
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the excited-state dynamics of low band gap, near-infrared absorbers via proquinoidal unit electronic structural modulation†
Yusong
Bai
,
Jeff
Rawson
 ,
Sean A.
Roget
,
Jean-Hubert
Olivier
,
Jiaxing
Lin
,
Peng
Zhang
,
David N.
Beratan
and
Michael J.
Therien
*
,
Sean A.
Roget
,
Jean-Hubert
Olivier
,
Jiaxing
Lin
,
Peng
Zhang
,
David N.
Beratan
and
Michael J.
Therien
*
Department of Chemistry, French Family Science Center, Duke University, 124 Science Drive, Durham, North Carolina 27708-0346, USA. E-mail: Michael.therien@duke.edu
First published on 7th June 2017
Abstract
While the influence of proquinoidal character upon the linear absorption spectrum of low optical bandgap π-conjugated polymers and molecules is well understood, its impact upon excited-state relaxation pathways and dynamics remains obscure. We report the syntheses, electronic structural properties, and excited-state dynamics of a series of model highly conjugated near-infrared (NIR)-absorbing chromophores based on a (porphinato)metal(II)-proquinoidal spacer-(porphinato)metal(II) (PM-Sp-PM) structural motif. A combination of excited-state dynamical studies and time-dependent density functional theory calculations: (i) points to the cardinal role that excited-state configuration interaction (CI) plays in determining the magnitudes of S1 → S0 radiative (kr), S1 → T1 intersystem crossing (kISC), and S1 → S0 internal conversion (kIC) rate constants in these PM-Sp-PM chromophores, and (ii) suggests that a primary determinant of CI magnitude derives from the energetic alignment of the PM and Sp fragment LUMOs (ΔEL). These insights not only enable steering of excited-state relaxation dynamics of high oscillator strength NIR absorbers to realize either substantial fluorescence or long-lived triplets (τT1 > μs) generated at unit quantum yield (ΦISC = 100%), but also crafting of those having counter-intuitive properties: for example, while (porphinato)platinum compounds are well known to generate non-emissive triplet states (ΦISC = 100%) upon optical excitation at ambient temperature, diminishing the extent of excited-state CI in these systems realizes long-wavelength absorbing heavy-metal fluorophores. This work highlights approaches to: (i) modulate low-lying singlet excited-state lifetime over the picosecond-to-nanosecond time domain, (ii) achieve NIR fluorescence with quantum yields up to 25%, (iii) tune the magnitude of S1–T1 ISC rate constant from 109 to 1012 s−1 and (iv) realize T1-state lifetimes that range from ∼0.1 to several μs, for these model PM-Sp-PM chromophores, and renders new insights to evolve bespoke photophysical properties for low optical bandgap π-conjugated polymers and molecules based on proquinoidal conjugation motifs.
Introduction
Low optical bandgap π-conjugated polymers and molecules have fuelled spectacular developments in optical and optoelectronic technologies.1 A common strategy to realize polymer bandgap reduction relies on augmenting the π-backbone quinoidal character. Introduction of so-called proquinoidal units into the conjugation main chain drives well understood perturbations to the ground-state electronic absorption spectrum, where augmented proquinoidal character diminishes both the optical (Eop) and potentiometric (Ep; E0/+1/2 − E−/01/2) band gaps of the material.2 In contrast to the ground-state absorptive and electronic structural control enabled by proquinoidal motifs,2c,d,g–j,3 considerably less insight exists concerning the impact of proquinoidal character on excited-state relaxation pathways and dynamics of extensively π-conjugated molecules and materials.Control over the excited-state relaxation dynamics of highly conjugated NIR-absorbing materials would broadly boost their utility for optical and optoelectronic applications. For instance, long-wavelength optical power limiting (OPL) relies upon reverse-saturable absorption, demanding materials whose photoexcitation is followed by rapid intersystem crossing (ISC) with near-unit quantum yield to a long-lived triplet excited state.1d,4 Triplet–triplet annihilation photon-upconversion (TTA-UC), which has applications that include solar energy harvesting, places a premium on enhancing near-infrared (NIR) absorptive oscillator strength and engineering ultrafast ISC.5 In contrast, deep tissue-penetrating in vivo imaging requires extending the absorptive and emissive wavelengths of NIR fluorophores while maintaining large S1 → S0 transition dipole moments.1e,6 Advancing these applications motivates the development of design approaches to regulate the natures of initially prepared photo-excited states and the subsequent relaxation dynamics of long-wavelength absorbers.
Here, we combine molecular design and synthesis, time-resolved spectroscopic methods, and quantum chemical analyses to delineate the excited-state properties of a series of model highly conjugated NIR-absorbing chromophores based on a (porphinato)metal(II)-spacer-(porphinato)metal(II) (PM-Sp-PM) structural motif2c in which: the porphyryl ligand is either electron rich (10,20-diarylporphyrin, ArP) or electron deficient (10,15,20-tris(perfluoroalkyl)porphyrin, Rf3P), the central metal ion is Zn(II) or Pt(II), and the proquinoidal Sp is either 4,7-diethynylbenzo[c][1,2,5]thiadiazole (E-BTD-E) or 4,9-diethynyl-6,7-dimethyl[1,2,5]thiadiazolo[3,4-g]quinoxaline (E-TDQ-E). Steady-state absorption, emission, and pump-probe transient absorption spectroscopic experiments, and time-dependent density functional theory (TD-DFT) computational studies, unambiguously correlate the extent of configuration interaction (CI) and charge transfer (CT) character of the initially prepared excited state with the relative magnitudes of S1 → S0 radiative (kr), S1 → T1 intersystem crossing (kISC), and S1 → S0 internal conversion (kIC) rate constants. Collectively, these studies demonstrate how electronic modulation of proquinoidal conjugation motifs, conventionally utilized to regulate the ground-state absorptivity of low optical bandgap π-conjugated polymers and molecules, can also be exploited to steer their excited-state relaxation pathways. This work: (i) underscores how modulation of the extent of excited-state CI for conjugation motifs that rely on proquinoidal units can produce either exceptional NIR fluorophores or chromophores that generate long-lived electronically excited state triplets at high quantum yield, and (ii) describes a roadmap via which the relative magnitudes of kr, kISC, and kIC rate constants may be engineered to tightly control photophysical function of highly conjugated NIR-absorbing materials.
Results and discussion
Synthesis
Scheme 1 outlines the synthetic strategies for the PM-Sp-PM species, including ArPM-Sp-ArPM [M = Zn(II), Pt(II); Sp = E-TDQ-E, E-BTD-E], Rf3PZnE-TDQ-ERf3PZn, and Rf3PZnArPPtE-TDQ-ERf3PZnArPPt. Generally, the desired PM-Sp-PM supermolecular chromophores were prepared by the metal-mediated cross-coupling7 of a mono meso-ethyne-functionalized (porphinato)metal(II) species with a dibrominated proquinoidal spacer moiety (Scheme 1). The key precursor compound Rf3PZnArPPtE was prepared via a Pd0-mediated coupling reaction involving Rf3PZnE and BrArPPtETIPS, followed by deprotection of the triisopropylsilyl group. Chromophores featuring ArPM-Sp-ArPM molecular structures were isolated via column chromatography on silica using 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 CH2Cl2:hexanes as the eluent, while for chromophores containing the Rf3PZn fragment, 3:7 THF:hexanes was employed due to their solubility in this mobile phase. Size exclusion column chromatography with THF eluent was employed for purification of each chromophore after silica column chromatography. Scheme 2 displays the chemical structures for previously established oligo(porphinato)metal(II) compounds8 that define key spectroscopic benchmarks for these newly designed PM-Sp-PM chromophores.
:3 CH2Cl2:hexanes as the eluent, while for chromophores containing the Rf3PZn fragment, 3:7 THF:hexanes was employed due to their solubility in this mobile phase. Size exclusion column chromatography with THF eluent was employed for purification of each chromophore after silica column chromatography. Scheme 2 displays the chemical structures for previously established oligo(porphinato)metal(II) compounds8 that define key spectroscopic benchmarks for these newly designed PM-Sp-PM chromophores.
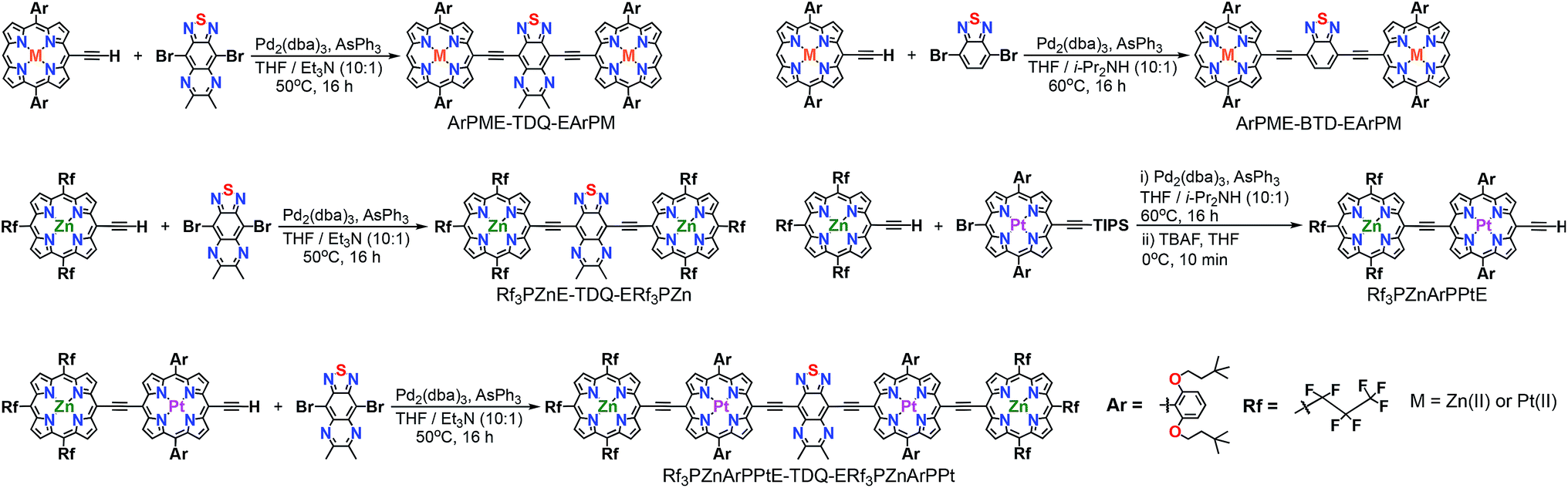 | ||
| Scheme 1 Synthetic routes and chemical structures for PM-Sp-PM compounds. | ||
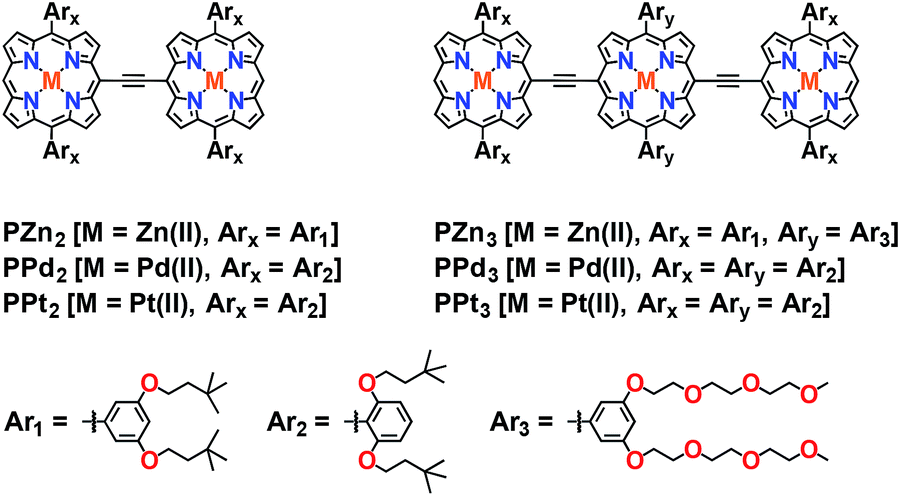 | ||
| Scheme 2 Chemical structures for previously established oligo(porphinato)metal(II) chromophores. | ||
Steady-state absorption and emission spectroscopy of ArPM-Sp-ArPM chromophores
Steady-state electronic absorption and fluorescence spectra recorded for ArPM-Sp-ArPM species (ArPZnE-BTD-EArPZn,2cArPZnE-TDQ-EArPZn,2cArPPtE-BTD-EArPPt, and ArPPtE-TDQ-EArPPt) are displayed in Fig. 1. The gross features of the electronic absorption spectra for these complexes resemble those of dimeric and multimeric porphyrin compounds that feature a meso-to-meso ethyne-linkage topology (PZnn compounds), manifesting two distinct absorption manifolds that are derived from the porphyrin B- (Soret) (S0 → S2) and Q-band (S0 → S1) transitions.8a–c,9 Previous investigations have demonstrated that Sp moieties modulate the respective ArPZn-Sp-ArPZn long axis-polarized Q state (Qx) absorption maxima from 689 to 1006 nm, depending upon the extent of the quinoidal resonance contribution to the electronically excited singlet state.2c Further, semi-empirical electronic structure calculations and electrochemical data underscored the cardinal role that ArPZn and Sp fragment orbital energy differences played in fixing the radical cation and anion state energy levels in these ArPZn-Sp-ArPZn structures.2c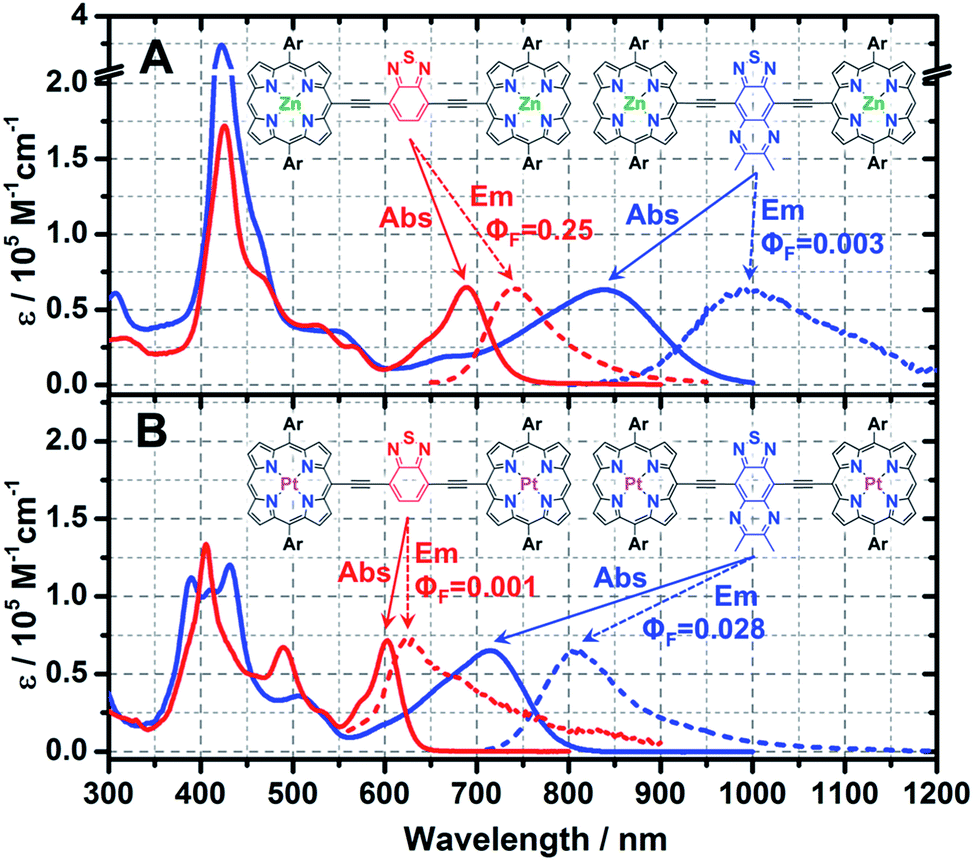 | ||
| Fig. 1 Steady-state electronic absorption and emission spectra for: (A) ArPZnE-TDQ-EArPZn (blue colored lines, λexc = 750 nm for emission) and ArPZnE-BTD-EArPZn (red colored lines, λexc = 640 nm for emission): and (B) ArPPtE-TDQ-EArPPt (blue colored lines, λexc = 650 nm for emission) and ArPPtE-BTD-EArPPt (red colored lines, λexc = 540 nm for emission). Steady-state spectra were all acquired in THF solvent at ambient condition. Ar = 2′,6′-bis(3,3-dimethyl-1-butyloxy)phenyl. | ||
A comparison of the Q-band regions in the electronic absorption spectra of ArPZnE-BTD-EArPZn, ArPZnE-TDQ-EArPZn, ArPPtE-BTD-EArPPt, and ArPPtE-TDQ-EArPPt reveals discrete effects correlated with the choice of spacer and (porphinato)metal(II) units. Compared with ArPME-BTD-EArPM, the ArPME-TDQ-EArPM analogues evince dramatically red-shifted Qx-state absorption maxima and substantial oscillator strength (f) redistribution into the low-energy spectral domain [compound (λmax(S0 → S1), f600–1000 nm): ArPZnE-BTD-EArPZn (689 nm, 0.455), ArPPtE-BTD-EArPPt (603 nm, 0.168), ArPZnE-TDQ-EArPZn (839 nm, 0.827), ArPPtE-TDQ-EArPPt (715 nm, 0.686); Table 1]. Furthermore, the Qx-state absorption manifolds of ArPME-TDQ-EArPM species manifest much broader full width at half maxima (FWHM) and less discernable vibrational structures relative to those evinced by ArPME-BTD-EArPM chromophores [Qx(FWHM): ArPME-TDQ-EArPM ∼ 2500 cm−1; ArPME-BTD-EArPM ∼ 1100 cm−1], suggesting that the TDQ-containing chromophores give rise to S1 states with greater CT character.2c,10 Compared to ArPZn-Sp-ArPZn compounds, the ArPPt-Sp-ArPPt chromophores display blue-shifted Qx-state transition manifold maxima; this phenomenon is typical of hypsoporphyrin species and has been attributed to the efficient mixing of metal ndπ and porphyrin π* orbitals.8e,11
| Chromophores | λ max/nm (FWHM/cm−1) | Stokes shifta/cm−1 | Oscillator strength in red-to-NIRb | |
|---|---|---|---|---|
| S0 → S1 | S1 → S0 | |||
a Stokes shift corresponds to the energy difference between the low-energy transition (Qx) absorption and fluorescence maxima.
b Integrated oscillator strengths (f) were calculated based on the following expression: 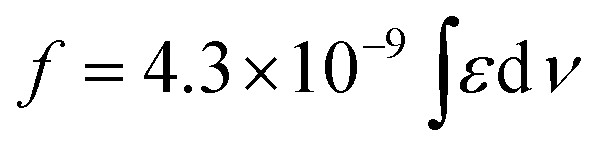 , where ε is the experimental extinction coefficient, and ν is the energy (in wave numbers) of the absorption. Oscillator strengths were calculated over the following spectral domains: ArPZnE-TDQ-EArPZn (600–1000 nm); ArPZnE-BTD-EArPZn (600–800 nm); ArPPtE-TDQ-EArPPt (600–850 nm); ArPPtE-BTD-EArPPt (600–700 nm); Rf3PZnE-TDQ-ERf3PZn (600–850 nm); Rf3PZnArPPtE-TDQ-EArPPtRf3PZn (600–950 nm). , where ε is the experimental extinction coefficient, and ν is the energy (in wave numbers) of the absorption. Oscillator strengths were calculated over the following spectral domains: ArPZnE-TDQ-EArPZn (600–1000 nm); ArPZnE-BTD-EArPZn (600–800 nm); ArPPtE-TDQ-EArPPt (600–850 nm); ArPPtE-BTD-EArPPt (600–700 nm); Rf3PZnE-TDQ-ERf3PZn (600–850 nm); Rf3PZnArPPtE-TDQ-EArPPtRf3PZn (600–950 nm).
|
||||
| ArPZnE-TDQ-EArPZn | 839 (2510) | 995 (1778) | 1869 | 0.827 |
| ArPZnE-BTD-EArPZn | 689 (1180) | 741 (1382) | 1019 | 0.455 |
| ArPPtE-TDQ-EArPPt | 715 (2415) | 810 (1530) | 1640 | 0.686 |
| ArPPtE-BTD-EArPPt | 603 (1061) | 629 (1579) | 686 | 0.168 |
| Rf3PZnE-TDQ-ERf3PZn | 705 (1163) | 750 (1370) | 851 | 0.451 |
| Rf3PZnArPPtE-TDQ-EArPPtRf3PZn | 755 (1738) | 813 (1432) | 945 | 1.809 |
ArPZnE-BTD-EArPZn and ArPZnE-TDQ-EArPZn exhibit markedly different Stokes shifts and fluorescence quantum yields (ΦF) (Fig. 1) in THF solvent. While ArPZnE-BTD-EArPZn displays a relatively small Stokes shift (Δν = 1019 cm−1) and a large fluorescence quantum yield (ΦF = 0.25) comparable to those of PZnn chromophoric benchmarks (e.g.PZn2: Δν = 711 cm−1, ΦF = 0.16. PZn3: Δν = 806 cm−1, ΦF = 0.22; Scheme 2),8d,9cArPZnE-TDQ-EArPZn exhibits a significant Stokes shift of 1869 cm−1 and negligible fluorescence (ΦF = 0.003). Furthermore, the Stokes shift and fluorescence quantum yield of ArPZnE-BTD-EArPZn show modest solvent dependence [e.g., toluene (Δν = 522 cm−1, ΦF = 0.27)] (Fig. S1†); in contrast, a substantial solvent-dependence of the Stokes shift and fluorescence quantum yield are evinced for ArPZnE-TDQ-EArPZn, with less polar solvent driving smaller Stokes shifts and significantly enhanced fluorescence quantum yields [toluene (ΔνStokes = 771 cm−1, ΦF = 0.02) vs. THF (ΔνStokes = 1869 cm−1, ΦF = 0.003)] (Fig. S2†). These observations underscore the differing CT characters of the ArPZnE-TDQ-EArPZn and ArPZnE-BTD-EArPZn Qx-states; as ArPZnE-TDQ-EArPZn exhibits substantial CT character, the reorganization energy associated with excited-state solvent relaxation is magnified.9c,12
Upon metal center substitution from zinc(II) to platinum(II), ArPPtE-BTD-EArPPt manifests weak fluorescence centered at 629 nm (Δν = 686 cm−1, ΦF = 0.001) in deoxygenated solution at ambient temperature, and phosphorescence centered at 845 nm (Δν = 4750 cm−1) (Fig. S3†). Such emissive properties are similar to those observed in oligomeric (porphinato)palladium(II)/platinum(II) compounds (PM3, M = Pd or Pt; Scheme 2), where phosphorescence and very weak fluorescence (77 K) are simultaneously evinced (PPd3: λS1→S0(max) = 747 nm; λT1→S0(max) = 828 nm; PPt3: λS1→S0(max) = 740 nm; λT1→S0(max) = 800 nm).8e In contrast, ArPPtE-TDQ-EArPPt maintains one constant emission band centered at 810 nm, regardless of the solution oxygen content (Fig. S4†). Note that no additional emission band can be observed in deoxygenated samples of ArPPtE-TDQ-EArPPt even at low temperature (77 K, Fig. S5†). In light of the magnitude of the Stokes shift (Δν = 1530 cm−1) and the measured lifetime of this chromophore (τ = 310 ps, Fig. S6E†), we denote the ArPPtE-TDQ-EArPPt emission band centered at 810 nm as fluorescence. Given the (porphinato)-platinum(II) components of this chromophore, it is recognized that the low-lying emissive state of ArPPtE-TDQ-EArPPt may be best described as a “sing–triplet”. In this regard, we emphasize that although the “fluorescence”, “phosphorescence”, “S1”, and “T1” labels are preserved throughout this report for describing the respective higher and lower energy emissive states of the platinum-containing PM-Sp-PM compounds, they suggest only the dominant characters of these excited states. Note that the fluorescence quantum yield determined for ArPPtE-TDQ-EArPPt (2.8%) is unusually large, considering that platinum atoms typically promote strong spin–orbit coupling, resulting in near-unit S1 → T1 ISC quantum yields (ΦISC values) in classic (porphinato)platinum(II) compounds.8e,13 The emissive properties observed for ArPPtE-TDQ-EArPPt suggest an unusually low ΦISC value relative to the ArPPtE-BTD-EArPPt chromophore (vide infra).
Femtosecond pump-probe transient absorption spectroscopy of ArPM-Sp-ArPM
The femtosecond transient absorption spectra recorded at selected time delays for ArPM-Sp-ArPM chromophores are displayed in Fig. 2. The early time-delay (<1 ps) transient spectra of these ArPM-Sp-ArPM chromophores share several common features that resemble the transient spectral signatures of ethyne-bridged multimeric (porphinato)zinc(II) structures (PZnn, n = 2, 3; Scheme 2).9a,c These include: (i) bleaching signals in the Soret and Qx band regions, (ii) weak transient absorptions between the two dominant ground-state absorption bleaching signatures, and (iii) intense NIR S1 → Sn transient absorption manifolds that feature extraordinary spectral breadth.8d,e,9c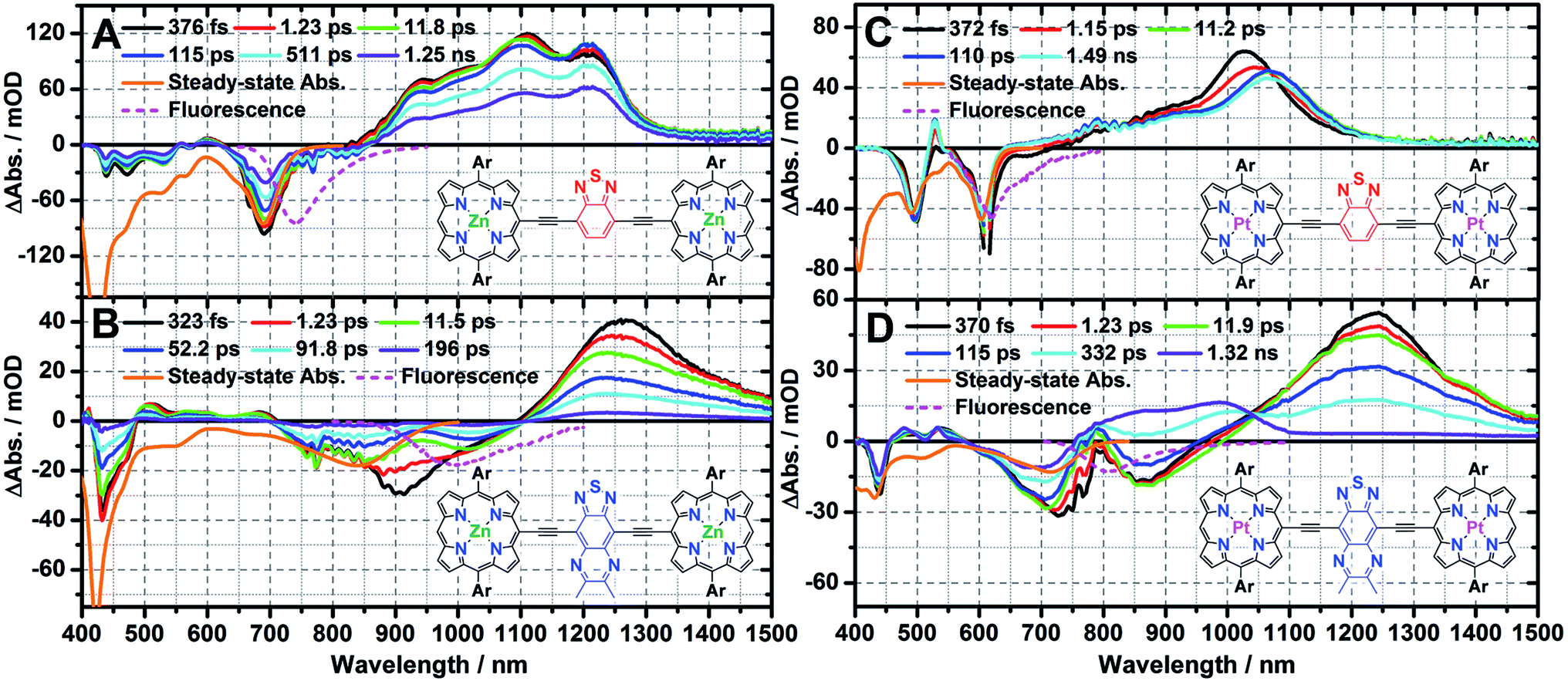 | ||
| Fig. 2 Pump-probe transient absorption spectra recorded at several time delays for (A) ArPZnE-BTD-EArPZn (λexc = 660 nm), (B) ArPZnE-TDQ-E ArPZn (λexc = 900 nm), (C) ArPPtE-BTD-EArPPt (λexc = 610 nm), and (D) ArPPtE-TDQ-EArPPt (λexc = 750 nm). Experimental conditions: solvent = THF; ambient temperature; magic angle polarization. Steady-state absorption (orange solid line) and fluorescence (magenta dash line) are provided as inverted spectra. Ar = 2′,6′-bis(3,3-dimethyl-1-butyloxy)phenyl. | ||
Conventional PZnn excited-state dynamics include fast S2 → S1 internal conversion (<1 ps),9a nanosecond timescale S1 state lifetimes,8e,9c and small Stokes shifts in virtually all solvents;8d,9c this latter point suggests minimal degrees of solvent repolarization and inner-sphere chromophore nuclear reorganization are associated with excited-state relaxation of these species. Furthermore, upon metal center substitution with palladium(II) or platinum(II), the corresponding PPdn, or PPtn chromophores (Scheme 2) manifest ultrafast intersystem crossing on sub-picosecond to picosecond timescales.8e For corresponding ArPM-Sp-ArPM species, however, it is clear that the proquinoidal spacer units regulate electronically excited-state electron density spatial distributions (Fig. 2).
Femtosecond transient absorption spectroscopy of ArPZn-Sp-ArPZn
A global fit of the NIR transient absorption dynamics observed following excitation (λexc = 660 nm), evinces three characteristic relaxation processes: 760 ± 90 fs, 24 ± 5 ps, 1.75 ± 0.1 ns (Fig. S8A†). The 760 fs and 24 ps components are assigned to respective solvent relaxation, and structural relaxation processes associated with torsional dynamic about the ethyne-Sp-ethyne ArPZn-to-ArPZn linkage; these dynamics have been discussed in detail for closely related structures.9a,c The 1.75 ns component corresponds with ground-state recovery and is assigned to the intrinsic S1 state lifetime, in close agreement with the fluorescence lifetime determined by streak-scope (τF = 1.67 ns, Fig. S6C†). Consistent with the insensitivity of the Stokes shift and fluorescence quantum yield to solvent polarity, the S1 state lifetime of ArPZnE-BTD-EArPZn manifests minimal variation with solvent dielectric constant [e.g. THF (τF = 1.67 ns) vs. toluene (τF = 1.61 ns, Fig. S6D†)], suggesting the electronically excited-state nature of ArPZnE-BTD-EArPZn features negligible CT character.The excited-state relaxation dynamics of ArPZnE-TDQ-EArPZn are also multiexponential in nature. Global fitting of the NIR spectral regime transient dynamical data acquired for ArPZnE-TDQ-EArPZn evinces three relaxation processes: 950 ± 120 fs, 12 ± 2 ps, and 87 ± 6 ps (Fig. S8B†). While the 950 fs and 12 ps components correspond, respectively, to the solvent relaxation and structural relaxation dynamics, the 87 ps component is very close to the streak-scope determined fluorescence lifetime (τF = 76 ps) of ArPZnE-TDQ-EArPZn (Fig. S6A†), and is thus assigned to the intrinsic singlet excited-state lifetime. Notably, the THF solvent relaxation component mirrors the timescale for the time-dependent red shift of the stimulated emission from 910 to 1020 nm (Δν = 1185 cm−1). This dynamic Stokes shift, absent for ArPZnE-BTD-EArPZn, indicates substantial displacement between the ground- and singlet excited-state potential energy surfaces along the solvation coordinate,10b,14 congruent with the marked solvent-dependent ArPZnE-TDQ-EArPZn Stokes shifts observed by steady-state emission spectroscopy (vide supra). Additionally, the S1 state lifetime (τS1 = 87 ps) of ArPZnE-TDQ-EArPZn is much shorter than that of ArPZnE-BTD-EArPZn, and depends strongly on solvent dielectric constant (εs). For example, the τF determined for ArPZnE-TDQ-EArPZn measured in toluene solvent (∼506 ps, Fig. S6B†) is nearly 1 order of magnitude larger than that found in THF. As such, the calculated S1 → S0 internal conversion rate constants for ArPZnE-TDQ-EArPZn in THF and toluene differ markedly [kIC(THF) = 1.15 × 1010 s−1vs. kIC(toluene) = 1.94 × 109 s−1], highlighting the acceleration of the nonradiative decay process in polar solvent: since the Qx-state transition features substantial CT character, solvent dipole reorientation dramatically stabilizes the polarized S1 state of ArPZnE-TDQ-EArPZn, decreasing the S1 → S0 transition oscillator strength [ΦF(THF) = 0.003 vs. ΦF(toluene) = 0.02].
Femtosecond transient absorption spectroscopy of ArPPt-Sp-ArPPt
A global fit of the time-dependent vis-NIR transient dynamical data acquired for ArPPtE-BTD-EArPPt following excitation at 610 nm evinces two relaxation processes: 960 ± 120 fs, 1.90 ± 0.13 ns (Fig. S8C†). Note that at an early time delay (tdelay ∼ 1 ps), the initially prepared ArPPtE-BTD-EArPPt electronically excited state has already evolved to a long-lived excited state that persists beyond the delay limit of the femtosecond pump-probe instrument. Nanosecond pump-probe transient absorption measurements determine that the lifetime of this long-lived excited state is 2.5 μs, which is reduced to 0.24 μs under oxygenated conditions (Fig. S7C and D†), suggesting that the long-lived excited state is triplet in nature. In this regard, the ultrafast component (960 fs) observed in the femtosecond transient dynamics corresponds to an ISC process, while the 1.90 ns component corresponds to the slow decay of ArPPtE-BTD-EArPPt triplet excited state. Furthermore, the decay-associated spectra (DAS, Fig. S8†) connected with the ultrafast component manifests an S-like shape in the 900–1200 nm domain, displaying a decay on the blue side and rise on the red side of the transient, suggesting unit quantum yield S1 → T1 conversion (i.e. τS1 ≈ τISC ∼ 960 fs, ΦISC ∼ 100%). The ultrafast S1 → T1 conversion and near-unit ISC quantum yield displayed by ArPPtE-BTD-EArPPt are similar to those evinced by PPtn compounds (Table 2).8e| Chromophores | τ S1/ns | Relaxation rate from S1a/s−1 | Φ F | Φ ISC | ||
|---|---|---|---|---|---|---|
| k IC(S1 → S0) | k 0F(S1 → S0) | k ISC(S1 → T1) | ||||
| a k IC denotes the S1 → S0 nonradiative decay rate constant, k0F denotes the intrinsic fluorescence rate constant, kISC denotes the S1 → T1 intersystem crossing rate constant. Excited-state relaxation rate constants were calculated based on the following equations: τS1 = 1/(kIC + k0F + kISC), ΦF = k0F × τS1, ΦISC = kISC × τS1, since τS1, ΦF, and ΦISC can all be experimentally acquired, therefore, rate constants kIC, k0F, and kISC can be calculated. b Fluorescence quantum yields were determined relative to bis[(porphinato)zinc(II)] (PZn2) in THF (0.16). c Φ ISC were determined based on femtosecond transient absorption spectra, for details see ESI. | ||||||
| ArPZnE-TDQ-EArPZn | 0.087 | 1.15 × 1010 | 3.10 × 107 | ≤107 | 0.003 | ∼0 |
| ArPZnE-BTD-EArPZn | 1.750 | 3.82 × 108 | 1.43 × 108 | 8.57 × 107 | 0.25 | 0.15 |
| ArPPtE-TDQ-EArPPt | 0.310 | 1.51 × 109 | 9.15 × 107 | 1.67 × 109 | 0.028 | 0.50 |
| ArPPtE-BTD-EArPPt | 0.001 | ≤108 | ∼108 | 1.04 × 1012 | 0.001 | ∼1 |
| Rf3PZnE-TDQ-ERf3PZn | 0.785 | 9.04 × 108 | 1.53 × 108 | 2.17 × 108 | 0.12 | 0.17 |
| Rf3PZnArPPtE-TDQ-EArPPtRf3PZn | 0.108 | 2.36 × 109 | 1.39 × 108 | 6.76 × 109 | 0.015 | 0.73 |
In contrast to the ultrafast ISC rate constant exhibited by ArPPtE-BTD-EArPPt, the ArPPtE-TDQ-EArPPt analogue manifests dramatically different excited-state relaxation dynamics. Three time constants were obtained from fitting the ArPPtE-TDQ-EArPPt transient data acquired following Q-band excitation (λexc = 750 nm): 1.05 ± 0.15 ps, 15 ± 2 ps, 330 ± 15 ps (Fig. S8D†). Analogous to the characteristic relaxation times evinced for ArPZn-Sp-ArPZn chromophores, the two fast time constants (1.05 ps and 15 ps) correspond respectively to established THF solvational and structural relaxation dynamics. The 330 ps component is comparable to the streak-scope determined fluorescence lifetime of ArPPtE-TDQ-EArPPt (τF = 310 ps, Fig. S6E†), and is thus assigned to the intrinsic S1 state lifetime. While the ArPZnE-TDQ-EArPZn transient spectra feature complete ground state recovery within 200 ps, those of ArPPtE-TDQ-EArPPt evolve at long-time delays to give rise to a new transient absorption band that spans the 800–1050 nm spectral domain. This transient absorption manifold persists beyond the delay limit of the femtosecond instrument (Fig. 2D). Nanosecond transient absorption spectroscopy determines the lifetime of this long-lived excited state to be 250 ns in deoxygenated solutions, while in ambient atmosphere saturated solutions it is reduced to 140 ns (Fig. S7A and B†), suggesting that the corresponding excited state is dominated by triplet character. Femtosecond timescale transient absorption spectral data allow direct evaluation of the S1 → T1 ISC quantum yield and rate constant (ΦISC ∼ 51%; kISC ∼ 1.67 × 109 s−1) for ArPPtE-TDQ-EArPPt (ESI; Table 2†). The excited-state relaxation dynamics of ArPPtE-TDQ-EArPPt thus contrast those evinced in classic oligomeric highly conjugated (porphinato)platinum(II) species where near-unit ISC quantum yields and sub-picosecond ISC time constants are manifested.8e Also note that the S1 state lifetime of ArPPtE-TDQ-EArPPt is about four times longer than that of its ArPZnE-TDQ-EArPZn analogue, whereas PPtn evince significantly diminished S1 state lifetimes relative to the corresponding PZnn chromophores due to ultrafast S1 → T1 ISC dynamics (Scheme 2).8d,e In this regard, the longer S1 state lifetime (Table 2) of ArPPtE-TDQ-EArPPt relative to ArPZnE-TDQ-EArPZn originates from two factors: (i) diminution of the nonradiative decay rate constant as the optical bandgap is widened when platinum(II) replaces zinc(II) in this chromophoric motif, and (ii) an unconventionally slow ISC rate constant. The origin of this slow ISC will be discussed in the context of TD-DFT calculations (vide infra).
These data (Fig. 1 and 2; Tables 1 and 2) demonstrate that varying the nature of the intervening proquinoidal spacer units drives substantially different absorptive and excited-state dynamical properties in ArPM-Sp-ArPM chromophores: (i) ArPZn-Sp-ArPZn chromophores, such as ArPZnE-TDQ-EArPZn, having significant excited-state proquinoidal character, exhibit broad long-wavelength absorptive manifolds and substantial NIR oscillator strength, negligible (ΦF < 0.01) fluorescence, and short-lived (∼tens of ps) singlet excited state lifetimes. (ii) ArPZn-Sp-ArPZn chromophores, such as ArPZnE-BTD-EArPZn, that feature less electronically excited-state proquinoidal character, exhibit narrower long-wavelength absorptive manifolds and less substantial NIR oscillator strength, long-lived (ns) S1 states and large (ΦF ∼ 0.25) fluorescence quantum yields. (iii) ArPPt-Sp-ArPPt chromophores, characterized by ArPPtE-TDQ-EArPPt and ArPPtE-BTD-EArPPt, evince similar dependences of absorptive oscillator strength distribution upon the nature of the proquinoidal spacer, but show an unusual dependence of the degree of electronically excited-state proquinoidal character upon the magnitude of kISC: ArPPtE-BTD-EArPPt, which features a modest extent of excited-sate proquinoidal character, features a S1 → T1 ISC rate constant (kISC = 1.04 × 1012 s−1) three orders of magnitude greater than that for ArPPtE-TDQ-EArPPt. The origins of these disparate photophysical properties modulated by these Sp units are explored below using TD-DFT calculations.
Computed electronic structures of ArPM-Sp-ArPM chromophores
The differing S1 state CT characters of these ArPM-Sp-ArPM chromophores are underscored by population analyses of their frontier orbitals (FOs) and the transition matrix eigenvectors computed by TD-DFT methods (Fig. 3). The S1 state of ArPZnE-TDQ-EArPZn is dominated (∼80%) by the HOMO → LUMO configuration, where ∼30% of the HOMO wavefunction amplitude and >70% of the LUMO amplitude is assigned to atoms in the TDQ spacer (see ESI† for molecular orbital population analyses). Photo-excitation of the ArPZnE-TDQ-EArPZn Qx-band region, thus, redistributes electron density from the ArPZn to the TDQ unit, resulting in a delocalized S1 state with substantial CT character. In contrast, ArPZnE-BTD-EArPZn manifests an S1 state composed of multiple single-excitation configurations that involve FOs from HOMO−3 to LUMO+3, where the shares of HOMO and LUMO wavefunction amplitudes that can be assigned to ArPZn and TDQ subunits are comparable. These data indicate that the S1 state for ArPZnE-BTD-EArPZn exhibits negligible CT character and substantial CI, more akin to classic porphyrin π → π* transitions and those characteristic of PZnn supermolecular chromophores.8a–c,9c These computational figures buttress the finding that the ArPZnE-TDQ-EArPZn Qx transition has greater quinoidal character than that of ArPZnE-BTD-EArPZn,2c as demonstrated by analyses of the Qx manifold FWHM and vibronic structures, fluorescence quantum yields, and the dynamic and solvent dependent Stokes shifts exhibited by these chromophores.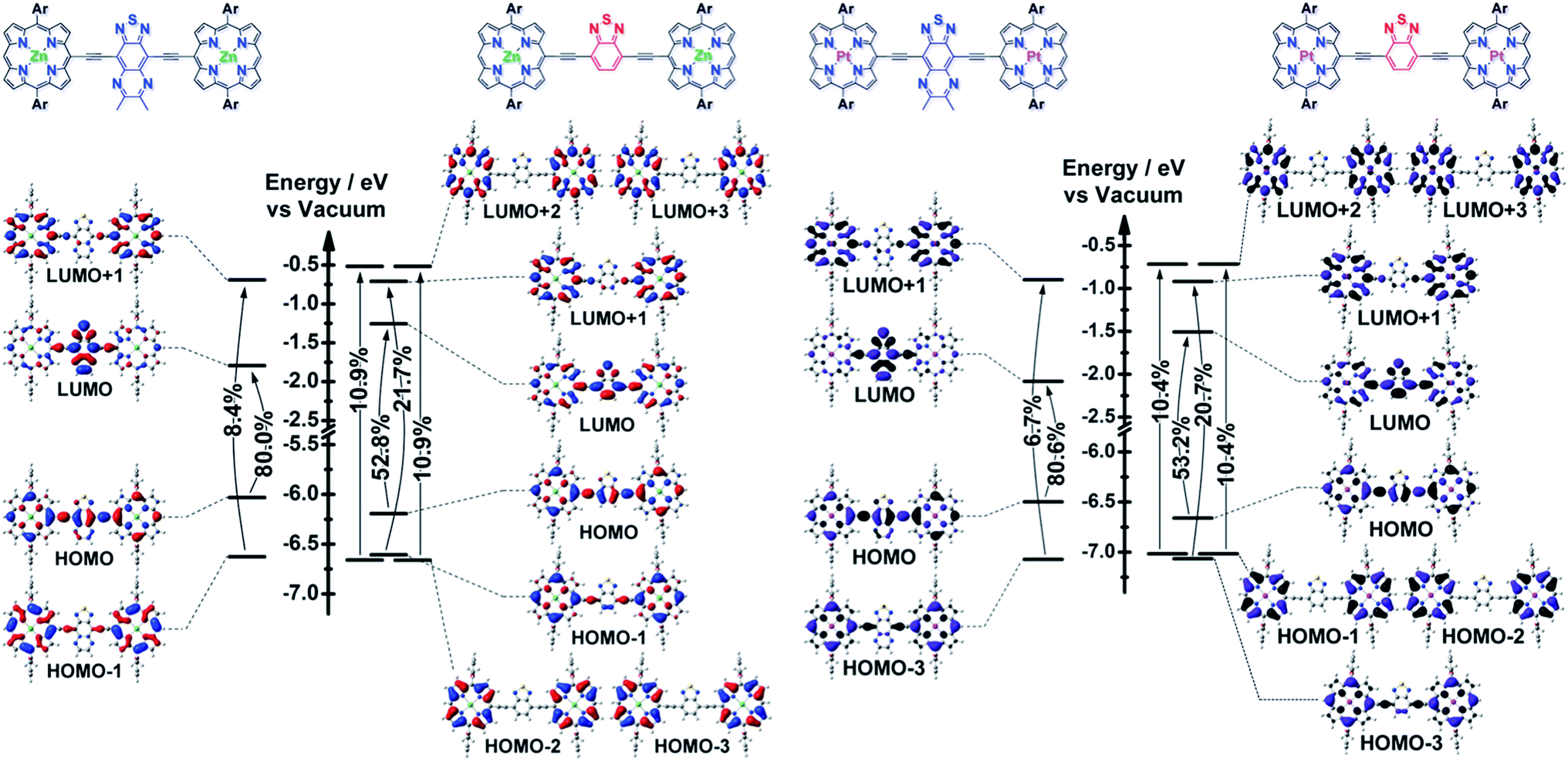 | ||
| Fig. 3 TD-DFT determined energy level diagrams and frontier molecular orbitals for ArPM-Sp-ArPM [M = Zn(II) or Pt(II), Sp = E-TDQ-E, or E-BTD-E], with arrows depicting the one-electron configurations that contribute most significantly to the low-energy (Qx) transitions. Calculations were performed at the M11/6-311g(d) theory level. | ||
The contrasts outlined above regarding the S1 state CT characters of the (porphinato)zinc(II) compounds are sustained in their (porphinato)platinum(II) counterparts. A further distinction that may be drawn for these latter ArPPtE-TDQ-EArPPt and ArPPtE-BTD-EArPPt chromophores is that a lesser share of the S1 state wavefunction amplitude lies proximal to the platinum atom for the TDQ-based compound than for the BTD-based analogue. Note that the ArPPtE-TDQ-EArPPt S0 → S1 transition is mainly described by the HOMO → LUMO configuration, and population analysis demonstrates that the (porphinato)platinum(II)-derived electron density contributes ∼23% to the ArPPtE-TDQ-EArPPt LUMO amplitude. In contrast, the ArPPtE-BTD-EArPPt S0 → S1 transition features the interaction of several configurations involving FOs beyond the HOMO and LUMO, and all of the virtual FOs evince dominant (>50%) wavefunction amplitude assigned to the (porphinato)platinum(II) fragment. These population analyses rationalize the correlation of large kISC for the ArPPtE-BTD-EArPPt S1 state with its strong CI character, and smaller kISC for the ArPPtE-TDQ-EArPPt S1 states with its strong CT character: as ISC can be induced by spin–orbit coupling (a local effect induced by wavefunction density proximal to a heavy atom),10a,15 it is clear that the wavefunction spatial distribution of the initially prepared singlet excited state of ArPPtE-BTD-EArPPt is responsible for its considerably greater S1–T1 mixing relative to that manifested for ArPPtE-TDQ-EArPPt.
Schematic potential energy surfaces summarizing the spectroscopic and dynamical data, and computed electronic structures for ArPME-Sp-EArPM chromophores
The spectroscopic, dynamical, and computational results for these ArPME-Sp-EArPM structures can be schematically illustrated using a series of qualitative potential energy surfaces as displayed in Scheme 3. In these schematic illustrations, the free energies of ArPM-Sp-ArPM chromophores in their ground states and low-lying singlet excited states are given as functions of a solvation coordinate, as spectroscopic and dynamical investigations indicate solvation dynamics play a prominent role in determining the excited-state relaxation pathways. TD-DFT calculations show that the ArPME-TDQ-EArPM S1 states feature stronger proquinoid contributions and greater CT character than those of ArPME-BTD-EArPM analogues (Fig. 3). The CT character of the ArPME-TDQ-EArPM Franck–Condon state initiates solvent dipolar reorientation, which in turn augments excited-state electronic polarization. The evolving solvent reorganization creates a ladder of stabilized states with progressively enhanced CT character relative to the initially prepared S1 state, funnelling excited-state energy through S1 → S0 nonradiative conversion that competes with S1 → T1 ISC (Scheme 3A). In this regard, note that the 1 ps characteristic solvent relaxation timescale (marked on potential energy surfaces for ArPME-TDQ-EArPM, Scheme 3A and C) corresponds to the timescale of the potential energy curve displacement (Δq) dynamics between S0 and S1 states that are driven along the solvent coordinate. The S0 → S1 vertical transition of ArPPtE-TDQ-EArPPt manifests a lower degree of CI relative to that for ArPPtE-BTD-EArPPt (vide supra), and the major single-excitation configuration involves virtual FOs featuring spacer-dominant (∼77%) electron density, resulting in diminished S1–T1 mixing (Scheme 3C), and hence the experimentally observed slow ISC rate constant (1.67 × 109 s−1). In contrast, the low-energy transition for ArPPtE-BTD-EArPPt exhibits a higher degree of CI, and (porphinato)platinum(II)-derived electron density makes prominent (>50%) contributions to the amplitudes of all the ArPPtE-BTD-EArPPt virtual orbitals involved in the corresponding transition, relative to that in the ArPPtE-BTD-EArPPt chromophore, thus giving rise to stronger S1–T1 mixing and the ultrafast ISC rate constant (1.04 × 1012 s−1) (Scheme 3D). Moreover, as the nature of the initially prepared S1 state of ArPPtE-BTD-EArPPt is barely perturbed by solvent dynamics, S1 → T1 ISC dominates excited-state relaxation processes, giving rise to the near-unity ΦISC in ArPPtE-BTD-EArPPt.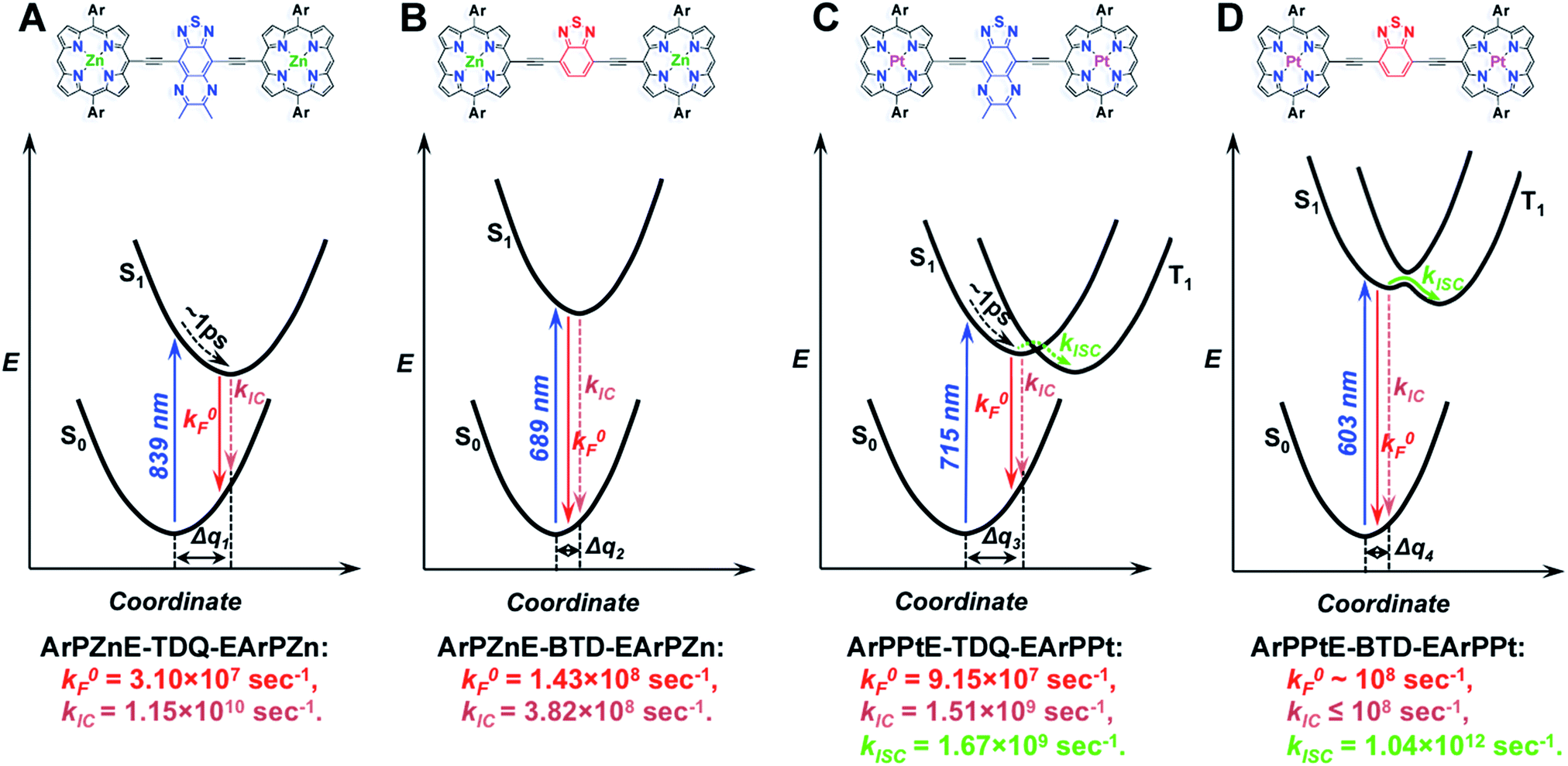 | ||
| Scheme 3 Schematic potential energy surfaces summarizing the spectroscopic and dynamical features, and computed electronic structures, for ArPME-Sp-EArPM chromophores [M = Zn(II) or Pt(II), Sp = thiadiazoloquinoxaline (TDQ), or benzothiadiazole (BTD)]. | ||
Relative frontier orbital energies of ArPM and spacers, and implications for new chromophore designs
The disparate CT characters of the ArPME-TDQ-EArPM and ArPME-BTD-EArPM S1 states trace their origins to relative (porphinato)metal and spacer fragment FO energies; DFT calculations thus suggest rules for the rational engineering of excited-state dynamics in these supermolecular chromophores (Fig. 4). Here, a critical distinction is the energetic alignment of the ArPM and Sp fragment LUMOs (ΔEL). For ArPME-BTD-EArPM compounds, which feature multiconfigurational S1 states of modest CT character, ΔEL between ArPME and E-BTD-E is smaller than 0.3 eV. For ArPME-TDQ-EArPM chromophores, which feature HOMO → LUMO-dominated S1 states of substantial CT character, ΔEL is larger than 1 eV. If the extent of CT character and degree of CI that describe the S1 states of ArPME-Sp-EArPM chromophores define the dominant factors that determine their ISC rate constants and fluorescence quantum yields, then minimizing ΔEL for ArPZnE-Sp-EArPZn chromophores will be the key to designing new long wavelength fluorophores, while minimizing ΔEL for ArPPtE-Sp-EArPPt chromophores will enable elucidation of NIR absorbers that manifest substantial ISC quantum yields.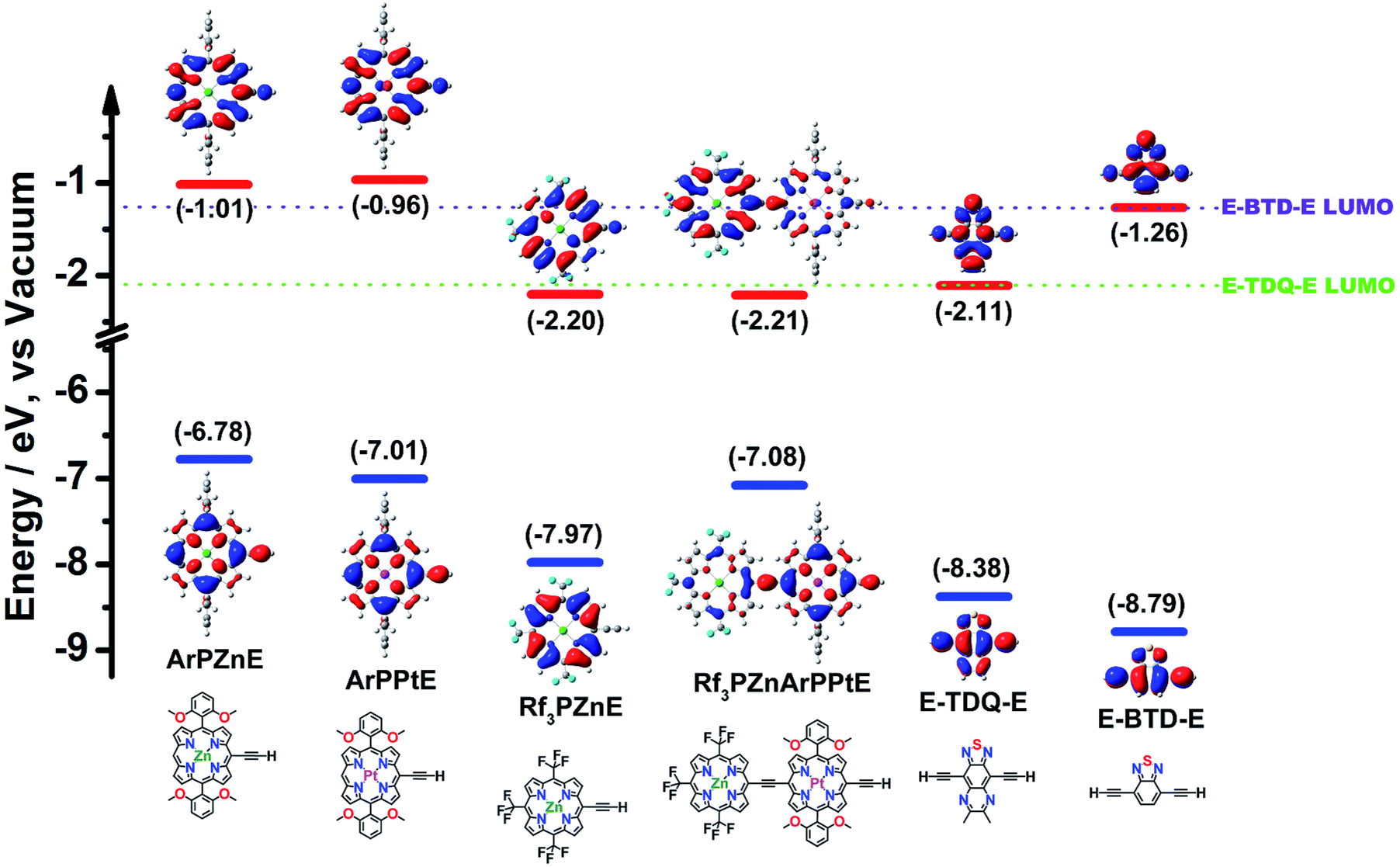 | ||
| Fig. 4 TD-DFT determined frontier molecular orbitals of the precursor fragments (ArPZnE, ArPPtE, Rf3PZnE, Rf3PZnArPPtE, E-TDQ-E, and E-BTD-E) plotted as 0.02 isodensity surfaces along with the corresponding calculated energy levels. Calculations were performed at the M11/6-311g(d) theory level. | ||
Supermolecules built from the TDQ spacer and porphyrin fragments endowed with LUMO energies that are substantially more stable than those of the meso-aryl derivatives mentioned so far permit the validation of this hypothesis. An established approach that uniformly stabilizes porphyrin macrocycle FOs by an inductive σ-electron withdrawing effect exploits meso-perfluoroalkyl substitution.8c,16 The recently described electron-deficient meso-tris(perfluoroalkyl)porphyrin Rf3PZnE is an ideal building for this purpose, as its HOMO and LUMO are both substantially stabilized relative to those of the [(5,-10,20-diphenylporphinato)zinc(II)]ethynyl (ArPZnE) unit (Fig. 4).17 Furthermore, as the computed Rf3PZnArPPtE LUMO is energetically close to that of Rf3PZnE (Fig. 4), the Rf3PZnArPPtE-TDQ-EArPPtERf3PZn compound was designed with the expectation that it would display attenuated electronically excited-state proquinoidal character compared to ArPPtE-TDQ-EArPPt, and thus an ISC rate constant substantially larger than that of the parent chromophore ArPPtE-TDQ-EArPPt. Likewise, Rf3PZnE-TDQ-ERf3PZn was designed with the expectation that this electronic structural engineering would provide a NIR fluorophore, and contrast those photophysics delineated above for ArPZnE-TDQ-EArPZn.
To that end, Rf3PZnE-TDQ-ERf3PZn and Rf3PZnArPPtE-TDQ-EArPPtERf3PZn were synthesized by methods closely analogous to those that provided the chromophores discussed above. The steady-state electronic absorption and emission spectra for Rf3PZnE-TDQ-ERf3PZn reveal features that significantly differ from those of ArPZnE-TDQ-EArPZn, while resembling those of ArPZnE-BTD-EArPZn (Fig. 5A, Table 1); Rf3PZnE-TDQ-ERf3PZn spectroscopic highlights include a narrow lowest-energy absorption manifold (Qx(FWHM) = 1163 cm−1), a modest Stokes shift (Δν = 851 cm−1), and a substantial fluorescence quantum yield (ΦF = 0.12) in THF solvent. Likewise, Rf3PZnArPPtE-TDQ-EArPPtRf3PZn manifests spectral signatures that reflect attenuated S1 state CT character relative to the parent chromophore ArPPtE-TDQ-EArPPt, and features a low-energy transition having a reduced Qx(FWHM) (1738 cm−1) and a smaller Stokes shift (Δν = 945 cm−1) relative to that evinced by ArPPtE-TDQ-EArPPt. These steady-state spectroscopic signatures evinced by the meso-perfluoroalkyl(porphinato)metal(II) supermolecules vindicate the prediction based on TD-DFT calculations: due to the minimization of ΔEL, Rf3PZnE-TDQ-ERf3PZn and Rf3PZnArPPtE-TDQ-EArPPtRf3PZn possess globally delocalized S1 states having reduced CT character relative to those of ArPME-TDQ-EArPM.
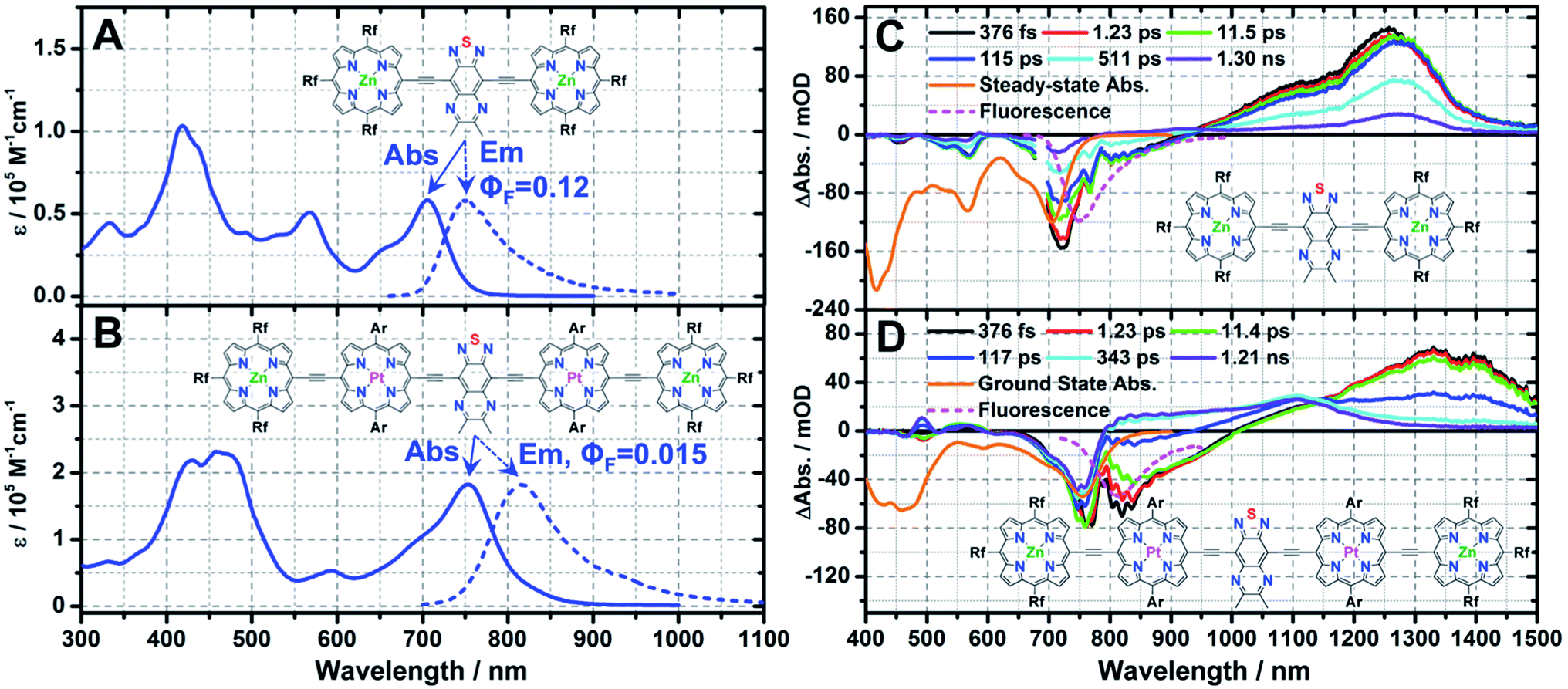 | ||
| Fig. 5 Steady-state and pump-probe transient absorption spectra recorded for Rf3PZnE-TDQ-ERf3PZn and Rf3PZnArPPtE-TDQ-EArPPtRf3PZn: steady-state electronic absorption and emission spectra for (A) Rf3PZnE-TDQ-ERf3PZn (λexc = 650 nm for emission) and (B) Rf3PZnArPPtE-TDQ-EArPPtRf3PZn (λexc = 700 nm for emission), solvent = THF, ambient condition. Pump-probe transient absorption spectra recorded at representative time delays for (C) Rf3PZnE-TDQ-ERf3PZn (λexc = 680 nm) and (D) Rf3PZnArPPtE-TDQ-EArPPtRf3PZn (λexc = 800 nm), solvent = THF, ambient temperature, magic angle polarization, steady-state absorption (orange solid line) and fluorescence (magenta dash line) are displayed as inverted spectra. Ar = 2′,6′-bis(3,3-dimethyl-1-butyloxy)phenyl; Rf = heptafluoropropyl. | ||
Representative transient absorption spectra recorded at selected time delays for Rf3PZnE-TDQ-ERf3PZn and Rf3PZnArPPtE-TDQ-EArPPtRf3PZn are displayed in Fig. 5C and D; key spectral data are highlighted in Table 2. Rf3PZnE-TDQ-ERf3PZn evinces no dynamic Stokes shift in the femtosecond transient absorption spectra, indicating modest CT character of the Franck–Condon state congruent with the minimal ΔEL calculated for this chromophore. As a result, S1 → S0 internal conversion along the solvation coordinate is greatly diminished, and the S1 state lifetime of Rf3PZnE-TDQ-ERf3PZn reaches ∼0.8 ns, comparable to that of ArPZnE-BTD-EArPZn. For Rf3PZnArPPtE-TDQ-EArPPtRf3PZn, because of the low ΔEL between the Rf3PZnArPPtE wings and the central TDQ unit, the S0 → S1 vertical transition involves less electron density redistribution than that of ArPPtE-TDQ-EArPPt. As a result, Rf3PZnArPPtE-TDQ-EArPPtRf3PZn manifests an ISC rate constant about four times as large as that of the parent chromophore ArPPtE-TDQ-EArPPt (6.76 × 1012 s−1vs. 1.67 × 1012 s−1), and now displays photophysics that approach those of ArPPtE-BTD-EArPPt. Note that these changes of ISC rate constant reflected in the evolution of supermolecular structure from ArPPtE-TDQ-EArPPt to Rf3PZnArPPtE-TDQ-EArPPtRf3PZn strikingly contrast the corresponding photophysics evinced in classic PZnn (Scheme 2) multimeric species, where ISC rate constants decrease dramatically with increasing PZnn conjugation length.8d The spectroscopic features displayed by ArPPtE-TDQ-EArPPt and Rf3PZnArPPtE-TDQ-EArPPtRf3PZn highlight the cardinal role of S1 state proquinoidal character in tuning the NIR ground-state absorption, fluorescence quantum yield, and ISC rate constant for long-wavelength absorbing (porphinato)metal(II) species.
Conclusion
While the influence of proquinoidal character upon the linear absorption spectrum of low optical bandgap π-conjugated polymers and molecules is well understood, less insight exists concerning its impact upon excited-state relaxation pathways and dynamics. Exploiting a series of model highly conjugated NIR-absorbing chromophores based on a (porphinato)metal(II)-spacer-(porphinato)metal(II) (PM-Sp-PM) structural motif in which the porphyryl ligand is either electron rich (10,20-diarylporphyrin, ArP) or electron deficient (10,15,20-tris(perfluoroalkyl)porphyrin, Rf3P), the central metal ion is Zn(II) or Pt(II), and the proquinoidal Sp is either 4,7-diethynylbenzo[c][1,2,5]thiadiazole (E-BTD-E) or 4,9-diethynyl-6,7-dimethyl[1,2,5]thiadiazolo[3,4-g]quinoxaline (E-TDQ-E), we elucidate design principles important for controlling the excited-state dynamics of highly conjugated NIR-absorbing materials that feature conjugation motifs that rely on proquinoidal units, and define strategies through which S1 → S0 radiative (kr), S1 → T1 intersystem crossing (kISC), and S1 → S0 internal conversion (kIC) rate constants may be manipulated over many orders of magnitude.A combination of excited-state dynamical studies and TD-DFT calculations: (i) points to the cardinal role that excited-state configuration interaction (CI) plays in determining the magnitudes of kr(S1 → S0), kISC(S1 → T1), and kIC(S1 → S0) in these PM-Sp-PM chromophores, and (ii) suggests that a primary determinant of CI magnitude derives from the energetic alignment of the PM and Sp fragment LUMOs. The chromophore ArPZnE-BTD-EArPZn, whose low-lying excited state is characterized by a substantial degree of CI, defines a NIR emitter having a substantial fluorescence quantum yield (λem(S1 → S0) = 740 nm; ΦF = 0.25); in contrast, ArPZnE-TDQ-EArPZn, whose low-lying excited state is described by a modest degree of CI and substantial charge transfer (CT) character, features an internal conversion rate constant, kIC, more than two orders of magnitude larger than its BTD analogue. Optical excitation of ArPPtE-BTD-EArPPt likewise generates a low-lying excited state having a substantial extent of CI, and gives rise to unit quantum yield formation of its electronically excited triplet state. In contrast, excitation of ArPPtE-TDQ-EArPPt demonstrates an unusually fluorescent Pt(II) complex, with an kISC three orders of magnitude smaller than its BTD analogue; the low CI/high CT character of the excited state provides a path to short-circuit the large magnitude spin–orbit coupling associated with heavy metal chromophores by minimizing S1-state wavefunction density proximal to the heavy Pt nuclei, thus realizing a rare if not unique example of a fluorescent (porphinato)platinum(II) chromophore at ambient temperature having ISC dynamics on the ns timescale.8e,13b,18
As TD-DFT computations and excited-state dynamical studies correlate high CI with modest energy gaps (ΔEL) between PM and Sp fragment LUMOs in PM-Sp-PM chromophores, Rf3PZnE-TDQ-ERf3PZn and Rf3PZnArPPtE-TDQ-EArPPtRf3PZn were synthesized: the σ-electron withdrawing effect of Rf3PZnEmeso-perfluoroalkyl moieties serves to align the Sp and PM LUMOs of these chromophores, defining NIR absorbers having low-lying excited states characterized by extensive CI. Rf3PZnE-TDQ-ERf3PZn thus contrasts ArPZnE-TDQ-EArPZn in that NIR fluorescence dominates its photophysics. Likewise, Rf3PZnArPPtE-TDQ-EArPPtRf3PZn, because of the low ΔEL between the Rf3PZnArPPtE wings and the central TDQ unit, features expansive excited-state CI and a large kISC, displaying photophysics congruent with large magnitude spin–orbit coupling.
For non-heavy-metal containing low optical bandgap π-conjugated structures that produce long-lived triplet states at high quantum yield, this work shows that these photophysics trace their genesis to the modest degree of CI that characterizes the low-lying excited states of these systems; however, if electronic structural modifications are introduced that give rise to high CI excited states, exceptional fluorophores may be produced. For heavy metal-containing low optical bandgap π-conjugated motifs, this work enables proquinoidal NIR chromophores having counter-intuitive properties to be evolved: for example, while (porphinato)platinum compounds are well known to generate non-emissive triplet states at unit quantum yield upon optical excitation at ambient temperature, diminishing the extent of excited-state CI in these systems realizes long-wavelength absorbing heavy-metal fluorophores. In sum, this work highlights approaches to: (i) modulate low-lying singlet excited-state lifetime over the picosecond-to-nanosecond time domain, (ii) achieve NIR fluorescence with quantum yields up to 25%, (iii) tune the magnitude of the S1–T1 ISC rate constant from 109 to 1012 s−1 and (iv) realize T1-state lifetimes that range from ∼0.1 to several μs, for these model PM-Sp-PM chromophores, and renders new insights to evolve bespoke photophysical properties for low optical bandgap π-conjugated polymers and molecules based on proquinoidal conjugation motifs.
Given the disparate photophysical requirements for NIR absorbers for applications ranging from optical power limiting to triplet–triplet annihilation photochemistry to in vivo bioimaging, this work underscores the photophysical diversity that may be engineered in supermolecular chromophores,2c,8a,c,d,12,19 and demonstrates how electronic modulation of proquinoidal conjugation motifs, conventionally utilized to regulate the ground-state absorptivity of low optical bandgap π-conjugated polymers and molecules, can also be exploited as powerful means to modulate their excited-state relaxation pathways.
Experimental section
Synthesis and characterization
The synthetic procedures and corresponding characterization data of all new compounds, complete with the reaction schemes, are given in the ESI.†Instrumentation
Electronic absorption spectra were recorded on a Shimadzu UV-1700 spectrophotometer. Steady-state emission spectra were recorded on a FLS920 spectrometer that utilized a xenon lamp (Xe900) as excitation light source and an extended red sensitive PMT (Hamamatsu R2658P side window photomultiplier, spectral range: 200–1010 nm) or NIR-PMT (Hamamatsu H10330-75, spectral range: 950–1700 nm) for detection. Emission spectra were corrected using a calibration curve supplied with the instrument.Picosecond fluorescence lifetime measurement system (streak-scope)
Time-resolved emission spectra were recorded using a Hamamatsu C4780 picosecond fluorescence lifetime measurement system. This system employs a Hamamatsu Streakscope C4334 as its photon-counting detector; a Hamamatsu C4792-01 synchronous delay generator electronically generated all time delays. The excitation light source chosen was a Hamamatsu 405 nm diode laser. All fluorescence data were acquired in single-photon-counting mode using Hamamatsu HPD-TA software. The data were analyzed using the Hamamatsu fitting module; both non-deconvoluted and deconvoluted data analyses were performed to ascertain whether or not any emissive processes were excitation pulse-limited.Femtosecond and nanosecond transient absorption experiments
The transient optical system utilized in this work has been discussed previously.20 All the samples for pump-probe experiments were deoxygenated via three successive freeze–pump–thaw cycles prior to measurement.Time-dependent density functional theory calculations
All electronic structure calculations were performed upon model compounds in which aliphatic chains were truncated to methyl groups (ESI†). Structure optimization and linear response calculations were performed with density functional theory (DFT) using Gaussian 09, revision C.1.21 The M11 (ref. 22) functional was employed for all calculations. Optimizations were performed with minimal symmetry constraints using tight optimization criteria; initial optimizations used smaller basis sets but the final optimizations and TD-DFT calculations employed the 6-311g(d) basis set23 as implemented in Gaussian 09. Selected frontier orbital wave functions were plotted as isosurfaces (iso = 0.02) using Gaussview 5.24 TD-DFT result files were post-processed using the GaussSum package;25 this software partitions the wave function amplitudes onto atomic components using Mulliken population analysis,26 and parses the electronic configurations contributing to each excitation.Conflict of interest
No competing financial interests have been declared.Acknowledgements
This work was funded by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, of the U.S. Department of Energy through Grant DE-SC0001517. J. L., P. Z., and D. N. B. acknowledge financial support from the National Institutes of Health through Grant GM048043.References
- (a) L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed; (b) Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed; (c) G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS; (d) C. W. Spangler, J. Mater. Chem., 1999, 9, 2013–2020 RSC; (e) J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef CAS PubMed.
- (a) T. D. Lash, P. Chandrasekar, A. T. Osuma, S. T. Chaney and J. D. Spence, J. Org. Chem., 1998, 63, 8455–8469 CrossRef CAS; (b) J. J. Piet, P. N. Taylor, H. L. Anderson, A. Osuka and J. M. Warman, J. Am. Chem. Soc., 2000, 122, 1749–1757 CrossRef CAS; (c) K. Susumu, T. V. Duncan and M. J. Therien, J. Am. Chem. Soc., 2005, 127, 5186–5195 CrossRef CAS PubMed; (d) C.-Y. Lin, Y.-C. Wang, S.-J. Hsu, C.-F. Lo and E. W.-G. Diau, J. Phys. Chem. C, 2010, 114, 687–693 CrossRef CAS; (e) H. J. Song, D. H. Kim, T. H. Lee and D. K. Moon, Eur. Polym. J., 2012, 48, 1485–1494 CrossRef CAS; (f) Y. Huang, L. Li, X. Peng, J. Peng and Y. Cao, J. Mater. Chem., 2012, 22, 21841–21844 RSC; (g) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed; (h) M. Morisue, Y. Hoshino, M. Nakamura, T. Yumura, S. Machida, Y. Ooyama, M. Shimizu and J. Ohshita, Inorg. Chem., 2016, 55, 7432–7441 CrossRef CAS PubMed; (i) N. Jiang, G. Zuber, S. Keinan, A. Nayak, W. Yang, M. J. Therien and D. N. Beratan, J. Phys. Chem. C, 2012, 116, 9724–9733 CrossRef CAS; (j) K. Susumu and M. J. Therien, J. Porphyrins Phthalocyanines, 2015, 19, 205–218 CrossRef CAS.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- (a) G.-J. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2011, 40, 2541–2566 RSC; (b) M. Calvete, G. Y. Yang and M. Hanack, Synth. Met., 2004, 141, 231–243 CrossRef CAS.
- (a) T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS; (b) J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937–950 RSC; (c) J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- (a) S. A. Hilderbrand and R. Weissleder, Curr. Opin. Chem. Biol., 2010, 14, 71–79 CrossRef CAS PubMed; (b) L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC; (c) P. P. Ghoroghchian, M. J. Therien and D. A. Hammer, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 156–167 CrossRef CAS PubMed.
- (a) S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627–630 CrossRef CAS; (b) S. G. DiMagno, V. S.-Y. Lin and M. J. Therien, J. Org. Chem., 1993, 58, 5983–5993 CrossRef CAS; (c) S. G. DiMagno, V. S.-Y. Lin and M. J. Therien, J. Am. Chem. Soc., 1993, 115, 2513–2515 CrossRef CAS; (d) S. M. LeCours, H.-W. Guan, S. G. DiMagno, C. H. Wang and M. J. Therien, J. Am. Chem. Soc., 1996, 118, 1497–1503 CrossRef.
- (a) V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105–1111 CAS; (b) V. S.-Y. Lin and M. J. Therien, Chem.–Eur. J., 1995, 1, 645–651 CrossRef CAS; (c) K. Susumu and M. J. Therien, J. Am. Chem. Soc., 2002, 124, 8550–8552 CrossRef CAS PubMed; (d) T. V. Duncan, K. Susumu, L. E. Sinks and M. J. Therien, J. Am. Chem. Soc., 2006, 128, 9000–9001 CrossRef CAS PubMed; (e) T. V. Duncan, P. R. Frail, I. R. Miloradovic and M. J. Therien, J. Phys. Chem. B, 2010, 114, 14696–14702 CrossRef CAS PubMed.
- (a) R. Kumble, S. Palese, V. S.-Y. Lin, M. J. Therien and R. M. Hochstrasser, J. Am. Chem. Soc., 1998, 120, 11489–11498 CrossRef CAS; (b) R. Shediac, M. H. B. Gray, H. T. Uyeda, R. C. Johnson, J. T. Hupp, P. J. Angiolillo and M. J. Therien, J. Am. Chem. Soc., 2000, 122, 7017–7033 CrossRef CAS; (c) I. V. Rubtsov, K. Susumu, G. I. Rubtsov and M. J. Therien, J. Am. Chem. Soc., 2003, 125, 2687–2696 CrossRef CAS PubMed.
- (a) N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, Calif., 2010 Search PubMed; (b) Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed; (c) P. Panda, D. Veldman, J. Sweelssen, J. J. A. M. Bastiaansen, B. M. W. Langeveld-Voss and S. C. J. Meskers, J. Phys. Chem. B, 2007, 111, 5076–5081 CrossRef CAS PubMed.
- (a) D. Eastwood and M. Gouterman, J. Mol. Spectrosc., 1970, 35, 359–375 CrossRef CAS; (b) M. Gouterman, Optical Spectra and Electronic Structure of Porphyrins, Academic Press, New York, 1978 Search PubMed; (c) A. Harriman, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 1281–1291 RSC.
- T. V. Duncan, T. Ishizuka and M. J. Therien, J. Am. Chem. Soc., 2007, 129, 9691–9703 CrossRef CAS PubMed.
- (a) R. S. Becker and J. B. Allison, J. Phys. Chem., 1963, 67, 2662–2669 CrossRef CAS; (b) J. R. Sommer, A. H. Shelton, A. Parthasarathy, I. Ghiviriga, J. R. Reynolds and K. S. Schanze, Chem. Mater., 2011, 23, 5296–5304 CrossRef CAS.
- (a) A. Samanta, J. Phys. Chem. B, 2006, 110, 13704–13716 CrossRef CAS PubMed; (b) M. Glasbeek and H. Zhang, Chem. Rev., 2004, 104, 1929–1954 CrossRef CAS PubMed.
- I. V. Khudyakov, Y. A. Serebrennikov and N. J. Turro, Chem. Rev., 1993, 93, 537–570 CrossRef CAS.
- (a) S. G. DiMagno, R. A. Williams and M. J. Therien, J. Org. Chem., 1994, 59, 6943–6948 CrossRef CAS; (b) J. G. Goll, K. T. Moore, A. Ghosh and M. J. Therien, J. Am. Chem. Soc., 1996, 118, 8344–8354 CrossRef CAS; (c) J. Rawson, A. C. Stuart, W. You and M. J. Therien, J. Am. Chem. Soc., 2014, 136, 17561–17569 CrossRef CAS PubMed.
- J. Rawson, Exploration of Porphyrin-based Semiconductors for Negative Charge Transport, Ph.D. thesis, Duke University, Durham, NC, 2014.
- (a) A. Vogler, H. Kunkely and B. Rethwisch, Inorg. Chim. Acta, 1980, 46, 101–105 CrossRef CAS; (b) T. Kobayashi, K. D. Straub and P. M. Rentzepis, Photochem. Photobiol., 1979, 29, 925–931 CrossRef CAS; (c) K. D. Straub, P. M. Rentzepis and D. Huppert, J. Photochem., 1981, 17, 419–425 CrossRef CAS.
- T. V. Duncan, I. V. Rubtsov, H. T. Uyeda and M. J. Therien, J. Am. Chem. Soc., 2004, 126, 9474–9475 CrossRef CAS PubMed.
- J. Park, P. Deria and M. J. Therien, J. Am. Chem. Soc., 2011, 133, 17156–17159 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. E. Hada, M. K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Rev. C.1., Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 2, 2810–2817 CrossRef CAS.
- (a) A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS; (b) P. J. Hay, J. Chem. Phys., 1977, 66, 4377–4384 CrossRef CAS; (c) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (d) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS; (e) K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062–1065 CrossRef; (f) R. C. Binning and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206–1216 CrossRef CAS; (g) M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511–516 CrossRef CAS; (h) L. A. Curtiss, M. P. McGrath, J.-P. Blaudeau, N. E. Davis, R. C. Binning and L. Radom, J. Chem. Phys., 1995, 103, 6104–6113 CrossRef CAS; (i) J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016–5021 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, GaussView Ver. 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02150j |
| This journal is © The Royal Society of Chemistry 2017 |