposed scandacyclopropene. The scandacyclopentadiene undergoes β,β′-C–C bond cleavage to generate the scandacyclopropene, which then dimerizes to afford the bridged bis-alkylidene complex via a cooperative double metathesis reaction. Reaction chemistry study of the bridged bis-alkylidene complex reveals their ligand-based reduction reactivity towards different oxidants such as hexachloroethane, disulfide and cyclooctatetraene.
Transition metal carbene and alkylidene complexes have been extensively studied because of their importance in organometallic chemistry, coordination chemistry and synthetic organic chemistry.1 In contrast, rare-earth metal carbene and alkylidene complexes are very limited mainly due to the energy mismatch between the rare-earth metals and ligand orbitals.2–12 Since the rare-earth alkylidene complex was first postulated in 1979,3 pioneering works have been made to isolate and characterize it. Some pincer-like rare-earth alkylidene complexes have been reported independently by Cavell,4 Liddle,5 and Mézailles.6 Very recently, Cui et al. reported the lutetium methanediide-alkyl complexes,7 and Chen et al. reported the non-pincer-type mononuclear scandium alkylidene complexes.8 Furthermore, rare-earth methylidene complexes were also stabilized by chloride bridges9 or Lewis-acids such as AlMe3.10 Interestingly, mixed methyl/methylidene complexes11 and cubane-like methylidene complexes12 have been reported. Despite these recent advances, the chemistry of rare-earth alkylidene complexes is still in its infancy, and the bridged bis-alkylidene complex remains scarce.
Reductive reaction of rare-earth organometallic compounds is a fundamental process in organometallic chemistry and coordination chemistry.13 Rare earth metal complexes (Ce, Sm, Eu and Yb) supported by redox-inert ligands tend to perform a single electron redox process. The utilization of redox-active ligands at the rare earth metal centers is an alternative strategy for affording multi-electron redox reactivity.14 Ligand-based reductive chemistry of trivalent rare-earth organometallic compounds has received much attention. Evans and coworkers have made great progress in studying the reductive reactivity of (C5Me5)3Ln (Ln = La, Nd, Sm, etc.) and provided a wide variety of new reductive chemistry for rare earth metals.13a,15
Herein, we report the first synthesis of the bridged bis-alkylidene complex featuring a 2-butene-1,1,4,4-tetraanion and four Sc–C(sp3) bonds from 1,4-dilithio-1,3-butadienes and ScCl3. This reaction proceeds via two key intermediates: scandacyclopentadiene16,17 and scandacyclopropene.18,19 DFT calculations indicate that the dimerization of scandacyclopropenes via the cooperative double metathesis is the key factor for the formation of the bridged bis-alkylidene complex. Interestingly, the bridged bis-alkylidene scandium(III) complex shows unexpected ligand-based two-electron or four-electron reduction reactivity towards different oxidants such as hexachloroethane, disulfide and cyclooctatetraene.
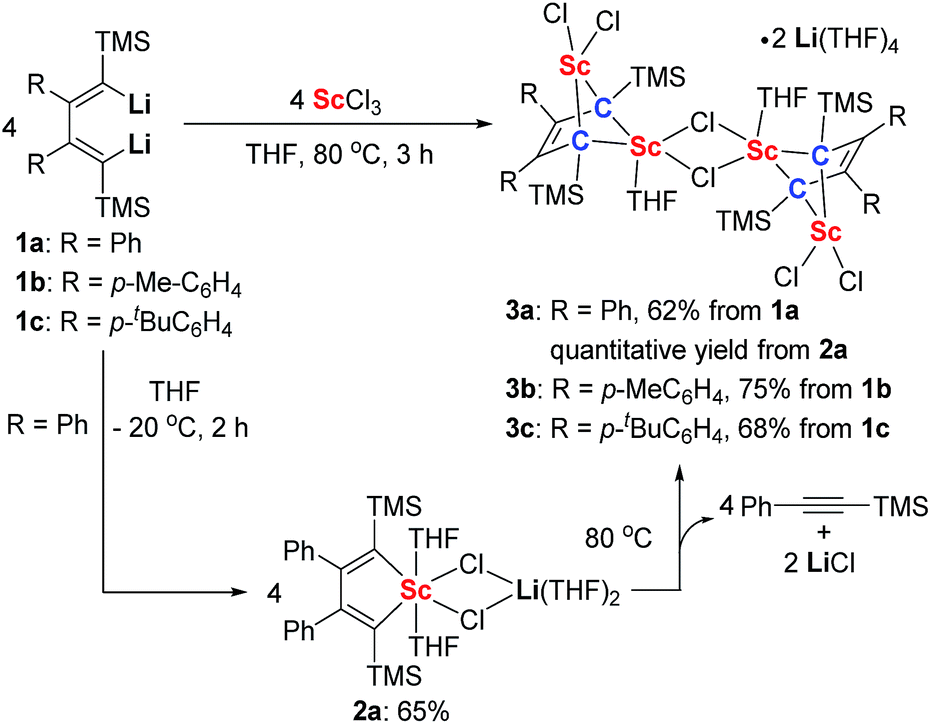
Silyl-substituted 1,4-dilithio-1,3-butadienes 1a–c were readily prepared according to our previous procedure.20 When the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of 1a and solvated ScCl3 in THF was conducted at −20 °C, the light yellow crystalline complex 2a could be isolated exclusively in 65% yield (Scheme 1). An X-ray analysis of 2a revealed that it is a LiCl-ligated scandacyclopentadiene (Fig. 1). The Sc(III) center adopts a distorted octahedral fashion bonded with two C(sp2) atoms, two chlorides and two THF molecules. The C1–C2 (1.348(4) Å) and C3–C4 (1.376(4) Å) bond lengths are within the range of standard C
:1 reaction of 1a and solvated ScCl3 in THF was conducted at −20 °C, the light yellow crystalline complex 2a could be isolated exclusively in 65% yield (Scheme 1). An X-ray analysis of 2a revealed that it is a LiCl-ligated scandacyclopentadiene (Fig. 1). The Sc(III) center adopts a distorted octahedral fashion bonded with two C(sp2) atoms, two chlorides and two THF molecules. The C1–C2 (1.348(4) Å) and C3–C4 (1.376(4) Å) bond lengths are within the range of standard C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths, and the C2–C3 bond length (1.520(3) Å) indicates a typical C–C single bond. These data of bond lengths clearly show the butadienyl dianionic structure in 2a.
C bond lengths, and the C2–C3 bond length (1.520(3) Å) indicates a typical C–C single bond. These data of bond lengths clearly show the butadienyl dianionic structure in 2a.
 | ||
| Scheme 1 Synthesis of scandacyclopentadiene 2a and bridged bis-alkylidene scandium(III) complexes 3a–c. | ||
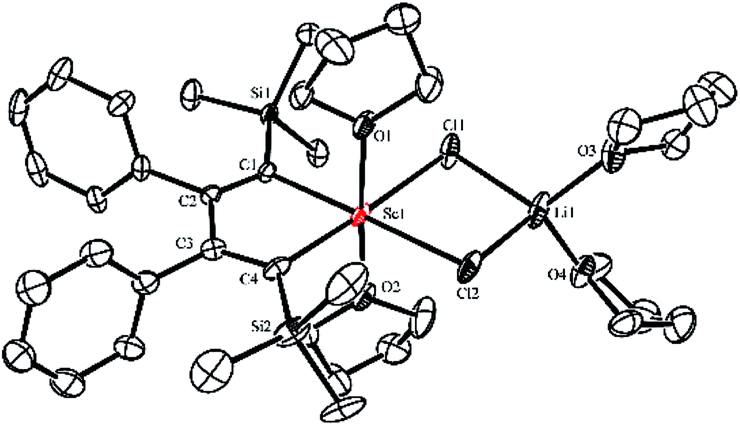 | ||
| Fig. 1 Molecular structure of complex 2a with thermal ellipsoids at 30% probability. H atoms are omitted for clarity. | ||
Complex 2a is sensitive to air and moisture but stable under dry N2 atmosphere. In the 1H NMR spectrum in THF-d8, a singlet at −0.38 ppm was observed and assigned to the proton resonance of TMS groups. Two β-C(sp2) atoms (C2 and C3) displayed a singlet at 167.6 ppm in the 13C NMR spectrum, while two α-C(sp2) atoms (C1 and C4) showed a broad peak at 203.8 ppm, probably due to the coupling with scandium (nuclear spin quantum number I = 7/2). The 1H NMR spectrum of 2a in THF-d8 showed no obvious change for 2 weeks at room temperature. However, when the THF-d8 solution of 2a was heated at 45 °C for 3 h or 80 °C for 10 min, the TMS proton resonance at −0.38 ppm completely disappeared in the 1H NMR spectrum, and two new singlets integrated to the same number of protons appeared at −0.23 ppm and 0.20 ppm (see ESI† for more details). The singlet at 0.20 ppm was assigned to the TMS proton resonance of PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CTMS by comparison with its standard spectrum. The GC retention time and molecular ion peak (m/z = 174) detected by GC-MS are also consistent with those of the standard sample of PhCCTMS. The other new singlet at −0.23 ppm was assigned to the TMS groups of a new complex 3a, which was obtained in almost quantitative yield by thermolysis of 2a. Furthermore, we found that the synthesis of 3a does not require isolation of 2a as the starting material. 3a could be conveniently prepared by the reaction of 1a with solvated ScCl3 in THF solution at 80 °C for 3 h. Similarly, 3b and 3c could be prepared from the corresponding 1,4-dilithio-1,3-butadienes and ScCl3 (Scheme 1).
CTMS by comparison with its standard spectrum. The GC retention time and molecular ion peak (m/z = 174) detected by GC-MS are also consistent with those of the standard sample of PhCCTMS. The other new singlet at −0.23 ppm was assigned to the TMS groups of a new complex 3a, which was obtained in almost quantitative yield by thermolysis of 2a. Furthermore, we found that the synthesis of 3a does not require isolation of 2a as the starting material. 3a could be conveniently prepared by the reaction of 1a with solvated ScCl3 in THF solution at 80 °C for 3 h. Similarly, 3b and 3c could be prepared from the corresponding 1,4-dilithio-1,3-butadienes and ScCl3 (Scheme 1).
An X-ray analysis of 3a reveals it is a bridged bis-alkylidene complex and adopts a dimeric ate complex via μ2-chloride bridges (Fig. 2). One scandium center (e.g. Sc1) is bonded with two carbon atoms and two terminal chlorides, while the other one (e.g. Sc2) is bonded with two carbon atoms, two bridged chlorides and one THF. The Sc1–Sc2 distance (3.1366(9) Å) is the shortest length found in the literature, which is notably shorter than those in dinuclear scandium hydride complexes (3.20–3.40 Å).21 Two lithium atoms act as counterions, and each lithium atom forms a distorted tetrahedron surrounded by four THF molecules. The bond lengths of C1–C2 (1.468(4) Å) and C3–C4 (1.465(5) Å) in 3a are significantly longer than those in 2a [C1–C2, 1.348(4) Å; C(3)–C(4), 1.376(4) Å]. The bond length of C2–C3 (1.430(4) Å) in 3a is significantly shorter than the corresponding C2–C3 (1.520(3) Å) in 2a. Thus, the bond lengths in the C1–C2–C3–C4 moiety in 3a are averaged and are not the classical bond lengths of C–C single and double bonds. These results show that 3a has a highly delocalized structure with a tetraanionic ligand. Most importantly, these results are in striking contrast with what was observed previously for the transmetalation reactions of 1,4-dilithio-1,3-butadienes with metal salts which gave 1,3-butadiene-1,4-dianion complexes.20
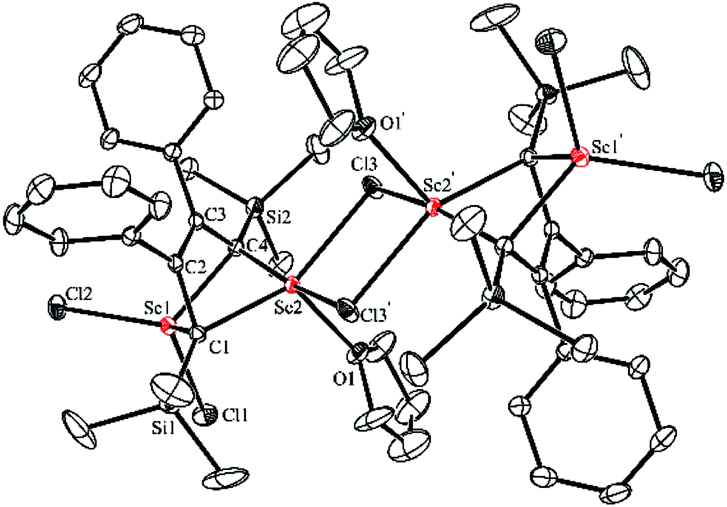 | ||
| Fig. 2 Molecular structure of complex 3a with thermal ellipsoids at 30% probability. H atoms and two [Li(THF)4]+ counterions are omitted for clarity. | ||
The formation of the asymmetric unit in 3a from two molecules of 2a along with elimination of two alkynes is a very interesting process and intrigued us to explore the reaction mechanism. The crossover reaction between 2a and 2a-D10 was carried out. When the reaction mixture was quenched with H2O, 4a, 4a-D5, and 4a-D10 could all be detected by HRMS (Scheme 2). This result unambiguously reveals that the 2-butene-1,1,4,4-tetraanion moiety in 3a should be originated from two molecules of scandacyclopentadienes instead of a simple reduction of a diene moiety in one scandacyclopentadiene. Thus, the crossover experiment excludes two possible pathways involving two eliminated alkynes from the same scandacyclopentadienes: (i) the cooperative intermolecular redox process, and (ii) stepwise intermolecular redox via the scandacyclopropene process (see ESI† for more details).
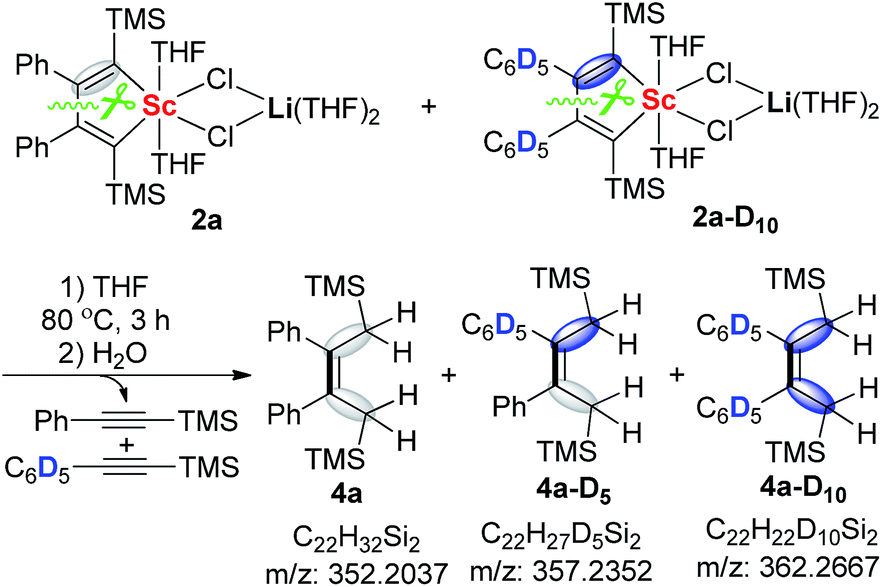 | ||
| Scheme 2 The crossover-reaction between 2a and 2a-D10. | ||
Based on the above information, we proposed a mechanism involving the scandacyclopropene intermediate. For a better understanding of the formation of 3a, DFT calculations were carried out using Gaussian 09 (Fig. 3).22 We chose the LiCl-free scandacyclopentadiene IM1 as a starting model compound and the THF-ligated monomer 3a-M as a targeted compound for simplicity.23 The structures of all of the minima and transition states were optimized at the B3LYP24/LANL2DZ (for Sc)/6-31+G* (for other elements) level in the gas phase. The effect of the solvent was examined by performing single-point self-consistent reaction field (SCRF) calculations based on the polarizable continuum model (PCM) for gas-phase optimized structures. Scandacyclopentadiene IM1 will undergo β,β′-C–C bond cleavage to generate scandacyclopropene IM2 by release of one equiv. of alkyne. The β,β′-C–C bond cleavage from IM1 to IM2 is the critical step with the highest energy barrier of 13.3 kcal mol−1 in the solution phase, which means that IM1 is isolable. Metallacyclopropenes, as an important class of reactive intermediate, have been isolated and characterized in transition and main group organometallic chemistry.18,19 The metallacyclopropene, e.g. aluminacyclopropene, can undergo dimerization to give a 1,4-dialuminacyclohexadiene.25 In contrast, rare-earth metallacyclopropenes are unknown. IM2 is the first optimized structure of a rare-earth metallacyclopropene by DFT calculations. Next, we tried to optimize the dimeric structure of IM3′ which is similar to 1,4-dialuminacyclohexadiene. However, the optimization of the structure of IM3′ to a local energy minimum failed, probably because of its high energy and instability. Rather than giving IM3′, a new intermediate, IM3, resulting from two IM2 species approaching each other via the weak Sc–C interaction, was optimized to a local minimal energy, 2.7 kcal mol−1 lower than IM2. Surprisingly, a cooperative double metathesis of IM3 gives 3a-Mvia the transition state TS2. In TS2, two scandacyclopropene rings adopt a triangular prism geometry, in which each Sc atom is coordinated to another carbon neighbouring TMS group. This geometry of TS2 could also explain the selectivity of C(Ph)–C(Ph) coupling.
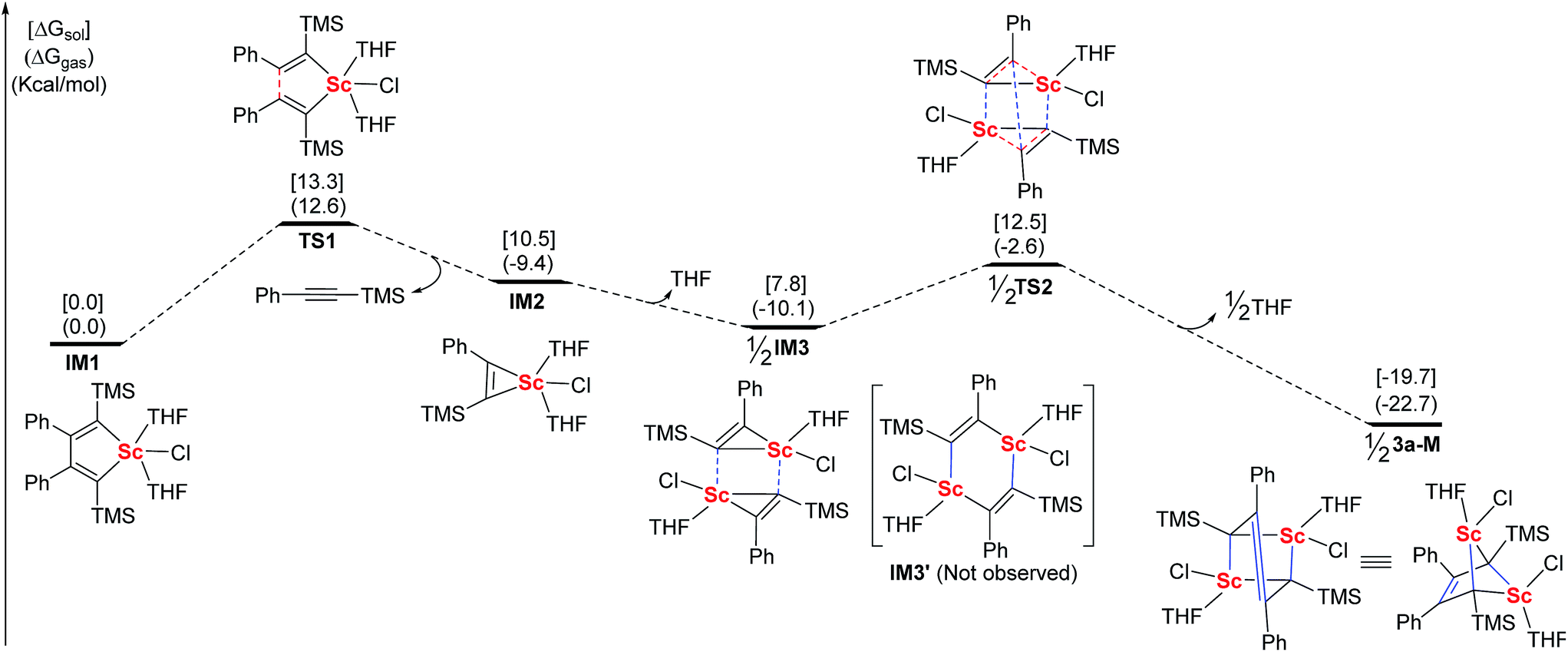 | ||
| Fig. 3 DFT calculated energy profiles of related intermediates and transition-states in the generation of 3a-M (red lines: broken bonds; blue lines: newly formed bonds). | ||
The structure of 3a features the 2-butene-1,1,4,4-tetraanion moiety and thus we thought it could be oxidized to generate the diene moiety in 2a, as illustrated in Scheme 3. As we expected, 2a was generated by treatment with two equivalents of hexachloroethane as an oxidant (Scheme 3a). This reaction resulted in the formation of ScCl3 which can be characterized as a ScCl3(THF)3 adduct by X-ray analysis, along with two equivalents of tetrachloroethylene which were identified using the 13C NMR spectrum and GC-MS. When four equivalents of hexachloroethane were used and the reaction mixture was heated at 80 °C, 3a was transformed to PhCCTMS and ScCl3 (Scheme 3b). Furthermore, when disulfide 5 served as an oxidant,26 the reaction of 3a with 5 provided complex 6 (see ESI† for the X-ray structure of 6, Scheme 3c) along with the formation of PhCCTMS. When 3a was treated with cyclooctatetraene at 80 °C, cyclooctatetraene was reduced to the cyclooctatetraene dianion. The corresponding complex 7 (see ESI† for the X-ray structure of 7, Scheme 3d) could be isolated after being recrystallized in DME (DME = 1,2-dimethoxyethane) in high yields along with the formation of PhCCTMS. These results clearly show that the bridged bis-alkylidene scandium(III) complex 3a can act as an efficient two-electron or four-electron reductant.
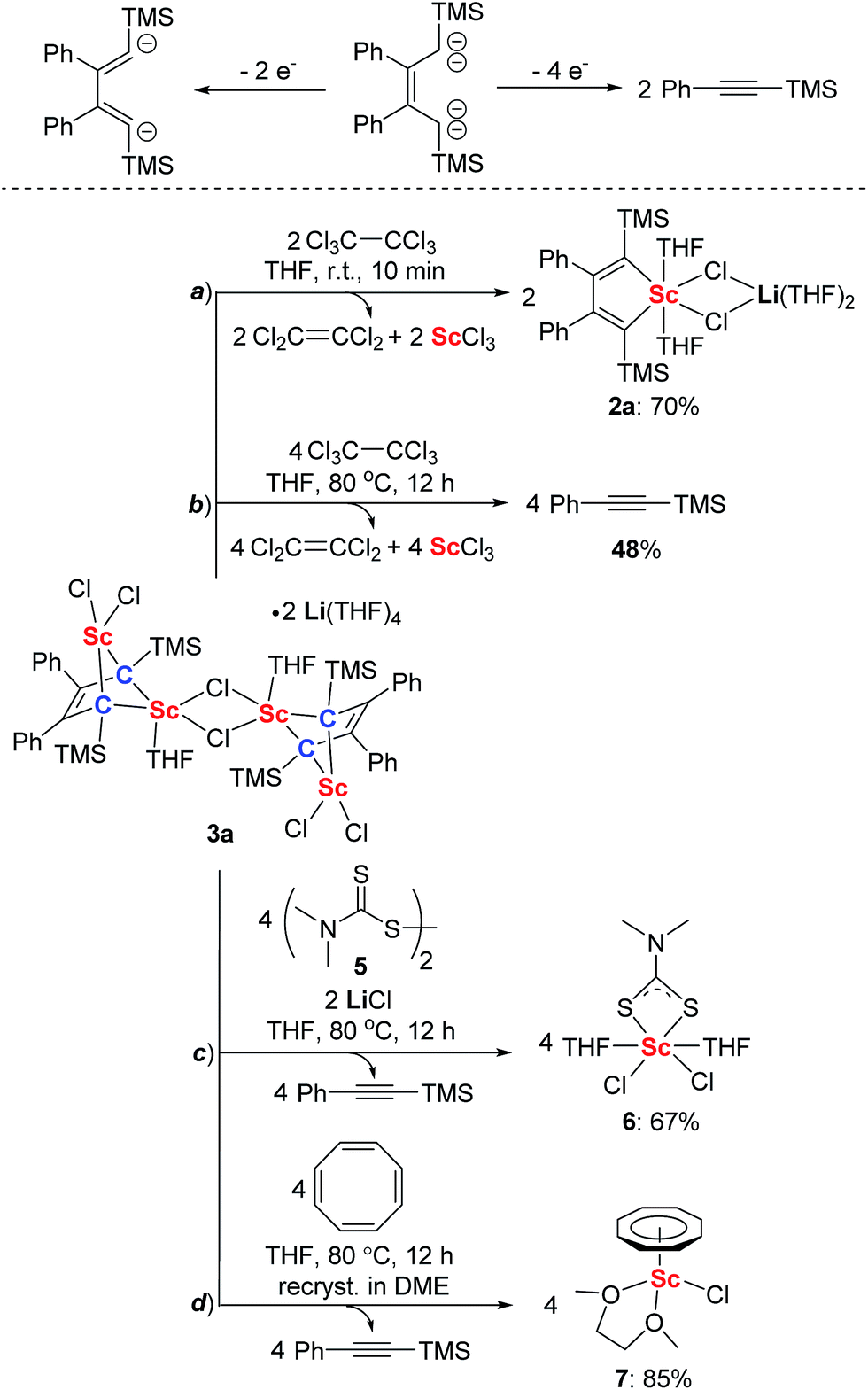 | ||
| Scheme 3 Ligand-based reduction reactivity of 3a towards different oxidants. | ||
Conclusions
In summary, we have developed a simple and efficient synthetic method for the first series of well-defined bridged bis-alkylidene scandium(III) complexes from 1,4-dilithio-1,3-butadienes and ScCl3. This reaction proceeds via two key intermediates: an isolable scandacyclopentadiene and a proposed scandacyclopropene. A mechanistic pathway of C–C bond recombination through the dimerization of scandacyclopropene intermediates is elucidated well by DFT calculations. Bridged bis-alkylidene scandium(III) complexes are found to show ligand-based reduction reactivity towards different kinds of oxidant. Further reaction chemistry of bis-alkylidene scandium(III) complexes and characterization of scandacyclopropenes are in progress.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (no. 21572005, 21372014).Notes and references
- (a) R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS PubMed; (b) R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed; (c) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS PubMed; (d) R. R. Schrock, J. Am. Chem. Soc., 1974, 96, 6796–6797 CrossRef CAS; (e) E. O. Fischer and A. Maasböl, Angew. Chem., Int. Ed., 1964, 3, 580–581 CrossRef.
- (a) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS PubMed; (b) P. L. Arnold and S. Pearson, Coord. Chem. Rev., 2007, 251, 596–609 CrossRef CAS; (c) S. T. Liddle, I. S. Edworthy and P. L. Arnold, Chem. Soc. Rev., 2007, 36, 1732–1744 RSC; (d) P. L. Arnold and S. T. Liddle, Chem. Commun., 2006, 3959–3971 RSC; (e) G. R. Giesbrecht and J. C. Gordon, Dalton Trans., 2004, 2387–2393 RSC; (f) R. G. Cavell, R. P. K. Babu and K. Aparna, J. Organomet. Chem., 2001, 617–618, 158–169 CrossRef CAS.
- H. Schumann and J. Müller, J. Organomet. Chem., 1979, 169, C1–C4 CrossRef CAS.
- K. Aparna, M. Ferguson and R. G. Cavell, J. Am. Chem. Soc., 2000, 122, 726–727 CrossRef CAS.
- D. P. Mills, L. Soutar, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2010, 132, 14379–14381 CrossRef CAS PubMed.
- M. Fustier, X. F. Le Goff, P. Le Floch and N. Mézailles, J. Am. Chem. Soc., 2010, 132, 13108–13110 CrossRef CAS PubMed.
- S. Li, M. Wang, B. Liu, L. Li, J. Cheng, C. Wu, D. Liu, J. Liu and D. Cui, Chem.–Eur. J., 2014, 20, 15493–15498 CrossRef CAS PubMed.
- (a) W. Mao, L. Xiang, C. A. Lamsfus, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2017, 139, 1081–1084 CrossRef CAS PubMed; (b) C. Wang, J. Zhou, X. Zhao, L. Maron, X. Leng and Y. Chen, Chem.–Eur. J., 2016, 22, 1258–1261 CrossRef CAS PubMed; (c) J. Zhou, T. Li, L. Maron, X. Leng and Y. Chen, Organometallics, 2015, 34, 470–476 CrossRef CAS.
- H. M. Dietrich, K. W. Törnroos and R. Anwander, J. Am. Chem. Soc., 2006, 128, 9298–9299 CrossRef CAS PubMed.
- (a) W. Huang, C. T. Carver and P. L. Diaconescu, Inorg. Chem., 2011, 50, 978–984 CrossRef CAS PubMed; (b) R. Litlabø, M. Zimmermann, K. Saliu, J. Takats, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2008, 47, 9560–9564 CrossRef PubMed; (c) M. Zimmermann, J. Takats, G. Kiel, K. W. Törnroos and R. Anwander, Chem. Commun., 2008, 612–614 RSC; (d) J. Scott, H. Fan, B. F. Wicker, A. R. Fout, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2008, 130, 14438–14439 CrossRef CAS PubMed.
- J. Hong, L. Zhang, X. Yu, M. Li, Z. Zhang, P. Zheng, M. Nishiura, Z. Hou and X. Zhou, Chem.–Eur. J., 2011, 17, 2130–2137 CrossRef CAS PubMed.
- (a) W.-X. Zhang, Z. Wang, M. Nishiura, Z. Xi and Z. Hou, J. Am. Chem. Soc., 2011, 133, 5712–5715 CrossRef CAS PubMed; (b) T. Li, M. Nishiura, J. Cheng, Y. Li and Z. Hou, Chem.–Eur. J., 2012, 18, 15079–15085 CrossRef CAS PubMed.
- For a selected review and book see: (a) W. J. Evans, Inorg. Chem., 2007, 46, 3435–3449 CrossRef CAS PubMed; (b) R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley Interscience, New York, 2014 Search PubMed.
- Selected examples of multielectron redox reactions of rare-earth metal complexes: (a) Y. Lv, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2013, 135, 14784–14796 CrossRef CAS PubMed; (b) C. Camp, V. Guidal, B. Biswas, J. Pécaut, L. Dubois and M. Mazzanti, Chem. Sci., 2012, 3, 2433–2448 RSC; (c) W. Huang, S. I. Khan and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 10410–10413 CrossRef CAS PubMed.
- Selected reviews on sterically induced reduction (SIR) of lanthanides: (a) W. J. Evans and B. L. Davis, Chem. Rev., 2002, 102, 2119–2136 CrossRef CAS PubMed; (b) W. J. Evans, J. Organomet. Chem., 2002, 647, 2–11 CrossRef CAS; (c) W. J. Evans, Coord. Chem. Rev., 2000, 206–207, 263–283 CrossRef CAS.
- Selected reviews of metallacyclopentadienes: (a) W. Ma, C. Yu, T. Chen, L. Xu, W.-X. Zhang and Z. Xi, Chem. Soc. Rev., 2017, 46, 1160–1192 RSC; (b) X. Yan and C. Xi, Acc. Chem. Res., 2015, 48, 935–946 CrossRef CAS PubMed; (c) T. Takahashi and Y. Li, in Titanium and Zirconium in Organic Synthesis, ed. I. Marek, Wiley-VCH, Weinheim, 2002, ch. 2 Search PubMed.
- Examples of rare-earth metallacyclopentadienes: (a) L. Xu, Y. Wang, Y.-C. Wang, Z. Wang, W.-X. Zhang and Z. Xi, Organometallics, 2016, 35, 5–8 CrossRef CAS; (b) L. Xu, J. Wei, W.-X. Zhang and Z. Xi, Chem.–Eur. J., 2015, 21, 15860–15866 CrossRef CAS PubMed; (c) L. Xu, Y.-C. Wang, J. Wei, Y. Wang, Z. Wang, W.-X. Zhang and Z. Xi, Chem.–Eur. J., 2015, 21, 6686–6689 CrossRef CAS PubMed.
- Selected reviews on metallacyclopropenes: (a) K. D. J. Parker and M. D. Fryzuk, Organometallics, 2015, 34, 2037–2047 CrossRef CAS; (b) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2003, 22, 884–900 CrossRef CAS.
- Selected examples of metallacyclopropenes: (a) L. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, J. Am. Chem. Soc., 2016, 138, 5130–5142 CrossRef CAS PubMed; (b) E. D. Jemmis, S. Roy, V. V. Burlakov, H. Jiao, M. Klahn, S. Hansen and U. Rosenthal, Organometallics, 2010, 29, 76–81 CrossRef CAS.
- (a) Z. Xi, Acc. Chem. Res., 2010, 43, 1342–1351 CrossRef CAS PubMed; (b) W.-X. Zhang and Z. Xi, Org. Chem. Front., 2014, 1, 1132–1139 RSC.
- (a) N. R. Halcovitch and M. D. Fryzuk, Organometallics, 2013, 32, 5705–5708 CrossRef CAS; (b) J. Oyamada, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 10720–10723 CrossRef CAS PubMed; (c) J. R. Hagadorn and J. Arnold, Organometallics, 1996, 15, 984–991 CrossRef CAS; (d) Y. Mu, W. E. Piers, D. C. MacQuarrie, M. J. Zaworotko and V. G. Young Jr, Organometallics, 1996, 15, 2720–2726 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09 (Revision C.01), Gaussian, Inc., Wallingford CT, 2010, for full reference, see ESI.†.
- The Sc–O interaction is much stronger than the Sc–Cl interaction because of the oxophilicity of rare-earth elements. In THF, LiCl in 2a will be easily replaced by THF to yield a LiCl-free complex IM1. Based on the effects of the THF atmosphere, we excluded LiCl from the calculation. In THF, 3a tends to be a monomer due to the solvent coordination interaction. Furthermore, the bond lengths and angles of the calculated monomeric structure are similar to those of the crystal dimeric structure. Thus, we think the calculation of monomer 3a-M is enough to describe the reaction pathway.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- (a) C. Üffing, A. Ecker, R. Köppe, K. Merzweiler and H. Schnöckel, Chem.–Eur. J., 1998, 4, 2142–2147 CrossRef; (b) H. Hoberg, V. Gotor, A. Milchereit, C. Krüger and J. C. Sekutowski, Angew. Chem., Int. Ed., 1977, 16, 539 CrossRef.
- I. V. Basalov, D. M. Lyubov, G. K. Fukin, A. S. Shavyrin and A. A. Trifonov, Angew. Chem., Int. Ed., 2012, 51, 3444–3447 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1555495, 1504242, 1519012 and 1539530. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02018j |
| This journal is © The Royal Society of Chemistry 2017 |