
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt-catalysed reductive C–H alkylation of indoles using carboxylic acids and molecular hydrogen†
Jose R.
Cabrero-Antonino
 ,
Rosa
Adam
,
Kathrin
Junge
and
Matthias
Beller
*
,
Rosa
Adam
,
Kathrin
Junge
and
Matthias
Beller
*
Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de
First published on 26th July 2017
Abstract
The direct CH-alkylation of indoles using carboxylic acids is presented for the first time. The catalytic system based on the combination of Co(acac)3 and 1,1,1-tris(diphenylphosphinomethyl)-ethane (Triphos, L1), in the presence of Al(OTf)3 as co-catalyst, is able to perform the reductive alkylation of 2-methyl-1H-indole with a wide range of carboxylic acids. The utility of the protocol was further demonstrated through the C3 alkylation of several substituted indole derivatives using acetic, phenylacetic or diphenylacetic acids. In addition, a careful selection of the reaction conditions allowed to perform the selective C3 alkenylation of some indole derivatives. Moreover, the alkenylation of C2 position of 3-methyl-1H-indole was also possible. Control experiments indicate that the aldehyde, in situ formed from the carboxylic acid hydrogenation, plays a central role in the overall process. This new protocol enables the direct functionalization of indoles with readily available and stable carboxylic acids using a non-precious metal based catalyst and hydrogen as reductant.
Introduction
Indoles are privileged scaffolds, acting as building blocks in multiple natural products.1 In fact, the essential amino acid tryptophan and the neurotransmitter serotonin contain the indole moiety in their structure. Inspired by nature, chemists have designed a myriad of indole based compounds with biological activities for its use as drugs or agrochemicals.2 For example, indometacin, sumatriptan or fluvastatin are currently marketed as drugs for the treatment of inflammation, migraine or hipercholesterolemia, respectively (Fig. 1). Hence, the development of new methodologies for the derivatization of this class of heterocycles continues to be an important topic in organic synthesis and catalysis.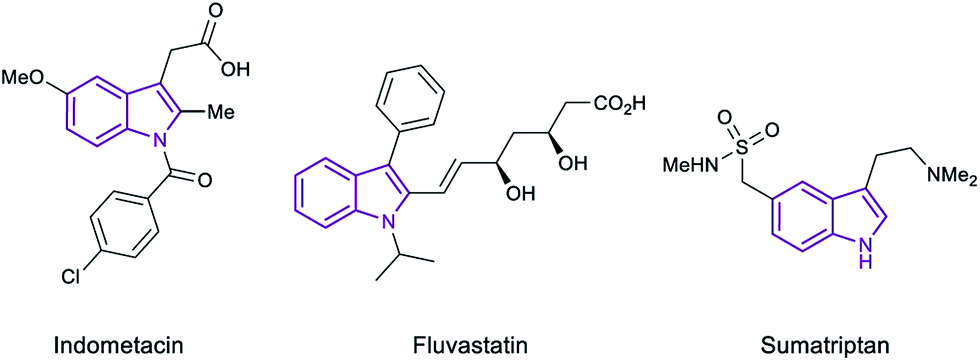 | ||
| Fig. 1 Examples of indole-based marketed drugs. | ||
Traditionally, the cyclisation of pre-functionalized benzoid precursors is the method of choice for the synthesis of substituted indoles.3 Since the development of the useful Fisher indole synthesis in the 19th century,4 many other catalytic and non-catalytic cyclisation protocols have been described along the years.5 In contrast, the direct functionalization of indole has emerged more recently as a preferred methodology as it is more practical and step economical.6 Among the several approaches to the direct substitution of indoles, transition metal catalysed CH activations are attractive.5n,7 While positions C2 and C3 are the most activated ones, direct C3 alkylations are still limited. Therefore, allylic alkylations,8 Baylis–Hillman9 and more frequently Friedel–Crafts type reactions10 have been especially useful to achieve this transformation. Intimately related with Friedel–Crafts functionalizations are reductive alkylations. Thus, reactions at indole C3 position have been successfully performed employing aldehydes or ketones11 in the presence of silanes,12 hydrogen13 or other reductants.14 Despite being a practical transformation, the use of carbonyl compounds limits its applicability as they are sometimes not easily available or undergo unwanted side-reactions, e.g., aldol condensations. Hence, the use of more stable carboxylic acids can be more convenient. However, to the best of our knowledge, this strategy has not been explored and only an example of a related indole methylation using CO2 was described by our group in 2014.15
Selective hydrogenation of carboxylic acids is a challenging transformation of interest for organic synthesis and catalysis. The first homogeneously catalysed examples of this reaction employed a ruthenium16 or an iridium17 based complex. Nowadays, the substitution of precious metals by earth abundant non-noble metals, less toxic and expensive, is an exciting goal in catalysis.18 In this direction, many examples of homogeneous hydrogenation catalysts based on cobalt have been reported recently.19,20 In 2015, the groups of de Bruin and Elsevier described the first base metal catalyst able to hydrogenate carboxylic acids using a system composed by [Co(BF4)2·6H2O/Triphos (L1)].21 Later on, our group reported the CO2 hydrogenation to methanol using a modified related catalyst [Co(acac)3/Triphos (L1)/HNTf2].22 Inspired by these precedents, we envisaged the development of a methodology for the alkylation of indole directly using carboxylic acids with a cobalt based catalytic system. Here we describe the first general reductive alkylation of indole C3 position with a variety of carboxylic acids).
Results and discussion
As a starting point of this project, we selected the reductive C–H alkylation of 2-methyl-1H-indole 1a with acetic acid 2a (4 eq. respect to 1a) as benchmark reaction (Table 1). Notably, functionalization of 2-substituted indoles is an interesting task for medicinal chemistry due to increased metabolic stability of the resulting products. Inspired by the previously known catalytic systems vide supra, Co(BF4)2·6H2O and Co(acac)3 (4 mol%) in combination with 1,1,1-tris(diphenylphosphinomethyl)-ethane, so-called Triphos (ligand L1, 2 eq. to Co), were tested under 60 bar of H2, at 160 °C, using THF as solvent during 18 h (Table 1, entries 1 and 2). Unfortunately, no activity was observed. Previous works involving carboxylic acid derivatives hydrogenation catalysed by a Ru/Triphos system showed the crucial effect of an acid additive for the catalytic activity.16a,23 Usually this additive provides a weakly coordinating counter anion, able to stabilize the active complex, as well as the optimal reaction medium pKa for the hydrogenation events to take place. Recently, we showed a similar effect in the Co/Triphos catalysed CO2 hydrogenation to methanol, where the presence of HNTf2 is required to form the catalytic active species.22 Hence, we decided to explore the reductive alkylation of indole 1a with the Co/Triphos system in the presence of an external acid additive.|
|
|||||
|---|---|---|---|---|---|
| Entrya | 2a (eq.) | [Co] | Additive | Conv.b (%) | 3a (%) |
| a Standard reaction conditions: 2-methyl-1H-indole 1a (67.0 mg, 0.5 mmol), Co precatalyst (0.02 mmol, 4 mol%), Triphos L1 (25.0 mg, 0.04 mmol, 8 mol%, 2 eq. to Co), additive (0.05 mmol, 10 mol%, 2.5 eq. to Co), THF (2 mL), acetic acid 2a (0.75–1.25 mmol, 1.5–2.5 eq.) and H2 (60 bar) at 160 °C. b Conversions of 1a and yields of product 3a were calculated by GC using hexadecane as internal standard. c Run with 2 mol% of Co(acac)3, 4 mol% of ligand L1 (2 eq. to Co) and 5 mol% of Al(OTf)3 (2.5 eq. to Co). d Run at 140 °C. e Run at 40 bar of H2. f Run at 30 bar of H2. g Run at 15 bar of H2. h Run without ligand L1. | |||||
| 1 | 4 | Co(BF4)2·6H2O | — | 15 | — |
| 2 | 4 | Co(acac)3 | — | — | — |
| 3 | 4 | Co(acac)3 | HNTf2 | 89 | 42 |
| 4 | 4 | Co(acac)3 | Al(OTf)3 | >99 | 52 |
| 5 | 2 | Co(acac)3 | Al(OTf)3 | >99 | 69 |
| 6c | 2 | Co(acac)3 | Al(OTf)3 | 74 | 53 |
| 7 | 1.5 | Co(acac)3 | Al(OTf)3 | 89 | 62 |
| 8d | 4 | Co(acac)3 | Al(OTf)3 | >99 | 44 |
| 9d | 3 | Co(acac)3 | Al(OTf)3 | 91 | 48 |
| 10e | 2 | Co(acac)3 | Al(OTf)3 | >99 | 68 |
| 11e | 1.75 | Co(acac)3 | Al(OTf)3 | >99 | 68 |
| 12e | 1.5 | Co(acac)3 | Al(OTf)3 | 94 | 67 |
| 13 | 1.75 | Co(acac) 3 | Al(OTf) 3 | >99 | 70 |
| 14f | 1.5 | Co(acac)3 | Al(OTf)3 | 90 | 63 |
| 15g | 1.75 | Co(acac)3 | Al(OTf)3 | 94 | 67 |
| 16h | 1.75 | Co(acac)3 | Al(OTf)3 | 35 | — |
Gratifyingly, moderate yields of 3-ethyl-2-methyl-1H-indole 3a were detected when catalytic amounts of HNTf2 or Al(OTf)3 (10 mol%, 2.5 eq. to Co) were added to the [Co(acac)3/Triphos (L1)] mixture (42–52%, Table 1, entries 3 and 4, respectively). Surprisingly, no traces of N-alkylated products or bis(indole) derivatives were observed in the reaction mixtures by GC analysis.
Next, a more detailed investigation regarding the effect of alkylating agent 2a amount, catalyst loading, pressure and temperature on the catalytic activity was carried out (Table 1, entries 5–15). In general, high conversions of 1a were detected, although only moderate yields of the desired C3-alkylated product 3a were obtained, indicating some degradation of the indoles. Nevertheless, the yield of the desired C3-alkylated indole derivative 3a could be improved to 70% using 1.75 eq. of acetic acid 2a under 30 bar of hydrogen and 160 °C (Table 1, entry 13). Milder reaction temperatures or lower catalysts loadings and hydrogen pressures did not afford better yields of 3a (Table 1, entries 6, 8, 9 and 15). In the absence of ligand L1, no desired product was detected and only some degradation of 1a was observed (Table 1, entry 16).
At this point we became interested in studying the influence of the acid co-catalyst. Fig. 2 (up) shows that, among the different Brönsted and Lewis acid additives tested for the CH-alkylation of the indole 1a, aluminium(III) trifluoromethanesulfonate [Al(OTf)3] afforded the best yield of 3a (70%). Other Lewis acid additives such as In(OTf)3, Ga(OTf)3 or Sn(OTf)2 also promoted the desired transformation and gave yields of alkylated product 3a above 60%. In order to improve the model reaction further on, we varied the relative Al(OTf)3 amount (with respect to the cobalt precatalyst). As shown in Fig. 2 (bottom) 10 mol% of Al(OTf)3 (2.5 eq. to Co) gave the optimal yield (70% of 3a).
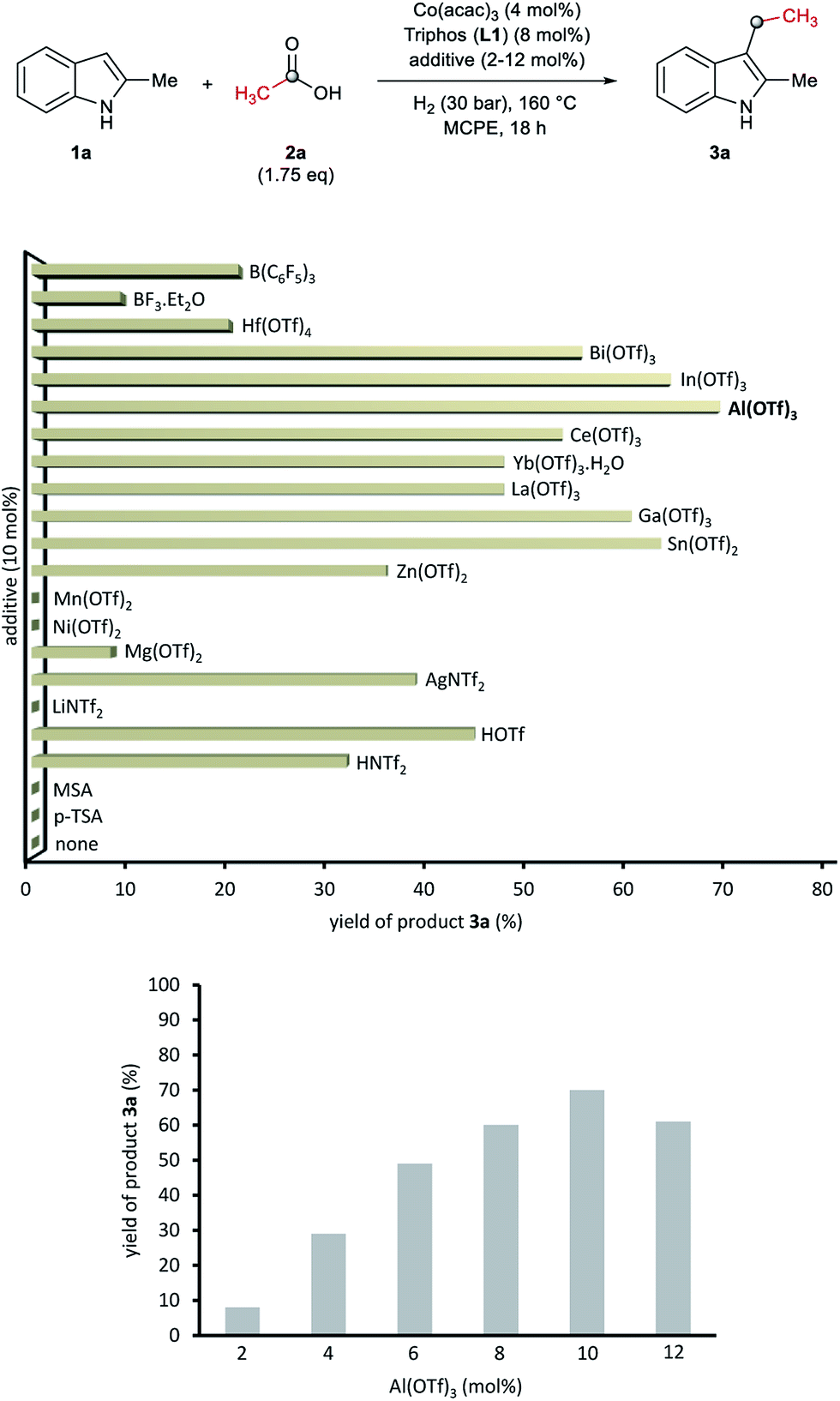 | ||
| Fig. 2 Up: Testing of additives in the cobalt-catalysed reductive C–H alkylation of 2-methyl-1H-indole 1a with acetic acid 2a and molecular hydrogen. Bottom: Influence of the Al(OTf)3 loading in the formation of product 3a from 1a and 2a. Standard reaction conditions: 2-methyl-1H-indole 1a (67.0 mg, 0.5 mmol), Co(acac)3 (7.2 mg, 0.02 mmol, 4 mol%), Triphos L1 (25.0 mg, 0.04 mmol, 8 mol%, 2 eq. to Co), additive (2–12 mol%, 0.5–3 eq. to Co), THF (2 mL), acetic acid 2a (50.3 μL, 0.875 mmol, 1.75 eq.) and H2 (30 bar) at 160 °C. The yields of product 3a were calculated by GC using hexadecane as internal standard. | ||
Next, the influence of the solvent was investigated in detail (see ESI, Table S1†). The presence of water (10% v/v respect to THF) was detrimental for the activity of the catalytic system, affording very poor yields of product 3a (7%, Table S1,† entry 2). In general, other ether-type solvents gave similar results compared to THF (Table S1,† entries 2–8), being methyl cyclopentyl ether (MCPE) the one that gave the best result (Table S1,† entry 6, 83% yield 3a). Also in toluene a good yield of the desired indole 3a was obtained (76%, Table S1,† entry 9). In contrast, the catalyst was totally inactive when DMF was used (Table S1,† entry 10).
To our delight, the employment of MCPE allowed to use a lower catalyst loading (2 mol% of cobalt) with excellent results (Table 2, entry 5, 89% yield of 3a). Blank experiments revealed that the presence of all the three components of the catalytic system, [Co(acac)3/Triphos (L1)/Al(OTf)3], are required for the reductive alkylation of indole (Table 2, entries 6–10). In addition, these experiments showed that the partial degradation of 1a can be attributed mainly to the presence of the acid additive (Table 2, entries 7 and 10).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | T (°C) | H2 (bar) | 2a (eq.) | [Co] (mol%) | Conv.b (%) | 3a (%) |
| a Standard reaction conditions: 2-methyl-1H-indole 1a (67.0 mg, 0.5 mmol), Co(acac)3 (1–4 mol%), Triphos L1 (2–8 mol%, 2 eq. to Co), Al(OTf)3 (2.5–10 mol%, 2.5 eq. to Co), MCPE (2 mL), acetic acid 2a (0.75–1.25 mmol, 1.5–2.5 eq.) and H2 (15–60 bar) at 140–160 °C. b Conversion of 1a and yield of product 3a were calculated by GC using hexadecane as internal standard. c Run without ligand L1 and Al(OTf)3. d Run without ligand L1. e Run without Al(OTf)3. f Run with 3 mol% of ligand L1 (1.5 eq. to Co). g Run with 2 mol% of ligand L1 (1 eq. to Co). h Run with 30 bar of N2. i Run at 6 h. | ||||||
| 1 | 160 | 30 | 1.75 | 4 | >99 | 83 |
| 2 | 160 | 30 | 1.5 | 4 | >99 | 79 |
| 3 | 160 | 30 | 1.25 | 4 | 86 | 62 |
| 4 | 160 | 30 | 2.5 | 2 | >99 | 85 |
| 5 | 160 | 30 | 1.75 | 2 | >99 | 89 |
| 6c | 160 | 30 | 1.75 | 2 | 24 | — |
| 7d | 160 | 30 | 1.75 | 2 | 57 | — |
| 8e | 160 | 30 | 1.75 | 2 | 14 | — |
| 9c | 160 | 30 | 1.75 | — | 12 | — |
| 10d | 160 | 30 | 1.75 | — | 54 | — |
| 11f | 160 | 30 | 1.75 | 2 | 64 | 42 |
| 12g | 160 | 30 | 1.75 | 2 | 47 | — |
| 13 | 160 | 30 | 1.5 | 2 | 87 | 76 |
| 14c | 160 | 30 | 2.5 | 1 | 47 | 31 |
| 15d | 160 | 30 | 1.75 | 1 | 40 | 23 |
| 16 | 160 | 15 | 1.75 | 2 | 56 | 27 |
| 17h | 160 | — | 1.75 | 2 | 15 | — |
| 18 | 140 | 30 | 1.75 | 4 | 96 | 68 |
| 19 | 140 | 30 | 2.5 | 4 | 96 | 70 |
| 20 | 140 | 60 | 1.75 | 2 | 69 | 41 |
| 21 | 140 | 60 | 2.5 | 2 | 83 | 49 |
| 22i | 160 | 30 | 1.75 | 2 | 70 | 64 |

Notably, the relative amounts of ligand L1 with respect to the cobalt precursor had a significant influence on the product yield. When the reaction was conducted using 1.5 and 1 equivalents of ligand, moderate yields (42%) of 3a or no product were detected, respectively (Table 2, entries 11 and 12), showing that 2 eq. of Triphos (L1) are the minimum required to perform the reaction efficiently.
Next, the catalytic activity of different metal pre-catalysts was evaluated under the optimized reaction conditions (30 bar of H2, 160 °C, 1.75 eq. of 2a, 2 mol% of cobalt and 18 h; see Table 1 and ESI, Fig. S1†). Among the different Co(III), Co(II), Ru(III), Mn(III), Fe(III) and Cu(II) acetylacetonate complexes tested, Co(acac)3 was the one that gave the best yields of the desired alkylated indole 3a (89%, see ESI, Fig. S1†). In addition, Co(acac)2·H2O afforded a slightly lower yield (78% yield of 3a, Fig. S1†), while other related metal salts were not active. These results indicate that cobalt is unique for this reaction.
Among the different cobalt-based precatalysts containing counteranions such as [BF4−], [ClO4−], [OAc−], [F−], [NO3−] and [SO42−] (see ESI, Fig. S1†) tested for the alkylation of indole 1a, only Co(BF4)2·6H2O and Co(SO4)·7H2O promoted the formation of the product 3a, albeit in lower yields (21% and 8% yield of 3a, respectively, Fig. S1†).
With regard to the ligand, we compared the activity of Triphos L1 with several multidentate (L2–L11) and one monodentate ligands (L11) (see ESI, Scheme S1†). All the other tested ligands showed lower activities than Triphos (L1), and only the tridentate ligands L2 and L3 or the tetradentate L5 afforded 3-ethyl-2-methyl-1H-indole 3a, though in low yields (18%, 17% and 3%, respectively, Scheme S1†).
At this point, we decided to explore the general applicability of the cobalt-based system in the C3-alkylation of 2-methyl-1H-indole 1a using a wide range of carboxylic acids (Table 3). In some cases, higher catalyst loadings (up to 4 mol%) or excess of the alkylating agent (up to 2.5 eq.) were needed in order to achieve a total conversion of indole 1a.
|
|
||||
|---|---|---|---|---|
| Entrya | T (°C) | [Co] (mol%) | Product 3 | Yieldb (%) |
| a Standard reaction conditions: 2-methyl-1H-indole 1a (67.0 mg, 0.5 mmol), Co(acac)3 (1–4 mol%), Triphos L1 (2–8 mol%, 2 eq. to Co), Al(OTf)3 (2.5–10 mol%, 2.5 eq. to Co), MCPE (2 mL), carboxylic acid (0.875 mmol, 1.75 eq.) and H2 (30 bar) at 140–160 °C. The selectivity to the desired product was >95% in all cases. In some examples degradation problems were observed. b Yield of isolated product after column chromatography on silica. c Run with (1.75 mmol, 2.5 eq.) of carboxylic acid. d Run at 5 h. | ||||
| 1 | 160 | 2 |
|
[80] |
| 2 | 140 | 2 |
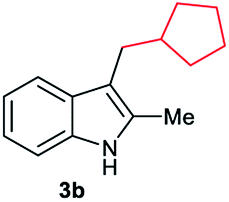
|
[80] |
| 3 | 160 | 2 |

|
[67] |
| 4 | 160 | 2 |

|
[74] |
| 5c | 140 | 2 |
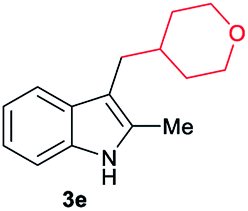
|
[59] |
| 6 | 160 | 2 |

|
[74] |
| 7c | 160 | 2 |

|
[70] |
| 8 | 140 | 4 |
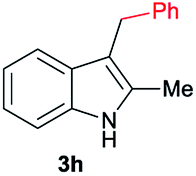
|
[75] |
| 9c | 160 | 4 |
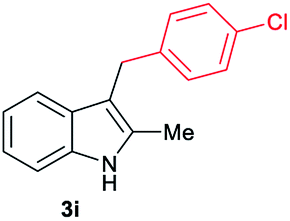
|
[64] |
| 10 | 140 | 2 |
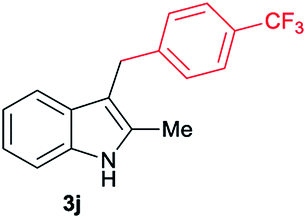
|
[49] |
| 11c | 160 | 3 |
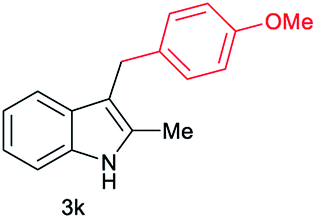
|
[46] |
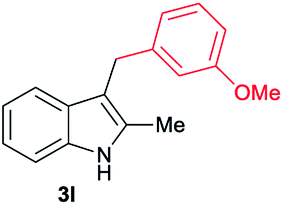
| 12 | 160 | 3 |
|
[68] |
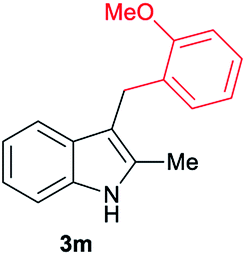
| 13d | 140 | 2 |
|
[47] |
| 14 | 140 | 1 |
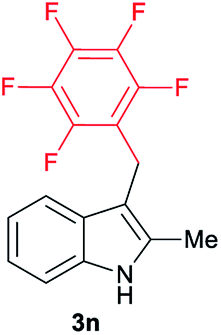
|
[35] |
| 15 | 140 | 2 |
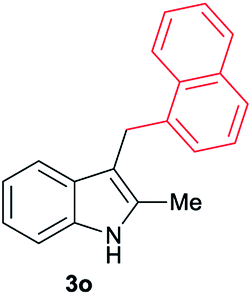
|
[73] |
| 16 | 140 | 2 |
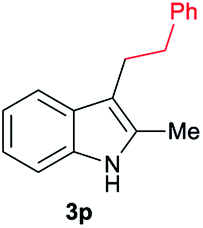
|
[78] |
| 17 | 160 | 3 |
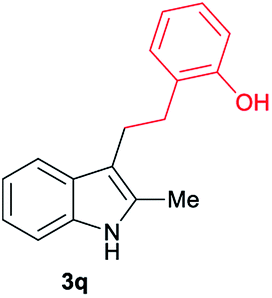
|
[70] |
| 18 | 140 | 2 |
|
[55] |
| 19 | 160 | 3 |
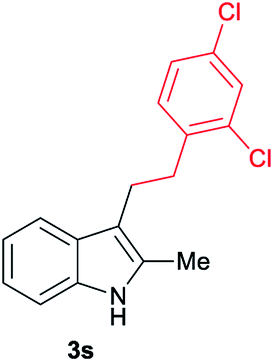
|
[72] |
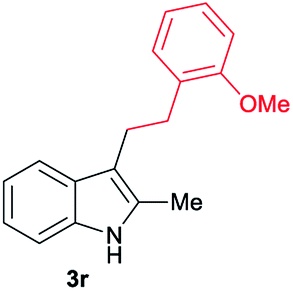
| 20 | 160 | 2 |
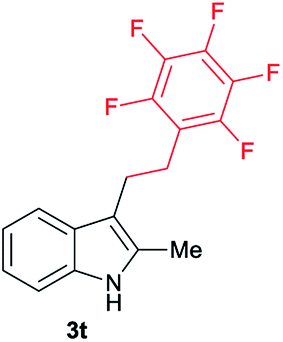
|
[42] |
| 21 | 140 | 2 |
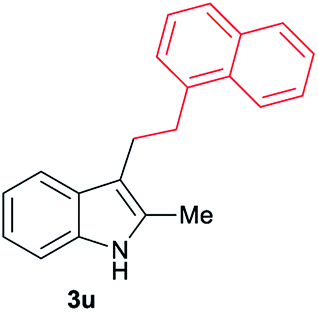
|
[64] |
| 22 | 140 | 2 |
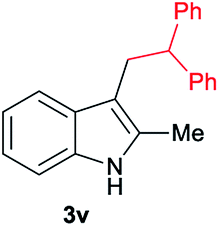
|
[70] |
| 23 | 140 | 2 |
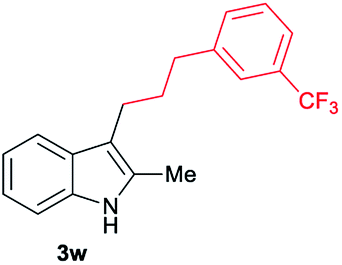
|
[72] |
| 24 | 140 | 2 |
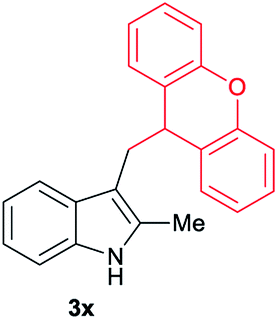
|
[55] |
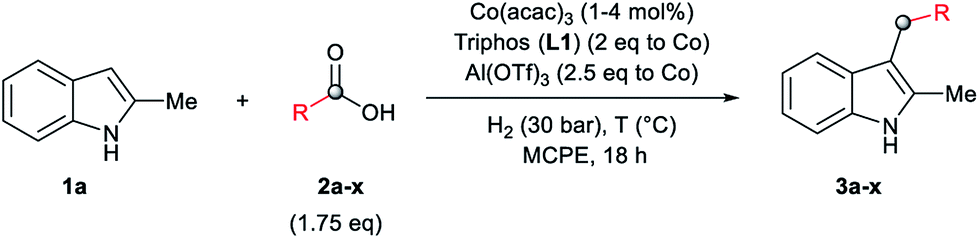
Aliphatic carboxylic acids 2a–g, including examples containing heteroatoms such as oxygen-(2e) or fluorine-(2f and 2g), afforded the corresponding C3-alkylated indoles 3a–g in good to very good isolated yields (59–80%, Table 3, entries 1–7). Moreover, benzoic acid 2h and different o-, m- and p-substituted benzoic acids containing chloro (2i), trifluoromethyl (2j) or methoxy groups (2k–m), could also be used as alkylating agents giving good isolated yields of the C3-benzylated indole derivatives 3h–m (46–75%, Table 3, entries 8–13). Gratifyingly, perfluorinated benzoic acid 2n and naphthoic acid 2o also gave the desired functionalized indole derivatives 3n and 3o in moderate to good yields after isolation (36 and 73%, Table 3, entry 14 and 15, respectively). In addition, more sensitive phenylacetic acid 2p, naphthaleneacetic acid 2u as well as 2-hydroxy-(2q), 2-methoxy-(2r), 2,4-dichloro-(2s) and perfluoro-(2t) substituted phenylacetic acids were successfully employed as alkylating agents of indole 1a (42–78%, Table 3, entries 16–21). In general, no clearly correlation between the reactivity and the electronic character of the substituent attached to the benzene ring of the carboxylic acid could be observed. Finally, diphenylacetic acid 2v, 3-(3-(trifluoromethyl)phenyl)propanoic acid 2w and 9-xanthene carboxylic acid 2x, as an example of (hetero)aromatic acid, exhibited good activity (55–70%, Table 3, entries 22–24). These examples illustrate the potential of the base metal catalysed methodology as a practical tool for introducing molecular diversity in the indole core using readily available carboxylic acids as alkylating agents.
After showing the reductive alkylation using different carboxylic acids, we investigated the reaction of several substituted indole derivatives (Scheme 1). Using the optimized conditions, a variety of indoles were reacted with acetic acid 2a, phenylacetic acid 2p or diphenylacetic acid 2v. In general, for the same indole, 2p was the most reactive carboxylic acid followed by 2v and 2a, as the less reactive one. As shown in Scheme 1, several 5-substituted 2-methyl-1H-indole derivatives – some of them previously synthetized by us (see ESI† for experimental details) – containing methyl, hydroxy, methoxy, chloro, amino, phenylamino, trifluoromethyl and thiophenyl groups were well tolerated. The desired C3-alkylated products 3y-ah were obtained in moderate to good yields after isolation (38–74%, Scheme 1). Interestingly, in the case of 5-amino-2-methyl-1H-indole reacting with acetic acid, the main product was the one corresponding to the simultaneous C3 alkylation and amidation, being possible to isolate the amide 3ad in moderate yields (38%, Scheme 1). This result indicates that the [Co(acac)3/Triphos (L1)/Al(OTf)3] catalysts exhibits selectivity towards the hydrogenation of a carboxylic acid in the presence of an amide. Moreover, 6-substituted as well as 5,7-substituted indole substrates were also successfully alkylated giving products 3ai–ak in 63–80% isolated yields. At this point it was interesting to explore the tolerance of our protocol towards different alkyl and aryl substituents in the C2 position as well as in the nitrogen atom of the indole. To our delight, the reductive alkylation of this indoles class was successfully achieved under different reaction conditions using phenylacetic acid 2p and diphenylacetic acid 2v as alkylating agents. Thus, the desired C3-alkylated products 3al–ap could be isolated in 49–70% yields (Scheme 1).
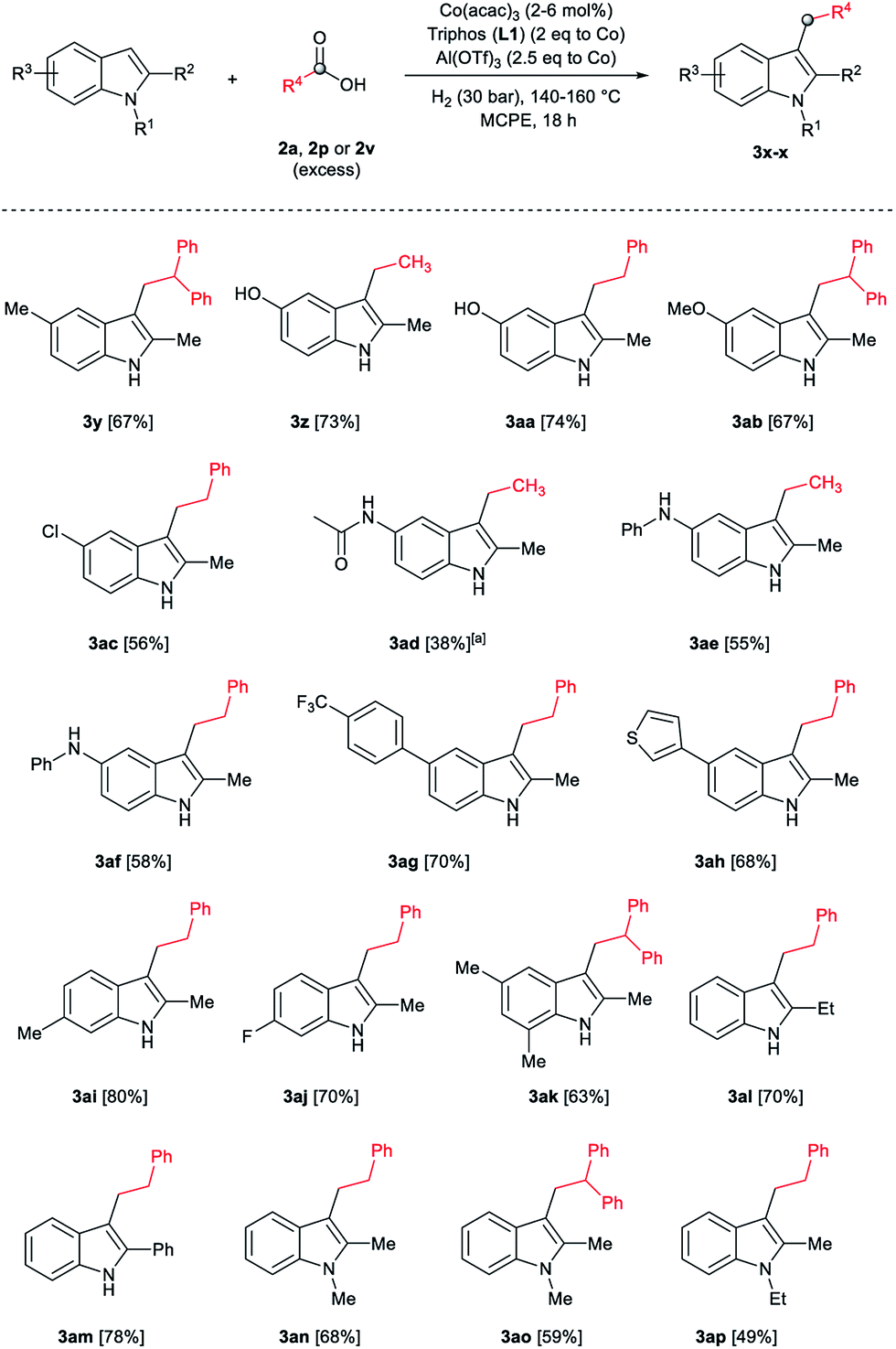 | ||
| Scheme 1 Standard reaction conditions: indole (0.5 mmol), Co(acac)3 (1–4 mol%), Triphos L1 (2–8 mol%, 2 eq. to Co), Al(OTf)3 (2.5–10 mol%, 2.5 eq. to Co), MCPE (2 mL), carboxylic acid (0.875–1.75 mmol, 1.75–3.5 eq.) and H2 (30 bar) at 140–160 °C during 18–60 h. Yield of isolated product after column chromatography on silica are given. The selectivity to the desired product, calculated by GC-MS, was >90% in all cases. In some examples degradation problems were observed. Specific reaction conditions for substrate 3y and 3ak: Co(acac)3 (4 mol%), carboxylic acid (0.875 mmol, 1.75 eq.), 160 °C, 18 h; for substrate 3z: Co(acac)3 (2 mol%), carboxylic acid (0.875 mmol, 1.75 eq.), 160 °C, 18 h; for substrates 3aa and 3ai: Co(acac)3 (2 mol%), carboxylic acid (0.875 mmol, 1.75 eq.), 140 °C, 18 h; for substrate 3ab: Co(acac)3 (3 mol%), carboxylic acid (0.875 mmol, 1.75 eq.), 160 °C, 48 h; for substrate 3ac: Co(acac)3 (6 mol%), carboxylic acid (1.5 mmol, 3.5 eq.), 160 °C, 18 h; for substrates 3ad–ae: Co(acac)3 (4 mol%), carboxylic acid (1.5 mmol, 3 eq.), 160 °C, 18 h; for substrate 3af: Co(acac)3 (3 mol%), carboxylic acid (0.875 mmol, 1.75 eq.), 160 °C, 18 h; for substrates 3ag–ah and 3aj: Co(acac)3 (5 mol%), carboxylic acid (1.25 mmol, 2.5 eq.), 160 °C, 18 h; for substrate 3al: Co(acac)3 (2 mol%), carboxylic acid (1.25 mmol, 2.5 eq.), 160 °C, 18 h; for substrate 3am: Co(acac)3 (6 mol%), carboxylic acid (1.25 mmol, 2.5 eq.), 160 °C, 18 h; for substrate 3an: Co(acac)3 (3 mol%), carboxylic acid (1.25 mmol, 2.5 eq.), 160 °C, 18 h; for substrate 3ao: Co(acac)3 (6 mol%), carboxylic acid (0.875 mmol, 1.75 eq.), H2 (60 bar), 160 °C, 60 h; for substrate 3ap: Co(acac)3 (6 mol%), carboxylic acid (1.25 mmol, 2.5 eq.), 160 °C, 18 h. [a] 2-Methyl-1H-indol-5-amine was used as starting material. | ||
As expected, in case of 2,3-dimethyl-1H-indole S1 (Scheme S2,† eqn (A)), no products were detected after its reaction with acetic acid 2a, discarding a possible N-alkylation catalysed by our system. In agreement with this observation, for 3-methyl-1H-indole 1b where the C3 position is blocked, only very low amounts of C2-ethylated product S3 (<2%) were detected (Scheme S2,† eqn (B)), confirming the lower nucleophilicity of C2 position in comparison with C3. Finally, when simple indole 1c or N-methyl derivative 1d were used, low conversions (<20%) and poor yields of the desired C3-ethylated products S3 and S5 were observed (10 and 8%, respectively, Scheme S2,† eqn (C) and (D)). Interestingly, in these two cases small amounts of bis(indole) compounds S4 and S6 (<2 and <5%, respectively, Scheme S2,† eqn (C) and (D)) were detected.
During the development of the alkylation reactions of different indoles using diphenylacetic acid 2v (see Scheme 1), we observed alkenylation of selected substrates as a side reaction.24 A fine control of the reaction parameters (temperature, pressure of hydrogen, catalyst loading, amount of alkylating agent and reaction time) allowed us to selectively stop the reaction at the alkene stage (Scheme 2). For example, 5-methyl- and 6-fluoro-2-methyl-1H-indole derivatives were selectively alkenylated (selectivity alkene vs. alkane >85%) affording the desired tri-substituted alkenes 4a and 4b in very good isolated yields (70 and 88%, respectively, Scheme 2). To the best of our knowledge such alkenylations using carboxylic acids have not described, yet.
 | ||
| Scheme 2 Cobalt-catalysed reductive selective C(3)–H diphenylvinylation of different indoles using diphenylacetic acid 2v and molecular hydrogen. Standard reaction conditions: indole (0.5 mmol), Co(acac)3 (1–6 mol%), Triphos L1 (2–12 mol%, 2 eq. to Co), Al(OTf)3 (2.5–15 mol%, 2.5 eq. to Co), MCPE (2 mL), diphenylacetic acid 2v (1.25–1.75 mmol, 2.5–3.5 eq.) and H2 (30 bar) at 120–160 °C over 18 h. Yield of isolated products after column chromatography on silica are given between brackets. Between parentheses is shown the selectivity to the desired alkene vs. alkane by-product, calculated by GC-MS. In some examples degradation problems were observed. Specific reaction conditions for substrate 4a: Co(acac)3 (2 mol%), carboxylic acid (1.75 mmol, 3.5 eq.), 120 °C; for substrate 4b: Co(acac)3 (2 mol%), carboxylic acid (1.75 mmol, 3.5 eq.), 130 °C; for substrates 4c–e: Co(acac)3 (4 mol%), carboxylic acid (1.25 mmol, 2.5 eq.), 160 °C; for substrate 4f: Co(acac)3 (6 mol%), carboxylic acid (1.75 mmol, 3.5 eq.), 160 °C; for substrate 4g: Co(acac)3 (1 mol%), carboxylic acid (2.25 mmol, 4.5 eq.), 140 °C; for substrate 4h: Co(acac)3 (4 mol%), carboxylic acid (1.75 mmol, 3.5 eq.), 120 °C. | ||
In addition, different C2-phenyl and/or nitrogen-alkyl substituted indole derivatives gave the corresponding alkene products 4c–f with high selectivities towards alkene vs. alkane (>94%) and good isolated yields (63–80%, Scheme 2). Notably, the simple indole 1c and the N-methyl substituted 1d, unreactive towards alkylation when acetic acid was used (vide supra), were also successfully vinylated in C3 position using diphenylacetic acid 2v. In these cases only traces of C2-alkenylated products were detected (<2%), observing a full selectivity to C3 vs. C2-position. In fact, C3-alkenylated products 4g and 4h were isolated in moderate to good yields (58 and 67%, respectively, Scheme 2) with selectivities alkene vs. alkane between 87–89%. Finally, despite the lower nucleophicility of the indole C2 position, it was possible to perform the cobalt catalysed C2 alkenylation of 3-methyl-1H-indole 1b by using an excess of diphenylacetic acid 2v as alkylating source. The desired C2-alkenylated product 4i could be isolated in 56% (Scheme 3). Hence, this novel non-precious metal based methodology opens the way to the development of further transformations for the C2 and C3-functionalization of indoles.
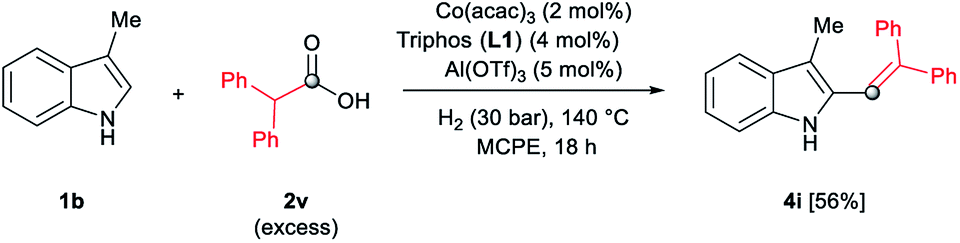 | ||
| Scheme 3 Cobalt-catalysed reductive C(2)–H diphenylvinylation of 3-methyl-1H-indole 1b using diphenylacetic acid 2v and molecular hydrogen. Standard reaction conditions: 3-methyl-1H-indole 1b (67.0 mg, 0.5 mmol), Co(acac)3 (3.6 mg, 0.01 mmol, 2 mol%), Triphos L1 (12.5 mg, 0.02 mmol, 4 mol%, 2 eq. to Co), Al(OTf)3 (11.9 mg, 0.025 mmol, 5 mol%, 2.5 eq. to Co), MCPE (2 mL), diphenylacetic acid 2v (486.0 mg, 2.25 mmol, 4.5 eq.) and H2 (30 bar) at 140 °C over 18 h. Yield of isolated product after column chromatography on silica is given. The selectivity to the alkene product 4i was 97%. | ||
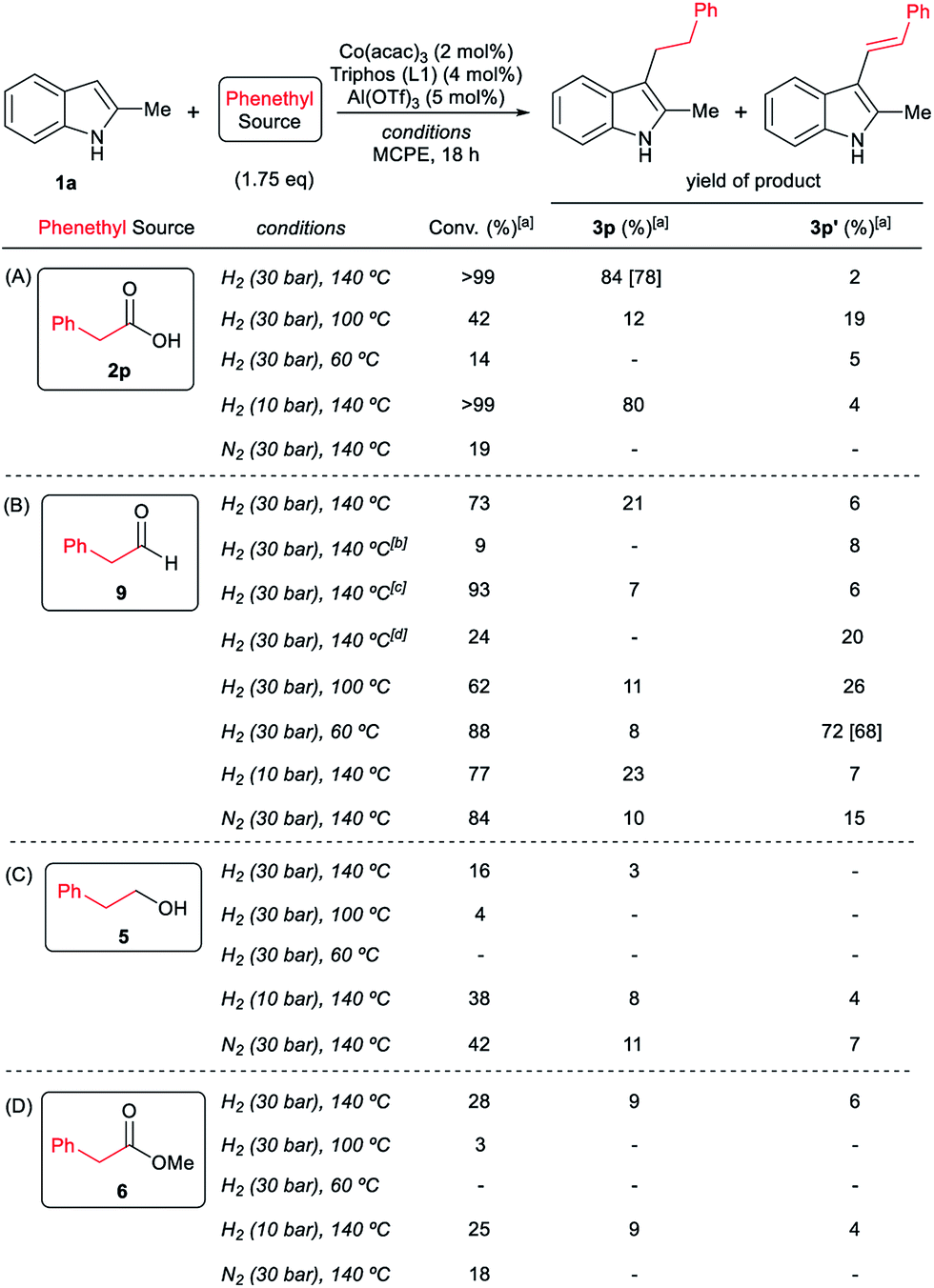
To gain insight into the mechanism of the cobalt-catalysed reductive alkylation of indoles, some kinetic studies (see Fig. 3 and 4) and control experiments were performed (see Schemes 4–6). Fig. 3 and 4 show the concentration/time profiles for the reductive alkylation of 2-methyl-1H-indole 1a with acetic acid 2a and phenylacetic acid 2p, respectively (optimized reaction conditions for each acid). It should be noted that no induction periods were observed in any of the experiments, indicating that the active catalytic species is easily formed. Interestingly, in the case of the reaction with phenylacetic acid 2p it is possible to detect low amounts of the intermediate (E)-alkene 3p′, observed in slightly higher concentrations at initial reaction times (see Fig. 4). This observation indicates that the hydrogenation of the alkene 3p′ is not the rate determining step of the process.
 | ||
| Fig. 3 Concentration/time profile for 2-methyl-1H-indole 1a (blue line) and 3-ethyl-2-methyl-1H-indole 3a (red line) in the reductive C–H alkylation of 1a with acetic acid 2a at 160 °C and 30 bar of molecular hydrogen. Standard reaction conditions: 2-methyl-1H-indole 1a (3.0 mmol, 402.0 mg), Co(acac)3 (21.6 mg, 0.06 mmol, 2 mol%), Triphos L1 (75.0 mg, 0.12 mmol, 4 mol%, 2 eq. to Co), Al(OTf)3 (71.4 mg, 0.15 mmol, 5 mol%, 2.5 eq. to Co), MCPE (12.0 mL), acetic acid 2a (301.8 μL, 5.25 mmol, 1.75 eq.) and H2 (30 bar) at 160 °C. Percentages of 1a and 3a were calculated by GC using hexadecane as internal standard. | ||
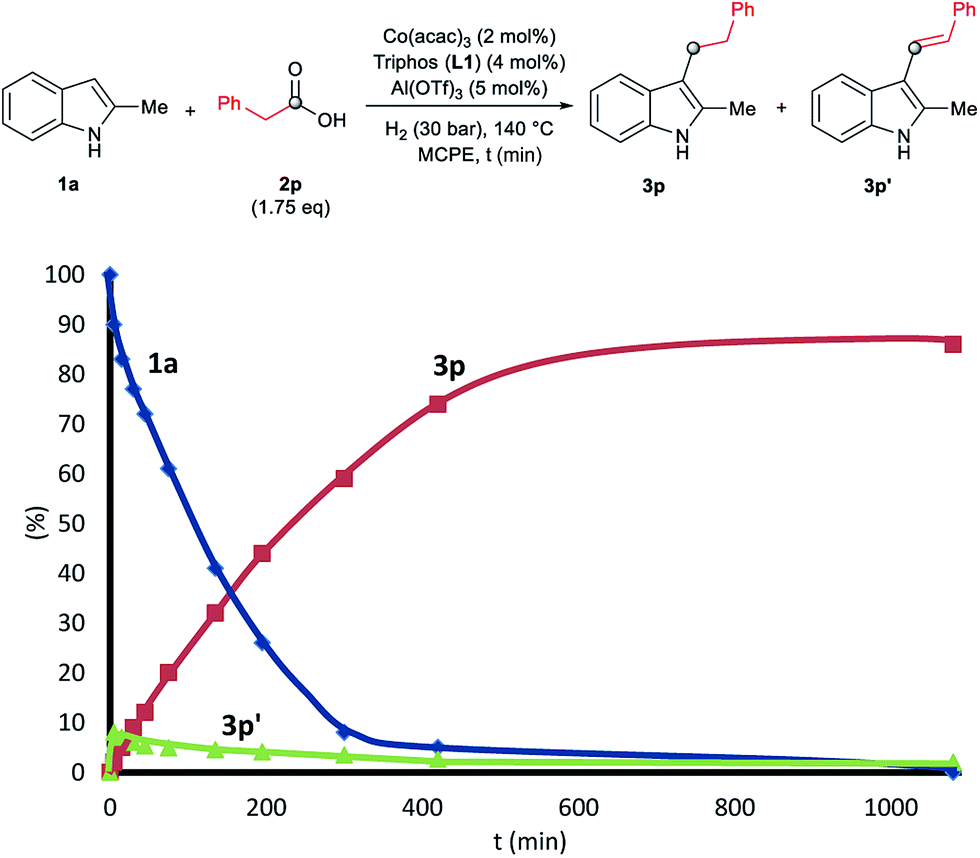 | ||
| Fig. 4 Concentration/time profile for 2-methyl-1H-indole 1a (blue line), 2-methyl-3-phenethyl-1H-indole 3p (red line) and (E)-2-methyl-3-styryl-1H-indole 3p′ (green line) in the reductive C–H alkylation of 1a with phenylacetic acid 2p at 140 °C and 30 bar of molecular hydrogen. Standard reaction conditions: 2-methyl-1H-indole 1a (3.0 mmol, 402.0 mg), Co(acac)3 (21.6 mg, 0.06 mmol, 2 mol%), Triphos L1 (75.0 mg, 0.12 mmol, 4 mol%, 2 eq. to Co), Al(OTf)3 (71.4 mg, 0.15 mmol, 5 mol%, 2.5 eq. to Co), MCPE (12.0 mL), phenylacetic acid 2p (720.0 mg, 5.25 mmol, 1.75 eq.) and H2 (30 bar) at 140 °C. Percentages of 1a, 3p and 3p′ were calculated by GC using hexadecane as internal standard. | ||
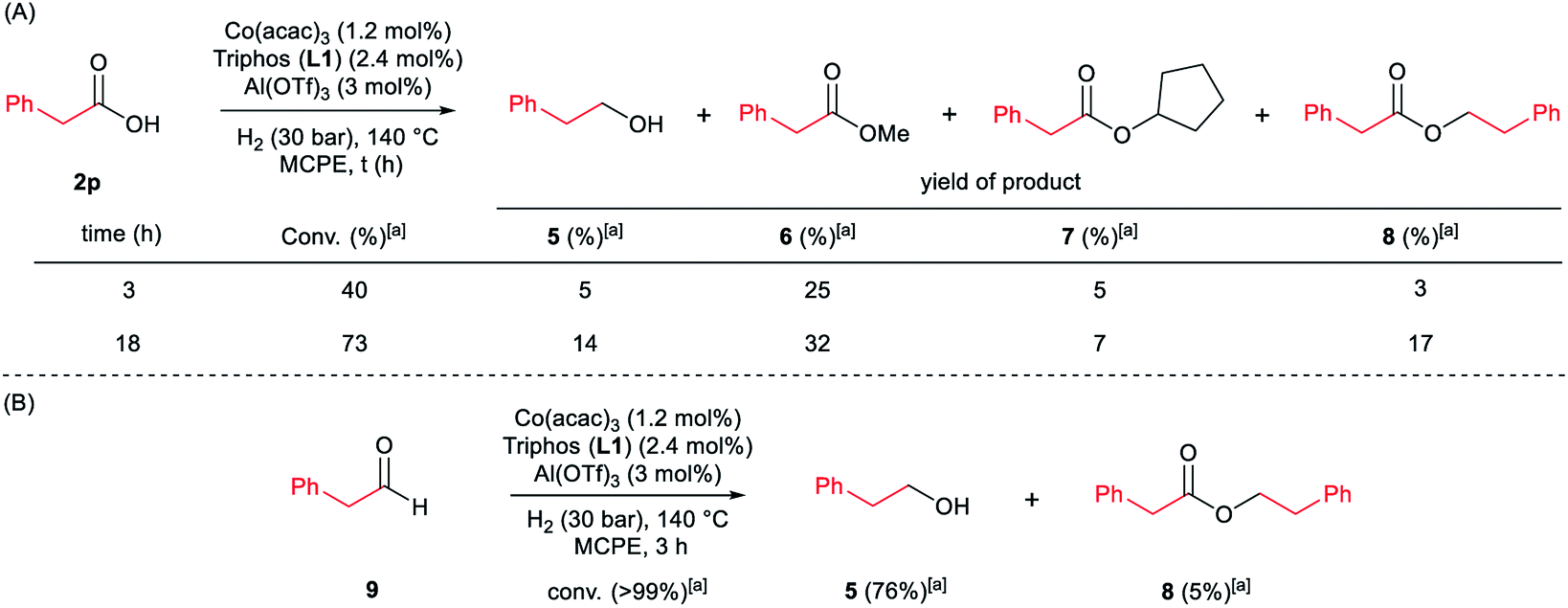 | ||
| Scheme 4 Control experiments in the hydrogenation of phenylacetic acid 2p (A) or phenylacetaldehyde 9 (B) in the absence of 2-methyl-1H-indole 1a. Standard reaction conditions: phenylacetic acid 2p or phenylacetaldehyde 9 (0.875 mmol), Co(acac)3 (3.6 mg, 0.01 mmol, 1.2 mol%), Triphos L1 (12.5 mg, 0.02 mmol, 2.4 mol%, 2 eq. to Co), Al(OTf)3 (11.9 mg, 0.025 mmol, 3 mol%, 2.5 eq. to Co), MCPE (2 mL) at 140 °C during 3–18 h. [a] Conversion of 2p and 9 and yield of products 5, 6, 7 and 8 were calculated by GC using hexadecane as internal standard. | ||
 | ||
| Scheme 5 Control experiments in the C–H phenethylation of 2-methyl-1H-indole 1a using phenylacetic acid 2p (A), phenylacetaldehyde 9 (B), phenethyl alcohol 5 (C) and methyl phenylacetate 6 (D) as alkylating agent. Standard reaction conditions: 2-methyl-1H-indole 1a (67.0 mg, 0.5 mmol), Co(acac)3 (3.6 mg, 0.01 mmol, 2 mol%), Triphos L1 (12.5 mg, 0.02 mmol, 4 mol%, 2 eq. to Co), Al(OTf)3 (11.9 mg, 0.025 mmol, 5 mol%, 2.5 eq. to Co), MCPE (2 mL), alkylating agent (0.875 mmol, 1.75 eq.) and H2 (10–30 bar) or N2 (30 bar) at 60–140 °C during 18 h. [a] Conversion of 1a and yield of products 3p and 3p′ were calculated by GC using hexadecane as internal standard. Between brackets is shown the isolated yield of the product after column chromatography on silica. [b] Run without Al(OTf)3. [c] Run without Co(acac)3 and Triphos L1. [d] Run without Co(acac)3, Triphos L1 and Al(OTf)3. | ||
 | ||
| Scheme 6 Control experiments in the hydrogenation of (E)-2-methyl-3-styryl-1H-indole 3p′ in the absence (A) or in the presence (B) of phenylacetic acid 2p. Standard reaction conditions: (E)-2-methyl-3-styryl-1H-indole 3p′ (116.7 mg, 0.5 mmol), phenylacetic acid 2p or not (25.7 mg, 0.375 mmol, 0.75 eq.), Co(acac)3 (3.6 mg, 0.01 mmol, 2 mol%), Triphos L1 (12.5 mg, 0.02 mmol, 4 mol%, 2 eq. to Co), Al(OTf)3 (11.9 mg, 0.025 mmol, 5 mol%, 2.5 eq. to Co), MCPE (2 mL) at 140 °C during 3–18 h. [a] Conversion of 3p′ and yield of products 3p and 1a were calculated by GC using hexadecane as internal standard. | ||
Next, we carried out the reaction under the optimized conditions for phenylacetic acid 2p but in the absence of any indole (Scheme 4A). This experiment revealed that our catalytic system is able to hydrogenate the carboxylic acid to the corresponding alcohol in moderate yields (14% of 2-phenylethanol 5 and 17% of 2-phenetyl phenylacetate 8 were detected, Scheme 4A). The experiment at shorter reaction times and using phenylacetaldehyde 9 as starting material, afforded 2-phenylethanol 5 in good yields (76%, Scheme 4B).
At this point it was interesting to compare the reactivity of several possible alkylating agents, all of them being related derivatives of phenylacetic acid 2p. Thus, reactions of indole 1a were performed in the presence of the [Co(acac)3/Triphos (L1)/Al(OTf)3] system at 140 °C and 30 bar of H2, using phenylacetic acid 2p, phenylacetaldehyde 9, phenethyl alcohol 5 and methyl phenylacetate 6 (usually formed from phenylacetic acid and methanol coming from MCPE cleavage) (Scheme 5A–D). Under these conditions, the best alkylating agent was the acid derivative (Scheme 5A), while the ester 6 and the alcohol 5 only afforded only traces of the alkylated product 3p (Scheme 5C and D, respectively). The aldehyde 9 gave the alkylated indole 3p in low yield (21%) together with traces of the alkenylated product 3p′ (Scheme 5B). This latter result is especially surprising considering the general use and high reactivity of aldehydes as alkylating agents. At milder reaction temperatures of 100 and 60 °C, a decrease in the catalytic activity was observed for phenylacetic acid 2p, while phenethyl alcohol 5 and methyl phenylacetate 6 were totally unreactive (Scheme 5A, C and D, respectively). Interestingly, the reaction with phenylacetaldehyde 9 at 100 and 60 °C gave moderate and good yields of the alkenylated product 3p′, respectively (Scheme 5B). This observation suggests that the aldehyde could be an important reaction intermediate that, when formed slowly from the carboxylic acid at the optimized conditions, is able to afford the alkylated product in good yields. The low yields of 3p obtained when the reaction was performed starting from the aldehyde at 140 °C, are partially explained by the aldehyde fast hydrogenation to the corresponding alcohol (Scheme 4B) and/or its degradation at this temperature. Notably, when the reaction was performed at low hydrogen pressure or in its total absence, low yields of the alkylated and alkenylated products were observed with phenethyl alcohol 5 and phenylacetaldehyde 9 as alkylating agents (Scheme 5B and C, respectively). This indicates that dehydrogenation pathways could also be partially contributing to the formation of the alkylated product, either by forming the aldehyde from the alcohol or by abstracting hydrogen from water or alcohols in the reaction media.
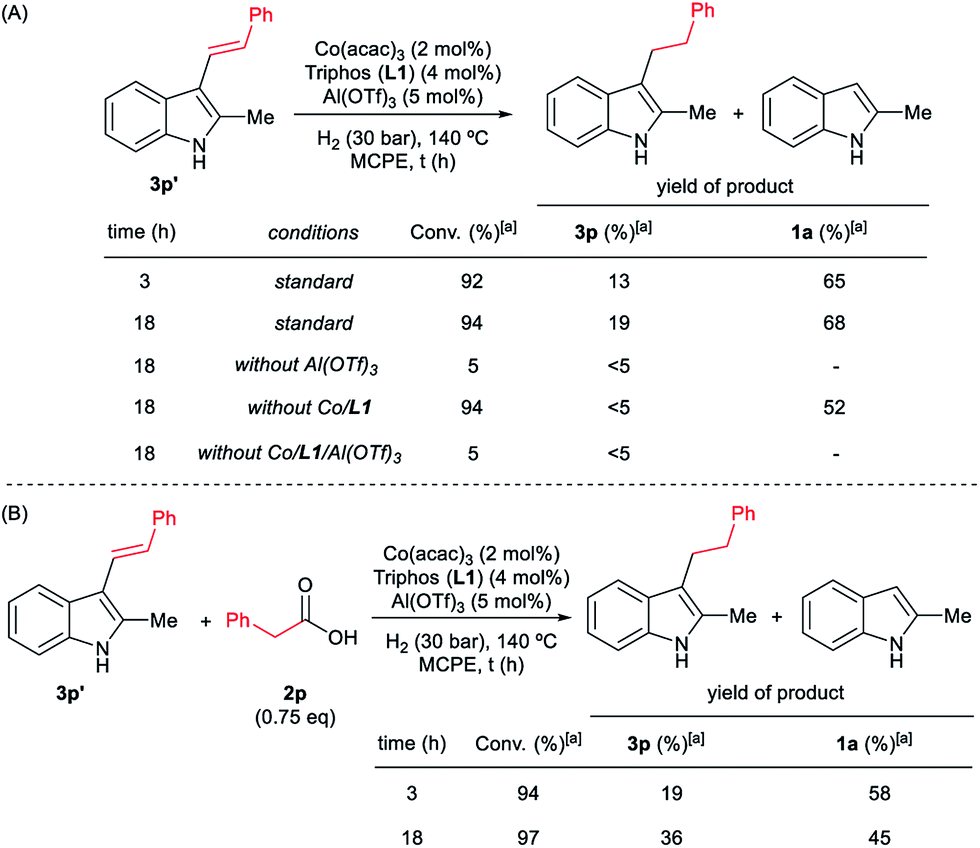
In addition, it was demonstrated that the presence of the three components of the catalytic system, [Co(acac)3/Triphos (L1)/Al(OTf)3], was required for the hydrogenation of the (E)-styryl indole 3p′ to the alkylated product 3p (Scheme 6A). Surprisingly, when 3p′ was submitted to the optimized reaction conditions, 2-methyl-1H-indole 1a was formed in 68% yield. In the presence of 0.75 eq. of phenylacetic acid 2p, lower amounts of indole 1a were detected (Scheme 6B).
Finally, to obtain some information about the catalytic system, the resting state of the reaction mixture after standard conditions was studied by 31P NMR (Fig. S2a†). In addition, the same experiment was repeated for several mixtures to see the effect of indole 1a, acetic acid 2a and Al(OTf)3 in the nature of resting state (Fig. S2b–d†). In all these experiments it was possible to detect signals between 18–32 ppm, corresponding to phosphine ligands coordinated to cobalt,25 and similar to the spectra obtained for the [Co(acac)3/Triphos (L1)/HNTf2] system.22 Interestingly, when the [Co(acac)3/Triphos (L1)/Al(OTf)3] system was employed, signals in the ranges of 18.7–20.2 ppm and 28.7–31.5 ppm were detected (Fig. S2a–c†). However, in the absence of Al(OTf)3 (Fig. S2d†), only signals around 30 ppm were observed, clearly indicating a main role of the additive in the formation of some of the active complexes.
With all these observations in hand, a plausible mechanism can be proposed for the cobalt catalysed reductive alkylation of indoles (Fig. 5). The major pathway involves in first place the hydrogenation of the carboxylic acid 2 to the corresponding aldehyde A or hemiacetal, which would react at the C3 nucleophilic position of indole 1 to afford the alkylated indole B. Subsequent dehydration leads to the captured alkene intermediate D, that finally gets hydrogenated to afford the alkylated indole. Minor pathways would involve the formation of an ester that can be also hydrogenated to the aldehyde, as well as a dehydrogenation mechanism from the formed alcohol. Scheme S6 (see ESI†) shows the extended version of the reaction mechanism containing additional possible minor pathways and secondary transformations involved in the overall process.
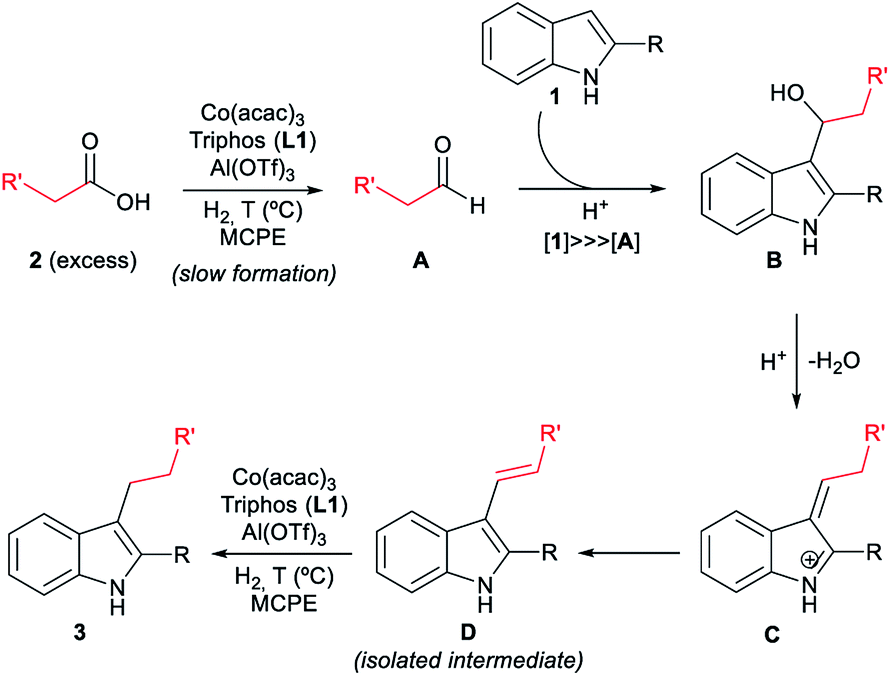 | ||
| Fig. 5 Possible reaction mechanism for the [Co/L1/Al(OTf)3]-catalysed reductive alkylation of indoles with carboxylic acids (simplified version). The extended version of the overall mechanism containing additional possible minor pathways and secondary transformations is depicted in Scheme S6 (see ESI†). | ||
Conclusions
For the first time, a general reductive C–H alkylation of indoles using carboxylic acids as alkylating agents is presented. This cobalt-catalysed methodology allows functionalization of different indoles at C3 in a straightforward manner. Using the [Co(acac)3/Triphos (L1)] system in combination with Al(OTf)3 as acid co-catalyst a wide range of carboxylic acids can be employed as alkylating agents in the presence of molecular hydrogen. In addition to alkylations, selective alkenylations of some substrates have been successfully performed. Control experiments revealed that the major reaction pathway involves the in situ formation of the corresponding aldehyde from the hydrogenation of the carboxylic acid, able to react with the indole. This novel protocol complements previously described reductive alkylations of indoles, mainly employing carbonyl compounds as alkylating agents. Advantageously, the use of carboxylic acids enlarges the potential molecular diversity introduced in the indole scaffold, due to the availability and stability of this class of compounds. Furthermore, the use of a catalyst based on a non-precious metal increases the interest of this transformation.Experimental details
General experimental procedure for the reductive C–H alkylation of 2-methyl-1H-indole (1a) with acetic acid (2a)
A 8 mL glass vial containing a stirring bar was sequentially charged with 2-methyl-1H-indole 1a (67.0 mg, 0.5 mmol), Co(acac)3 (3.6 mg, 0.01 mmol, 2 mol%), Triphos L1 (12.5 mg, 0.02 mmol, 4 mol%, 2 eq. to Co), Al(OTf)3 (11.9 mg, 0.025 mmol, 5 mol%, 2.5 eq. to Co), n-hexadecane (50.0 mg) as an internal standard, MCPE (2.0 mL) as solvent and acetic acid 2a (50.3 μL, 0.875 mmol, 1.75 eq.). Afterwards, the reaction vial was capped with a septum equipped with a syringe and set in the alloy plate, which was then placed into a 300 mL autoclave. Once sealed, the autoclave was purged three times with 30 bar of hydrogen, then pressurized to 30 bar and placed into an aluminium block, which was preheated at 160 °C. After 18 h, the autoclave was cooled in an ice bath, and the remaining gas was carefully released. Finally, the reaction mixture was diluted with ethyl acetate and analysed by GC.General experimental procedure for the reductive C–H alkylation of indoles with carboxylic acids
A 8 mL glass vial containing a stirring bar was sequentially charged with the indole substrate (0.5 mmol), Co(acac)3 (2–6 mol%), Triphos L1 (4–12 mol%, 2 eq. to Co), Al(OTf)3 (5–15 mol%, 2.5 eq. to Co), MCPE (2.0 mL) as solvent and the carboxylic acid (0.875–1.75 mmol, 1.75–3.5 eq.). Afterwards, the reaction vial was capped with a septum equipped with a syringe and set in the alloy plate, which was then placed into a 300 mL autoclave. Once sealed, the autoclave was purged three times with 30–60 bar of hydrogen, then pressurized to 30 bar and placed into an aluminium block, which was preheated at 120–160 °C. After 18–48 h, the autoclave was cooled in an ice bath, and the remaining gas was carefully released. Finally, the reaction mixture was diluted with ethyl acetate and purified by silica gel column chromatography (n-heptane/ethyl acetate mixtures) obtaining the desired C3-substituted indole derivatives.Acknowledgements
J. R. C.-A. and R. A. thanks Ramon Areces Foundation for a postdoctoral fellowship. This work was supported by the state of Mecklenburg-Vorpommern and the BMBF.Notes and references
- (a) G. W. Gribble, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees, E. F. V. Scriven and C. W. Bird, Pergamon Press, Oxford (UK), 1996, vol. 2, p. 207 Search PubMed; (b) R. J. Sundberg, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees, E. F. V. Scriven and C. W. Bird, Pergamon Press, Oxford (UK), 1996, vol. 2, p. 119 Search PubMed; (c) M. Somei and F. Yamada, Nat. Prod. Rep., 2005, 22, 73–103 RSC; (d) S. R. Walker, E. J. Carter, B. C. Huff and J. C. Morris, Chem. Rev., 2009, 109, 3080–3098 CrossRef CAS PubMed; (e) R. J. Melander, M. J. Minvielle and C. Melander, Tetrahedron, 2014, 70, 6363–6372 CrossRef CAS PubMed; (f) N. Netz and T. Opatz, Mar. Drugs, 2015, 13, 4814–4914 CrossRef CAS PubMed; (g) E. Stempel and T. Gaich, Acc. Chem. Res., 2016, 49, 2390–2402 CrossRef CAS PubMed.
- (a) J. Poojitha, K. N. Mounika, G. N. Raju and R. R. Nadendla, World J. Pharm. Res., 2015, 4, 656–666 CAS; (b) M.-Z. Zhang, Q. Chen and G.-F. Yang, Eur. J. Med. Chem., 2015, 89, 421–441 CrossRef CAS PubMed; (c) T. Saini, S. Kumar and B. Narasimhan, Cent. Nerv. Syst. Agents Med. Chem., 2016, 16, 19–28 CrossRef CAS; (d) T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed; (e) D. Sunil and P. R. Kamath, Curr. Top. Med. Chem., 2017, 17, 959–985 CrossRef CAS PubMed.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195–7210 CrossRef CAS PubMed.
- (a) E. Fischer and F. Jourdan, Ber. Dtsch. Chem. Ges., 1883, 16, 2241–2245 CrossRef; (b) B. Robinson, Chem. Rev., 1963, 63, 373–401 CrossRef.
- For some reviews dealing with the synthesis of indoles by cyclization protocols: (a) G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075 RSC; (b) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2310 CrossRef CAS PubMed; (c) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed; (d) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641–652 RSC; (e) S. A. Patil, R. Patil and D. D. Miller, Curr. Med. Chem., 2011, 18, 615–637 CrossRef CAS PubMed; (f) M. Platon, R. Amardeil, L. Djakovitch and J.-C. Hierso, Chem. Soc. Rev., 2012, 41, 3929–3968 RSC; (g) M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC; (h) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466–478 CrossRef CAS; (i) T. Guo, F. Huang, L. Yu and Z. Yu, Tetrahedron Lett., 2015, 56, 296–302 CrossRef CAS; (j) L. L. Anderson, M. A. Kroc, T. W. Reidl and J. Son, J. Org. Chem., 2016, 81, 9521–9529 CrossRef CAS PubMed; (k) G. Chelucci, Coord. Chem. Rev., 2017, 331, 37–53 CrossRef CAS; (l) R. B. Susick, L. A. Morrill, E. Picazo and N. K. Garg, Synlett, 2017, 28, 1–11 CrossRef CAS; (m) J. J. Song, J. T. Reeves, D. R. Fandrick, Z. Tan, N. K. Yee and C. H. Senanayake, ARKIVOC, 2010, 390–449 CAS; (n) S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215–PR283 CrossRef PubMed.
- For some reviews dealing with the direct functionalization of indole: (a) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; (b) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449–4465 RSC; (c) C. C. J. Loh and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 46–48 CrossRef CAS PubMed; (d) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742–778 RSC.
- For some reviews dealing with functionalization of indoles by CH activation: (a) G. Broggini, E. M. Beccalli, A. Fasana and S. Gazzola, Beilstein J. Org. Chem., 2012, 8, 1730–1746 CrossRef CAS PubMed; (b) L. Ackermann, J. Org. Chem., 2014, 79, 8948–8954 CrossRef CAS PubMed; (c) A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 CrossRef CAS; (d) N. Della Ca, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef CAS PubMed.
- For selected examples of allylic alkylation of indole C3 position: (a) M. Bandini, A. Melloni and A. Umani-Ronchi, Org. Lett., 2004, 6, 3199–3202 CrossRef CAS PubMed; (b) B. M. Trost and J. Quancard, J. Am. Chem. Soc., 2006, 128, 6314–6315 CrossRef CAS PubMed; (c) Q.-F. Wu, H. He, W.-B. Liu and S.-L. You, J. Am. Chem. Soc., 2010, 132, 11418–11419 CrossRef CAS PubMed; (d) L. Du, P. Cao, J. Xing, Y. Lou, L. Jiang, L. Li and J. Liao, Angew. Chem., Int. Ed., 2013, 52, 4207–4211 CrossRef CAS PubMed; (e) Y. Liu and H. Du, Org. Lett., 2013, 15, 740–743 CrossRef CAS PubMed; (f) Q.-L. Xu, L.-X. Dai and S.-L. You, Chem. Sci., 2013, 4, 97–102 RSC; (g) X. Zhang, W.-B. Liu, H.-F. Tu and S.-L. You, Chem. Sci., 2015, 6, 4525–4529 RSC; (h) R.-D. Gao, Q.-L. Xu, L.-X. Dai and S.-L. You, Org. Biomol. Chem., 2016, 14, 8044–8046 RSC.
- For selected examples of C3 alkylation of indole using Baylis–Hillman reaction: (a) J. S. Yadav, B. V. S. Reddy, A. K. Basak, A. V. Narsaiah, A. Prabhakar and B. Jagadeesh, Tetrahedron Lett., 2005, 46, 639–641 CrossRef CAS; (b) Z. Shafiq, L. Liu, Z. Liu, D. Wang and Y.-J. Chen, Org. Lett., 2007, 9, 2525–2528 CrossRef CAS PubMed; (c) C. Ramesh, V. Kavala, B. R. Raju, C.-W. Kuo and C.-F. Yao, Tetrahedron Lett., 2009, 50, 4037–4041 CrossRef CAS; (d) C. Ramesh, P.-M. Lei, D. Janreddy, V. Kavala, C.-W. Kuo and C.-F. Yao, J. Org. Chem., 2012, 77, 8451–8464 CrossRef CAS PubMed; (e) P. Goswami, A. J. Borah and P. Phukan, J. Org. Chem., 2015, 80, 438–446 CrossRef CAS PubMed.
- For selected examples of C3 alkylation of indole by Friedel–Crafts reaction: (a) J. F. Austin and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 1172–1173 CrossRef CAS PubMed; (b) C. Liu, X. Han, X. Wang and R. A. Widenhoefer, J. Am. Chem. Soc., 2004, 126, 3700–3701 CrossRef CAS PubMed; (c) H. D. King, Z. Meng, D. Denhart, R. Mattson, R. Kimura, D. Wu, Q. Gao and J. E. Macor, Org. Lett., 2005, 7, 3437–3440 CrossRef CAS PubMed; (d) J. Itoh, K. Fuchibe and T. Akiyama, Angew. Chem., Int. Ed., 2008, 47, 4016–4018 CrossRef CAS PubMed; (e) G. Blay, I. Fernandez, A. Monleon, M. C. Munoz, J. R. Pedro and C. Vila, Adv. Synth. Catal., 2009, 351, 2433–2440 CrossRef CAS; (f) M. Veguillas, M. Ribagorda and M. C. Carreno, Org. Lett., 2011, 13, 656–659 CrossRef CAS PubMed; (g) X. Liang, S. Li and W. Su, Tetrahedron Lett., 2012, 53, 289–291 CrossRef CAS; (h) F. de Nanteuil, J. Loup and J. Waser, Org. Lett., 2013, 15, 3738–3741 CrossRef CAS PubMed; (i) H.-M. Ko, K. K.-Y. Kung, J.-F. Cui and M.-K. Wong, Chem. Commun., 2013, 49, 8869–8871 RSC; (j) C. García-García, L. Ortiz-Rojano, S. Álvarez, R. Álvarez, M. Ribagorda and M. C. Carreño, Org. Lett., 2016, 18, 2224–2227 CrossRef PubMed; (k) X.-W. Wang, Y.-Z. Hua and M.-C. Wang, J. Org. Chem., 2016, 81, 9227–9234 CrossRef CAS PubMed; (l) P. C. Rao and S. Mandal, ChemCatChem, 2017, 9, 1172–1176 CrossRef CAS.
- Reductive alkylation employing alcohols and alkynes has also been described: (a) T. Tsuchimoto and M. Kanbara, Org. Lett., 2011, 13, 912–915 CrossRef CAS PubMed; (b) X. Han and J. Wu, Angew. Chem., Int. Ed., 2013, 52, 4637–4640 CrossRef CAS PubMed.
- (a) J. E. Appleton, K. N. Dack, A. D. Green and J. Steele, Tetrahedron Lett., 1993, 34, 1529–1532 CrossRef CAS; (b) A. Mahadevan, H. Sard, M. Gonzalez and J. C. McKew, Tetrahedron Lett., 2003, 44, 4589–4591 CrossRef CAS; (c) J. A. Campbell, V. Bordunov, C. A. Broka, J. Dankwardt, R. T. Hendricks, J. M. Kress, K. A. M. Walker and J.-H. Wang, Tetrahedron Lett., 2004, 45, 3793–3796 CrossRef CAS; (d) J. R. Rizzo, C. A. Alt and T. Y. Zhang, Tetrahedron Lett., 2008, 49, 6749–6751 CrossRef CAS; (e) S. Chandrasekhar, S. Khatun, G. Rajesh and C. Raji Reddy, Tetrahedron Lett., 2009, 50, 6693–6697 CrossRef CAS; (f) N. D. Shashikumar, G. N. Krishnamurthy, S. R. Rao, K. K. Shridhara, H. S. B. Naik and K. Nagarajan, Org. Process Res. Dev., 2010, 14, 918–920 CrossRef CAS; (g) S. Nomiyama, T. Hondo and T. Tsuchimoto, Adv. Synth. Catal., 2016, 358, 1136–1149 CrossRef CAS.
- (a) A. P. Gray, J. Org. Chem., 1958, 23, 1453–1454 CrossRef CAS; (b) L.-L. Cao, D.-S. Wang, G.-F. Jiang and Y.-G. Zhou, Tetrahedron Lett., 2011, 52, 2837–2839 CrossRef CAS; (c) Y. Duan, M.-W. Chen, Z.-S. Ye, D.-S. Wang, Q.-A. Chen and Y.-G. Zhou, Chem.–Eur. J., 2011, 17, 7193–7197 CrossRef CAS PubMed.
- (a) A. T. Gillmore, M. Badland, C. L. Crook, N. M. Castro, D. J. Critcher, S. J. Fussell, K. J. Jones, M. C. Jones, E. Kougoulos, J. S. Mathew, L. McMillan, J. E. Pearce, F. L. Rawlinson, A. E. Sherlock and R. Walton, Org. Process Res. Dev., 2012, 16, 1897–1904 CrossRef CAS; (b) A. Taheri, B. Lai, C. Cheng and Y. Gu, Green Chem., 2015, 17, 812–816 RSC.
- Y. Li, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 10476–10480 CrossRef CAS PubMed.
- (a) F. M. A. Geilen, B. Engendahl, M. Hölscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2011, 133, 14349–14358 CrossRef CAS PubMed; (b) T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed; (c) X. Cui, Y. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed.
- T. P. Brewster, A. J. M. Miller, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2013, 135, 16022–16025 CrossRef CAS PubMed.
- R. M. Bullock, Science, 2013, 342, 1054 CrossRef CAS PubMed.
- For examples involving homogeneous cobalt catalysed hydrogenation of alkenes, alkynes, carbonyl compounds, nitriles, carboxylic acid derivatives, CO2 and N-heteroarenes: (a) C. Federsel, C. Ziebart, R. Jackstell, W. Baumann and M. Beller, Chem.–Eur. J., 2012, 18, 72–75 CrossRef CAS PubMed; (b) S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561–4564 CrossRef CAS PubMed; (c) G. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed; (d) M. Amezquita-Valencia and A. Cabrera, J. Mol. Catal. A: Chem., 2013, 366, 17–21 CrossRef CAS; (e) M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076–1080 CrossRef CAS PubMed; (f) D. Gärtner, A. Welther, B. R. Rad, R. Wolf and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2014, 53, 3722–3726 CrossRef PubMed; (g) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS PubMed; (h) A. Mukherjee, D. Srimani, S. Chakraborty, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2015, 137, 8888–8891 CrossRef CAS PubMed; (i) S. Rösler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef PubMed; (j) D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed; (k) R. Xu, S. Chakraborty, H. Yuan and W. D. Jones, ACS Catal., 2015, 5, 6350–6354 CrossRef CAS; (l) A. Z. Spentzos, C. L. Barnes and W. H. Bernskoetter, Inorg. Chem., 2016, 55, 8225–8233 CrossRef CAS PubMed; (m) K. Tokmic and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 13700–13705 CrossRef CAS PubMed; (n) D. Zhang, E.-Z. Zhu, Z.-W. Lin, Z.-B. Wei, Y.-Y. Li and J.-X. Gao, Asian J. Org. Chem., 2016, 5, 1323–1326 CrossRef CAS; (o) R. Adam, C. B. Bheeter, J. R. Cabrero-Antonino, K. Junge, R. Jackstell and M. Beller, ChemSusChem, 2017, 10, 842–846 CrossRef CAS PubMed; (p) R. Adam, J. R. Cabrero-Antonino, A. Spannenberg, K. Junge, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 3216–3220 CrossRef CAS PubMed; (q) J. Guo, X. Shen and Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 615–618 CrossRef CAS PubMed.
- For reviews dealing with cobalt catalysed hydrogenation: (a) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed; (b) J.-L. Renaud and S. Gaillard, Synthesis, 2016, 48, 3659–3683 CrossRef CAS.
- T. J. Korstanje, J. Ivar van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298 CrossRef CAS PubMed.
- J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 1890–1893 CrossRef CAS PubMed.
- For examples of hydrogenation of carboxylic acids employing the [Ru/Triphos/acid additive] catalytic system: (a) A. A. Núñez Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154–3156 RSC; (b) D. L. Dodds, J. Coetzee, J. Klankermayer, S. Brosinski, W. Leitner and D. J. Cole-Hamilton, Chem. Commun., 2012, 48, 12249–12262 RSC; (c) K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed; (d) J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinski, W. Leitner, A. M. Z. Slawin and D. J. Cole-Hamilton, Chem.–Eur. J., 2013, 19, 11039–11050 CrossRef CAS PubMed; (e) Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed; (f) K. Beydoun, G. Ghattas, K. Thenert, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2014, 53, 11010–11014 CrossRef CAS PubMed; (g) S. Savourey, G. Lefevre, J.-C. Berthet and T. Cantat, Chem. Commun., 2014, 50, 14033–14036 RSC; (h) T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hoelscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed; (i) X. Cui, Y. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed; (j) E. J. Derrah, M. Hanauer, P. N. Plessow, M. Schelwies, M. K. da Silva and T. Schaub, Organometallics, 2015, 34, 1872–1881 CrossRef CAS; (k) Y. Li, C. Topf, X. Cui, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 5196–5200 CrossRef CAS PubMed; (l) I. Sorribes, J. R. Cabrero-Antonino, C. Vicent, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 13580–13587 CrossRef CAS PubMed; (m) S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. v. Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC; (n) R. Adam, J. R. Cabrero-Antonino, K. Junge, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 11049–11053 CrossRef CAS PubMed; (o) K. Beydoun, K. Thenert, E. S. Streng, S. Brosinski, W. Leitner and J. Klankermayer, ChemCatChem, 2016, 8, 135–138 CrossRef CAS; (p) J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Catal. Sci. Technol., 2016, 6, 7956–7966 RSC; (q) J. R. Cabrero-Antonino, E. Alberico, K. Junge, H. Junge and M. Beller, Chem. Sci., 2016, 7, 3432–3442 RSC; (r) J. R. Cabrero-Antonino, I. Sorribes, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 387–391 CrossRef CAS PubMed; (s) M. L. Yuan, J. H. Xie and Q. L. Zhou, ChemCatChem, 2016, 8, 3036–3040 CrossRef CAS.
- Alkenylation of indole C3 position has been reported using carbonyl compounds: (a) G. Fridkin, N. Boutard and W. D. Lubell, J. Org. Chem., 2009, 74, 5603–5606 CrossRef CAS PubMed; (b) A. Taheri, C. Liu, B. Lai, C. Cheng, X. Pan and Y. Gu, Green Chem., 2014, 16, 3715–3719 RSC.
- B. Capelle, A. L. Beauchamp, M. Dartiguenave, Y. Dartiguenave and H. F. Klein, J. Am. Chem. Soc., 1982, 104, 3891–3897 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General information concerning experimental procedures, additional tables, figures, schemes, characterization data and NMR spectra of the isolated compounds are available. See DOI: 10.1039/c7sc02117h |
| This journal is © The Royal Society of Chemistry 2017 |