
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst-controlled regioselectivity in phosphine catalysis: the synthesis of spirocyclic benzofuranones via regiodivergent [3 + 2] annulations of aurones and an allenoate†
Huanzhen
Ni
ab,
Zhaoyuan
Yu
 c,
Weijun
Yao
d,
Yu
Lan
*c,
Nisar
Ullah
*e and
Yixin
Lu
*abf
c,
Weijun
Yao
d,
Yu
Lan
*c,
Nisar
Ullah
*e and
Yixin
Lu
*abf
aGraduate School for Integrative Sciences & Engineering (NGS), National University of Singapore, #05-01, 28 Medical Drive, 117456, Singapore. E-mail: chmlyx@nus.edu.sg
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore
cSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400030, P. R. China. E-mail: lanyu@cqu.edu.cn
dDepartment of Chemistry, Zhejiang Sci-Tech University, 310018, P. R. China
eChemistry Department, King Fahd University of Petroleum and Materials, Dhahran 31261, Saudi Arabia. E-mail: nullah@kfupm.edu.sa
fNational University of Singapore (Suzhou) Research Institute, 377 Lin Quan Street, Suzhou Industrial Park, Suzhou, Jiangsu 215123, P. R. China
First published on 12th June 2017
Abstract
Catalyst-controlled regiodivergent [3 + 2] annulations of aurones and allenoates have been developed. When a dipeptide phosphine catalyst with an L-D- configuration was employed, α-selective [3 + 2] annulation products could be obtained with good regioselectivities and enantioselectivities. With the employment of L-L- dipeptide phosphines, γ-selective annulation products could be selectively obtained with excellent enantioselectivities. By simply tuning the catalyst configurations, a wide range of α-selective or γ-selective spirocyclic benzofuranones with either aryl or alkyl substitutions could be readily prepared. DFT calculations suggest that the conformation of the dipeptide phosphines influences the hydrogen bonding interactions or the distortion energy, resulting in delicate energy differentiation in the transition states, and accounting for the observed regioselectivity.
1. Introduction
The past decade has witnessed the blossoming of enantioselective nucleophilic phosphine catalysis.1 Among the wide range of phosphine-mediated asymmetric processes, phosphine-catalyzed annulations2–4 are arguably the most important reactions in synthetic organic chemistry. Ever since Lu’s seminal discovery of the phosphine-catalyzed [3 + 2] annulation of electron-deficient allenes with activated olefins in 1995,2a this powerful mode of cyclization has attracted enormous attention from synthetic organic chemists and has now become a common method for the construction of 5-membered ring systems. In a typical phosphine-catalyzed [3 + 2] annulation reaction5 between an allenoate and an activated alkene, the phosphine adds on to the allene and forms a zwitterionic intermediate, which has two resonance forms, and their reactions with activated olefins lead to the formation of α- or γ- regioisomers (Scheme 1). In the reported phosphine-catalyzed [3 + 2] annulation reactions, α-adducts and γ-adducts are often mixed. In most cases, the α-adducts can be obtained as the major or sole regioisomer. There are only a handful of examples describing the asymmetric formation of γ-selective regioisomers in [3 + 2] annulation processes.2g,2j,2k While the issue of regioselectivity is not particularly attended, it appears that the employment of different activated olefin substrates is the key to the observed γ-selectivity in those studies.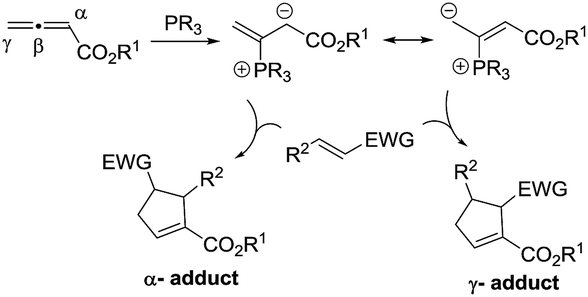 | ||
| Scheme 1 Regioselectivity in Lu’s [3 + 2] annulation between allenoates and activated alkenes. | ||
Given the widespread use of phosphine-mediated annulation reactions for ring construction, and the fact that obtaining different regioisomers in annulation reactions in an uncontrolled manner impedes the efficiency of these processes, it was quite surprising to note that the regioselectivity issue in phosphine-catalyzed [3 + 2] cycloadditions has not drawn much investigation. The only report2u devoting efforts to obtain both α- and γ- regioisomers is a study by Shi and co-workers, in which they employed either simple or γ-substituted allenoates as annulation partners in order to obtain different regioisomers (Scheme 2). It is certainly not very desirable that different substrates have to be prepared in order to access different regioisomers. Moreover, the requirement of employing different allene/olefin reaction partners limits the general applicability of the annulation methods. We aimed to address this challenging issue by developing a general strategy to access different regioisomers from the same starting materials, i.e. without varying the allenes and olefins in phosphine-triggered [3 + 2] annulation reactions. Building upon our previous success of dipeptide-based phosphines,6 we hypothesize that different regioisomers may be obtained by employing different diastereomeric dipeptide phosphine catalysts (Scheme 2). We envision that the ready tunability of dipeptide structures in phosphine catalysts may be utilized to provide not only efficient stereochemical control, but also serve as an effective means to differentiate pathways leading to the divergent formation of regioisomers.
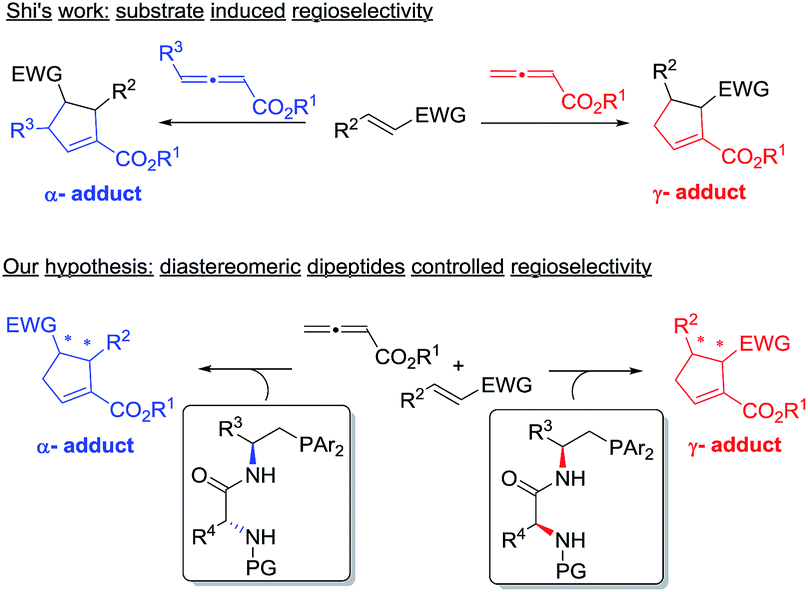 | ||
| Scheme 2 Controlling regioselectivity in [3 + 2] annulations. | ||
Aurones are an important class of unique natural products exhibiting remarkable biological activities,7 and they are widely used as synthetic building blocks.8 However, the applications of aurones in phosphine catalysis are unknown. As part of our continuous efforts in asymmetric phosphine catalysis,9 we envisioned that aurones could be used as C2 synthons in [3 + 2] annulation with allenes, for the creation of structurally unique spiroaurone motifs. In this article, we document the first catalyst-controlled regiodivergent [3 + 2] annulations of aurones; by employing dipeptide phosphines with either an L-D- or an L-L- configuration, the annulation of aurones with allenoates yielded either α- or γ-selective spirobenzofuranones in a highly enantioselective and diastereoselective manner.
2. Results and discussion
2.1 Tuning the configurations of dipeptide phosphines for different regioselectivity
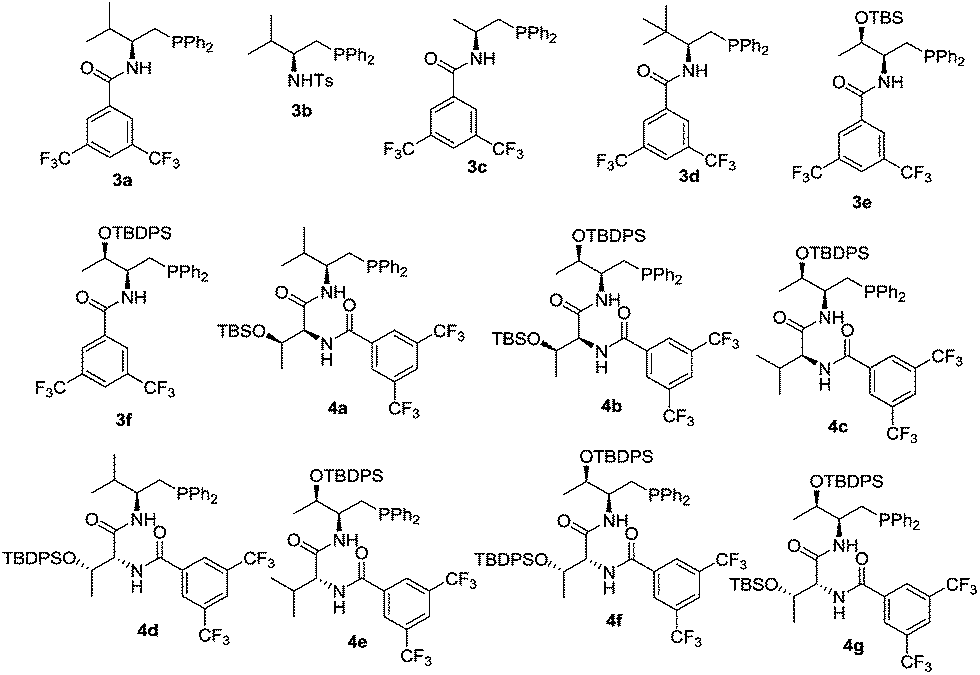
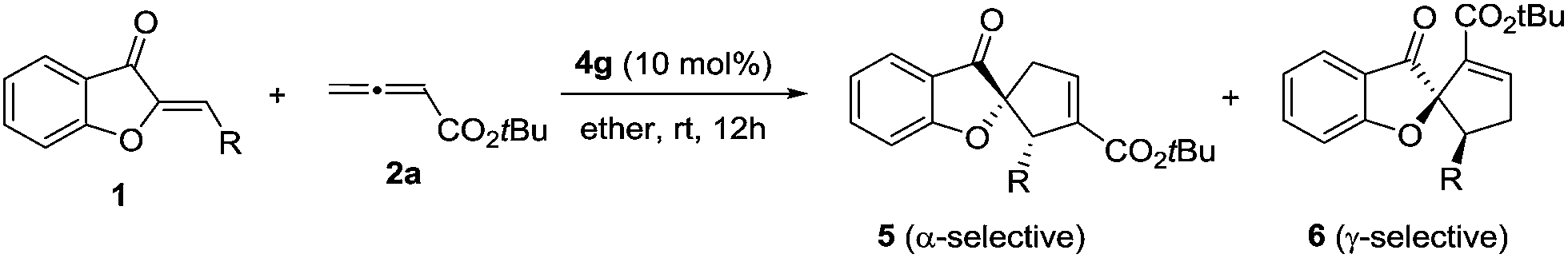
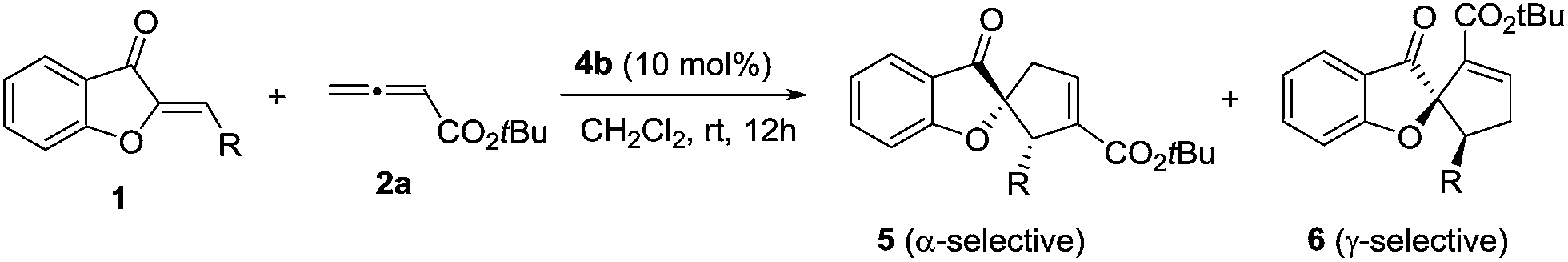
We initiated our investigation by examining the catalytic effects of a number of amino acid-derived phosphines on the annulation reaction between aurone 1a and allenoate 2a, and the results are summarized in Table 1. Mono-amino acid-derived phosphines led to the formation of products with a certain degree of regioselectivity and moderate enantioselectivity (entries 1–6). We were delighted to discover that dipeptide phosphine catalysts were efficient in promoting regiodivergent cyclizations. L-Val-L-thr-derived 4a, L-thr-L-thr-based 4b, and L-thr-L-val-derived 4c all favored the formation of γ-adducts, which were obtained with excellent enantioselectivity (entries 7–9). Interestingly, when dipeptide catalysts 4d, 4e, 4f, and 4g with an L-D- configuration were used, α-selective cycloaddition products were constantly obtained as the major regioisomer with excellent enantioselectivity (entries 10–13). The regioselectivity of the annulations was further enhanced by performing a quick solvent screening (entries 14–18). Under optimal conditions, the [3 + 2] annulation reaction catalyzed by L-thr-D-thr-based 4g in ether afforded regioisomers in a ratio of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, favoring the α-isomer (94% ee) (entry 14), whereas the reaction promoted by L-thr-L-thr-derived 4b in CH2Cl2 led to the selective formation of the α-annulation product (α/γ = 1:6, 98% ee for γ-isomer) (entry 17).
:1, favoring the α-isomer (94% ee) (entry 14), whereas the reaction promoted by L-thr-L-thr-derived 4b in CH2Cl2 led to the selective formation of the α-annulation product (α/γ = 1:6, 98% ee for γ-isomer) (entry 17).
|
|
|||||
|---|---|---|---|---|---|
|
|
|||||
| Entry | Cat. | Solvent |
5a:6ab |
Yieldc (%) | eed (%) |
| a Reactions were performed with 1a (0.10 mmol), 2a (0.12 mmol) and the catalyst (0.01 mmol) in the solvent specified (1 mL) at room temperature. b Determined by crude 1H NMR analysis. c Isolated yield for the major regioisomer. d The ee value for the major regioisomer, determined by HPLC analysis on a chiral stationary phase. | |||||
| 1 | 3a | Toluene | 3:1 |
68 | 52 |
| 2 | 3b | Toluene | 2:1 |
42 | 1 |
| 3 | 3c | Toluene | 1:1 |
23 | 34 |
| 4 | 3d | Toluene | 2:1 |
58 | 79 |
| 5 | 3e | Toluene | 3:1 |
70 | 64 |
| 6 | 3f | Toluene | 3:1 |
69 | 74 |
| 7 | 4a | Toluene | 1:4 |
72 | 97 |
| 8 | 4b | Toluene | 1:4 |
74 | 98 |
| 9 | 4c | Toluene | 1:3.5 |
70 | 92 |
| 10 | 4d | Toluene | 2:1 |
52 | 79 |
| 11 | 4e | Toluene | 6:1 |
78 | 93 |
| 12 | 4f | Toluene | 5:1 |
76 | 94 |
| 13 | 4g | Toluene | 6:1 |
80 | 96 |
| 14 | 4g | Ether |
13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
88 | 94 |
| 15 | 4g | CH2Cl2 | 4:1 |
73 | 91 |
| 16 | 4g | EtOAc | 19:1 |
92 | 90 |
| 17 | 4b | CH 2 Cl 2 |
1:6
|
80 | 98 |
| 18 | 4b | CHCl3 | 1:5 |
76 | 97 |
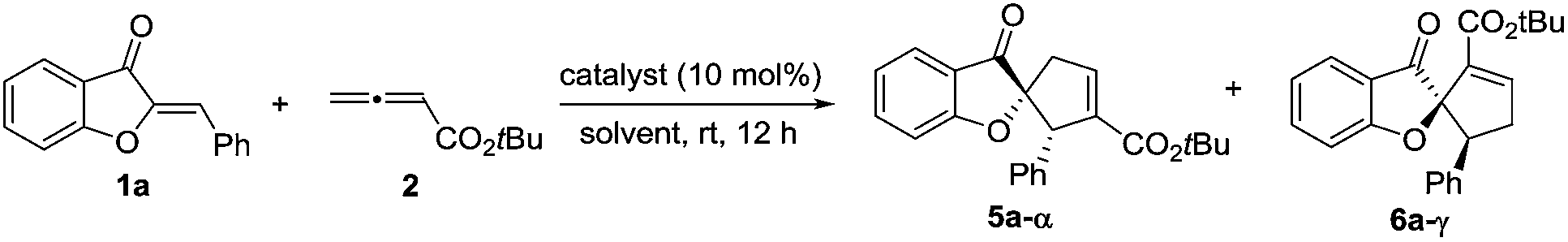
2.2 The α-selective and γ-selective [3 + 2] annulations: the substrate scope
The scope of this regiodivergent [3 + 2] annulation was subsequently probed. When different substituted aurones 1 were reacted with allenoate 2a in the presence of L-thr-D-thr-based 4g in ether, the α-adducts were formed selectively (Table 2). The reaction was applicable to various aryl-substituted aurones, regardless of the substitution patterns and electronic properties of the aryl moiety, and excellent enantioselectivities and very good regioselectivities were attainable (entries 1–12). Aurones with an aliphatic substituent could also be used, albeit lower α-selectivities were observed (entries 13–16). In all the examples examined, the pure α-adducts could be isolated mostly in good yields and with excellent enantioselectivities.|
|
|||||
|---|---|---|---|---|---|
| Entry | R (1) |
5:6b |
5 | Yieldc (%) | eed (%) |
| a Reactions were performed with 1 (0.10 mmol), 2a (0.12 mmol) and 4g (0.01 mmol) in ether (1 mL) at room temperature. b Determined by crude 1H NMR analysis. c Isolated yield for the pure α-regioisomer. d The ee value for the α-regioisomer, determined by HPLC analysis on a chiral stationary phase. e The catalyst loading was 20 mol%. | |||||
| 1 | Ph (1a) | 13:1 |
5a | 88 | 94 |
| 2 | 4-Cl-C6H4 (1b) | 9:1 |
5b | 81 | 93 |
| 3 | 3-Cl-C6H4 (1c) | 6:1 |
5c | 75 | 91 |
| 4 | 2-Cl-C6H4 (1d) | 7:1 |
5d | 76 | 91 |
| 5 | 4-F-C6H4 (1e) | 9:1 |
5e | 83 | 94 |
| 6 | 4-OMe-C6H4 (1f) | 10:1 |
5f | 87 | 96 |
| 7 | 4-Me-C6H4 (1g) | 8:1 |
5g | 76 | 95 |
| 8 | 2-Me-C6H4 (1h) | 15:1 |
5h | 85 | 97 |
| 9 | 4-CN-C6H4 (1i) | 13:1 |
5i | 69 | 93 |
| 10 | 2-Naphthyl (1j) | 5:1 |
5j | 74 | 94 |
| 11 | 3,4-(OMe)2-C6H4 (1k) | 5:1 |
5k | 80 | 96 |
| 12 | 2-Thienyl (1l) | 12:1 |
5l | 73 | 95 |
| 13e | Cyclohexyl (1m) | 3:1 |
5m | 62 | 95 |
| 14e | Isopropyl (1n) | 3:1 |
5n | 53 | 94 |
| 15e | nBu (1o) | 5:1 |
5o | 40 | 94 |
| 16e | Ethyl (1p) | 6:1 |
5p | 60 | 96 |
The scope of γ-selective [3 + 2] annulation between substituted aurones 1 and allenoate 2a in the presence of L-thr-L-thr-based 4b was next investigated (Table 3). Similarly, aurones with simple/fused aryl and heterocyclic substituents (entries 1–12) and alkyl substituents (entries 13–16) were shown to be suitable, and pure γ-adducts were generally isolated in good yields. Notably, the enantioselectivities for the above γ-selective [3 + 2] cyclization were extremely high, between 96% to 99% ee. The absolute configurations of the α-selective and γ-selective products were assigned based on the X-ray crystallographic analysis of the products 5b10 and 6b,11 respectively.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R (1) |
5:6b |
6 | Yieldc (%) | eed (%) |
| a Reactions were performed with 1 (0.10 mmol), 2a (0.12 mmol) and 4g (0.01 mmol) in CH2Cl2 (1 mL) at room temperature. b Determined by crude 1H NMR analysis. c Isolated yield for the pure γ-regioisomer. d The ee value for the γ-regioisomer, determined by HPLC analysis on a chiral stationary phase. e Catalyst loading was 20 mol%. | |||||
| 1 | Ph (1a) | 1:6 |
6a | 80 | 98 |
| 2 | 4-Cl-C6H4 (1b) | 1:5 |
6b | 72 | 98 |
| 3 | 3-Cl-C6H4 (1c) | 1:3 |
6c | 63 | 98 |
| 4 | 2-Me-C6H4 (1k) | 1:4 |
6d | 67 | 99 |
| 5 | 4-Br-C6H4 (1s) | 1:4 |
6e | 70 | 98 |
| 6 | 4-OMe-C6H4 (1f) | 1:5 |
6f | 80 | 99 |
| 7 | 4-Me-C6H4 (1g) | 1:5 |
6g | 74 | 99 |
| 8 | 3-Me-C6H4 (1r) | 1:6 |
6h | 78 | 98 |
| 9 | 4 F-C6H4 (1e) | 1:3 |
6i | 64 | 96 |
| 10 | 2-Naphthyl (1j) | 1:5 |
6j | 75 | 98 |
| 11 | 3,4-(OMe)2-C6H4 (1k) | 1:6 |
6k | 70 | 99 |
| 12 | 2-Thienyl (1l) | 1:6 |
6l | 70 | 99 |
| 13e | Cyclohexyl (1m) | 1:7 |
6m | 68 | 98 |
| 14e | Isopropyl (1n) | 1:6 |
6n | 74 | 98 |
| 15e | nBu (1o) | 1:3 |
6o | 35 | 97 |
| 16e | Ethyl (1p) | 1:4 |
6p | 40 | 98 |
It is noteworthy that the spiro[benzofuran-2,1′-cyclopentane] motif prepared in the above [3 + 2] annulation reaction is widely present in many natural products and bioactive molecules, and thus is of great significance in medicinal chemistry.12 As an illustration (Scheme 3), the γ-adduct 6n was readily converted in a highly diastereoselective and enantio-retentive manner to 8, a close analogue of a bioactive natural product extracted from fungi.12a
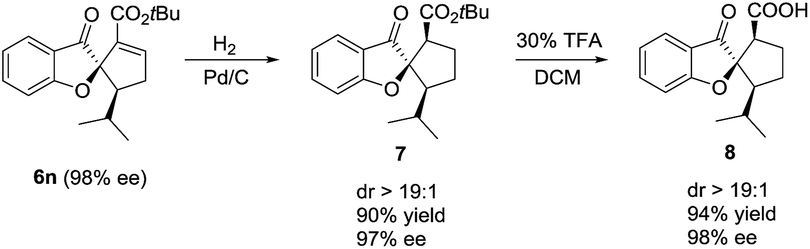 | ||
| Scheme 3 Deriving an anti-fungi analog from the annulation product. | ||
2.3 Theoretical studies to understand the origin of the observed regioselectivities
The mechanism of the phosphine-catalyzed [3 + 2] annulation reaction between aurone and allene is shown in Scheme 4, which follows the general pathways commonly accepted in the literature.5 The nucleophilic attack of the phosphine catalyst A on allene 2a yields zwitterionic intermediate B, which has two resonance forms with the negative charge either delocalized on the α-carbon (C) or the γ-carbon (G). The subsequent [3 + 2] annulation of C or G with aurone 1 then affords advanced intermediate E or I. The following proton transfer process, regeneration of the phosphine catalyst, and formation of the desired α-selective (5) or γ-selective (6) products complete the catalytic cycle.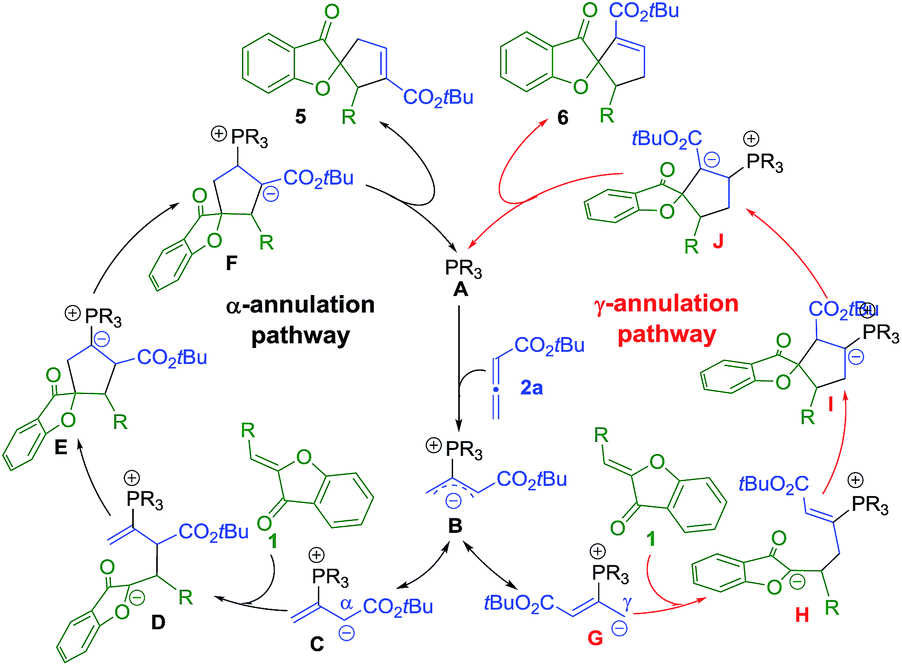 | ||
| Scheme 4 Proposed mechanism for the phosphine-catalyzed [3 + 2] annulation of aurones with allenoate 2a. | ||
Density functional theory (DFT) calculations were performed to gain insight into the catalyst-controlled regioselectivity in bifunctional phophine-catalyzed [3 + 2] annulation.13 Aurone 1a and allene 2a were chosen for our theoretical studies, and the phosphines 4c and 4e were selected since they offered similar regioselectivities to those of 4b and 4g in the annulation reactions, but possess slightly simpler structures. The Gibbs free energy profiles of the 4c or 4e-catalyzed [3 + 2] cycloaddition of aurone 1a to allenoate 2a were calculated, and we focused on the addition step of the phosphonium zwitterionic intermediate C or G to aurone 1a to understand the observed regioselectivity.
Initially, we suspected that the electron density of the phosphonium enolate may influence the regioselectivity, therefore we calculated the electrostatic potential (ESP) surface and nature population analysis (NPA) charge distribution for the 4e-derived INT-1 and 4c-derived INT-2 zwitterionic intermediates. Both the ESP and NPA calculations showed that the negative charges of C-α and C-γ in INT-1/INT-2 are close, therefore the difference of reactivity for C-α and C-γ is not the reason behind the observed regioselectivitiy (Fig. 1).
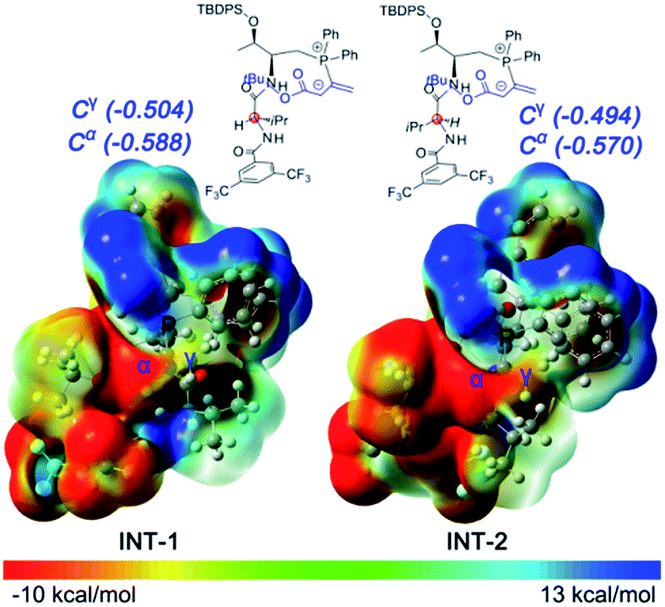 | ||
| Fig. 1 The B3LYP calculated NPA charge distributions for intermediate INT-1 and INT-2. | ||
We then applied a distortion/interaction model14 (ΔE≠act = ΔE≠dist + ΔE≠int) utilizing phosphonium allenoate and aurone as two fragments to gain more mechanistic insights. For the annulation reaction catalyzed by L-D-dipeptide phosphine 4e (Fig. 2a), the difference of the distortion energy terms (ΔE≠dist) between Ts-1 and Ts-2 is only 1.7 kcal mol−1. However, the difference of the interaction energy terms (ΔE≠int) between those two transition states is 3.4 kcal mol−1, which suggests that the interaction energy played a key role in determining the regioselectivity of the reaction. In the α-attack pathway (Ts-1), the aurone is activated by two hydrogen bonds with bond lengths of 1.89 Å and 1.99 Å, respectively. However, in the γ-attack pathway (Ts-2), the two bond distances become 1.88 Å and 2.10 Å, suggesting that one hydrogen bond is weaker. The strength of the hydrogen bond is determined by the conformation of the L-D- dipeptide. In Ts-1, the dihedral angle of O1-C1-C2-C3 is 78.5°, indicating that the isopropyl group is almost perpendicular to the amide moiety when the H2⋯O2 hydrogen bond is formed. On the other hand, a smaller dihedral angle of 74.7° is observed in Ts-2, and the strain of the isopropyl group in the valine residue results in the H2 atom in the valine residue rotating far away from the O2 atom of the aurone moiety, thus leading to a weaker H2⋯O2 hydrogen bond. The more favorable hydrogen bonding interactions, resulting from the conformation of the L-D- dipeptide moiety in the advanced phosphonium enolate intermediate, account for the observed α-selectivity in the annulation reaction.
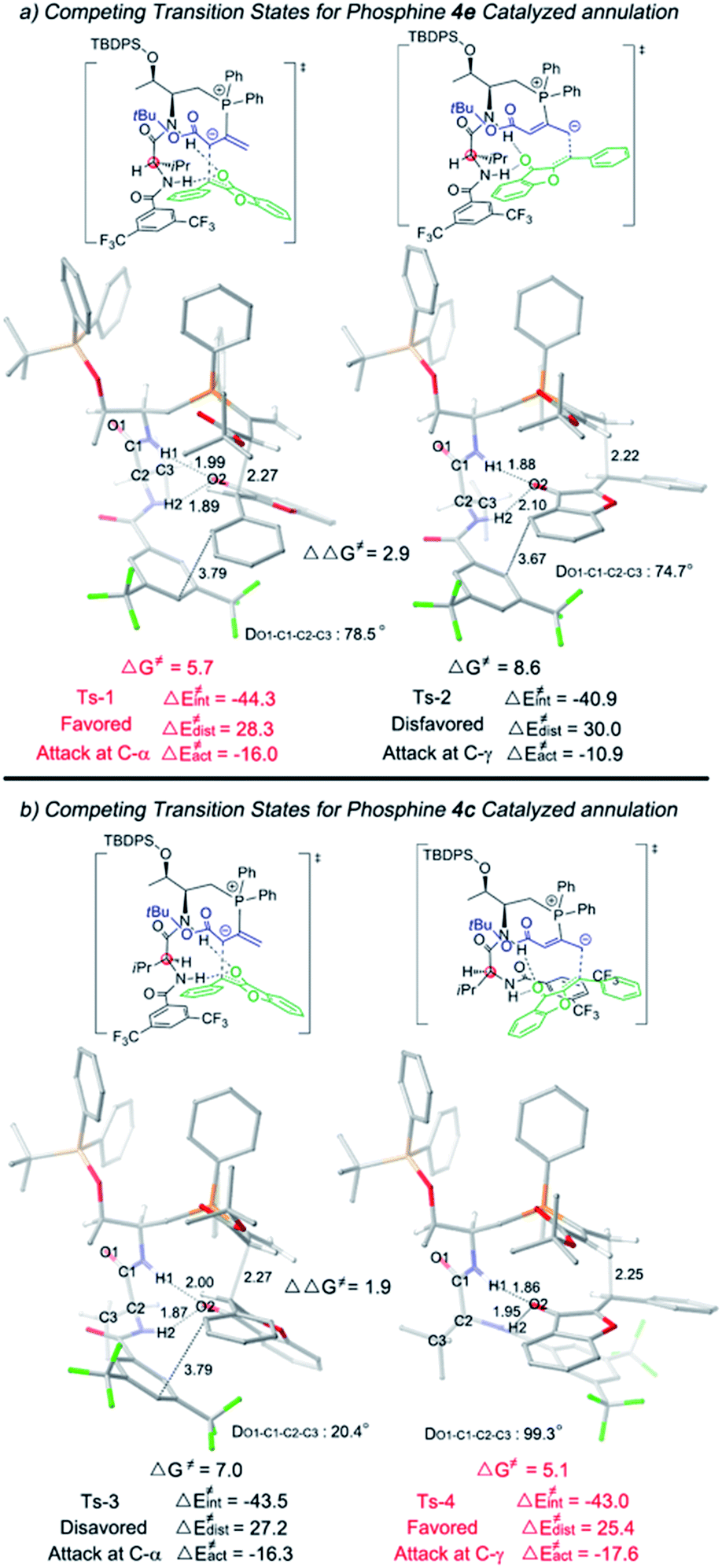 | ||
| Fig. 2 Optimized transition states Ts-1, Ts-2, Ts-3 and Ts-4. The relative free energies are given in kilocalories per mole. | ||
In the [3 + 2] annulation promoted by the L-L- dipeptide phosphine 4c (Fig. 2b), the activation energy of the γ-addition pathway (Ts-4) is more favored than the α-addition (Ts-3) by 1.9 kcal mol−1. The conformation of the L-L- dipeptide phosphine again accounts for the energy difference in the two transition states. In Ts-3, the dihedral angle of O1-C1-C2-C3 is only 20.4°, which exhibits a strong steric repulsion between the O1 atom and isopropyl group. Whereas in Ts-4, the amino moiety is rotated clockwise about 80° to form the H2⋯O2 hydrogen bond, thus the isopropyl group is perpendicular to the amide moiety, leading to an O1-C1-C2-C3 dihedral angle of 99.3° and a smaller distortion energy, meaning that the γ-isomer is selectively formed in the cyclization reaction.
3. Conclusions
In conclusion, we have utilized aurones as C2 synthons in phosphine catalysis for the first time. We have also successfully developed the first catalyst-controlled regiodivergent [3 + 2] annulation reaction. By simply utilizing dipeptide phosphines with either an L-L- or L-D- configuration, the γ-selective or α-selective annulation products could be readily obtained with excellent enantioselectivity. DFT calculations suggest that the observed catalyst-controlled α/γ- regioselectivity is determined by the conformation of the dipeptide phosphine catalysts, which differentiates the distortion energy or hydrogen bonding interactions in the competing transition state pathways, thus favoring the formation of specific regioisomers. Currently, we are extending our findings in this report to the discovery of other regiodivergent processes in asymmetric phosphine catalysis.Acknowledgements
Yixin Lu thanks the National University of Singapore (R-143-000-599-112) and the National Natural Science Foundation of China (21672158) for the generous financial support. Z. Y. thanks the China Scholarship Council for a Research Scholarship. N. U. and Yixin Lu are grateful for the KFUPM–NUS Collaborative Fund support (NUS15103NUS15103, R-143-004-617-597).Notes and references
- For selected reviews on phosphine catalysis, see: (a) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed; (b) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS; (c) X. Lu, Y. Du and C. Lu, Pure Appl. Chem., 2005, 77, 1985 CrossRef CAS; (d) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140 RSC; (e) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102 RSC; (f) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed; (g) A. Marinetti and A. Voituriez, Synlett, 2010, 174 CrossRef CAS; (h) S.-X. Wang, X. Han, F. Zhong, Y. Wang and Y. Lu, Synlett, 2011, 2766 CAS; (i) Q.-Y. Zhao, Z. Lian, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 1724 RSC; (j) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588 RSC; (k) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927 RSC; (l) Y. Xiao, Z. Sun, H. Guo and O. Kwon, Beilstein J. Org. Chem., 2014, 10, 2089 CrossRef PubMed; (m) Y. Wei and M. Shi, Chem.–Asian J., 2014, 9, 2720 CrossRef CAS PubMed; (n) W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657 RSC; (o) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Acc. Chem. Res., 2016, 49, 1369 CrossRef CAS PubMed.
- For selected phosphine catalyzed [3 + 2] annulations of allenes, see: (a) C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CrossRef CAS; (b) G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Am. Chem. Soc., 1997, 119, 3836 CrossRef CAS; (c) Y. Du, X. Lu and Y. Yu, J. Org. Chem., 2002, 67, 8901 CrossRef CAS PubMed; (d) Y. Du and X. Lu, J. Org. Chem., 2003, 68, 6463 CrossRef CAS PubMed; (e) T. Q. Pham, S. G. Pyne, B. W. Skelton and A. H. White, J. Org. Chem., 2005, 70, 6369 CrossRef CAS PubMed; (f) X. Lu, Z. Lu and X. Zhang, Tetrahedron, 2006, 62, 457 CrossRef CAS; (g) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426 CrossRef CAS PubMed; (h) C. E. Henry and O. Kwon, Org. Lett., 2007, 9, 3069 CrossRef CAS PubMed; (i) B. J. Cowen and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 10988 CrossRef CAS PubMed; (j) A. Voituriez, A. Panossian, N. Fleury-Brégeot, P. Retailleau and A. Marinetti, J. Am. Chem. Soc., 2008, 130, 14030 CrossRef CAS PubMed; (k) A. Voituriez, N. Pinto, M. Neel, P. Retailleau and A. Marinetti, Chem.–Eur. J., 2010, 16, 12541 CrossRef CAS PubMed; (l) H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467 CrossRef CAS PubMed; (m) Y. Fujiwara and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 12293 CrossRef CAS PubMed; (n) X. Han, Y. Wang, F. Zhong and Y. Lu, J. Am. Chem. Soc., 2011, 133, 1726 CrossRef CAS PubMed; (o) X.-C. Zhang, S.-H. Cao, Y. Wei and M. Shi, Chem. Commun., 2011, 47, 1548 RSC; (p) Q. Zhao, X. Han, Y. Wei, M. Shi and Y. Lu, Chem. Commun., 2012, 48, 970 RSC; (q) P.-Y. Dakas, J. A. Parga, S. Höing, H. R. Schöler, J. Sterneckert, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 9576 CrossRef CAS PubMed; (r) J. Marco-Martínez, V. Marcos, S. Reboredo, S. Filippone and N. Martín, Angew. Chem., Int. Ed., 2013, 52, 5115 CrossRef PubMed; (s) C. Gomez, M. Gicquel, J.-C. Carry, L. Schio, P. Retailleau, A. Voituriez and A. Marinetti, J. Org. Chem., 2013, 78, 1488 CrossRef CAS PubMed; (t) S. S. Bruna and M. V. D. P. Tetesa, Eur. J. Org. Chem., 2013, 3901 Search PubMed; (u) D. Wang, G.-P. Wang, Y.-L. Sun, S.-F. Zhu, Y. Wei, Q.-L. Zhou and M. Shi, Chem. Sci., 2015, 6, 7319 RSC; (v) M. Gicquel, Y. Zhang, P. Aillard, P. Retailleau, A. Voiturie and A. Marinetti, Angew. Chem., Int. Ed., 2015, 54, 5470 CrossRef CAS PubMed.
- For selected phosphine catalyzed [4 + 2] annulations of allenoates, see: (a) X.-F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716 CrossRef CAS PubMed; (b) R. P. Wurz and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 12234 CrossRef CAS PubMed; (c) Y. S. Tran and O. Kwon, Org. Lett., 2005, 7, 4289 CrossRef CAS PubMed; (d) Y. S. Tran and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12632 CrossRef CAS PubMed; (e) H. Xiao, Z. Chai, H.-F. Wang, X.-W. Wang, D.-D. Cao, W. Liu, Y.-P. Lu, Y.-Q. Yang and G. Zhao, Chem.–Eur. J., 2011, 17, 10562 CrossRef CAS PubMed; (f) Y. S. Tran, T. J. Martin and O. Kwon, Chem.–Asian J., 2011, 6, 2101 CrossRef CAS PubMed; (g) X.-Y. Chen and S. Ye, Eur. J. Org. Chem., 2012, 5723 CrossRef CAS; (h) F. Zhong, X. Han, Y. Wang and Y. Lu, Chem. Sci., 2012, 3, 1231 RSC; (i) E. Li, Y. Huang, L. Liang and P. Xie, Org. Lett., 2013, 15, 3138 CrossRef CAS PubMed; (j) M. Gicquel, C. Gomez, P. Retailleau, A. Voituriez and A. Marinetti, Org. Lett., 2013, 15, 4002 CrossRef CAS PubMed; (k) H. Liu, Y. Liu, C. Yuan, G.-P. Wang, S.-F. Zhu, Y. Wu, B. Wang, Z. Sun, Y. Xiao, Q.-L. Zhou and H. Guo, Org. Lett., 2016, 18, 1302 CrossRef CAS PubMed.
- For phosphine catalyzed [4 + 1] annulations of allenoates, see: (a) Q. Zhang, L. Yang and X. Tong, J. Am. Chem. Soc., 2010, 132, 2550 CrossRef CAS PubMed; (b) X. Han, W. Yao, T. Wang, Y. R. Tan, Z. Yan, J. Kwiatkowski and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 5643 CrossRef CAS PubMed; (c) D. T. Ziegler, L. Riesgo, T. Ikeda, Y. Fujiwara and G. C. Fu, Angew. Chem., Int. Ed., 2014, 53, 13183 CrossRef CAS PubMed; (d) S. Kramer and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 3803 CrossRef CAS PubMed.
- For mechanistic studies, see: (a) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS PubMed; (b) Y. Liang, S. Liu, Y. Xia, Y. Li and Z.-X. Yu, Chem.–Eur. J., 2008, 14, 4361 CrossRef CAS PubMed; (c) G.-T. Huang, T. Lankau and C.-H. Yu, J. Org. Chem., 2014, 79, 1700 CrossRef CAS PubMed; (d) M. C. Holland, R. Gilmour and K. N. Houk, Angew. Chem., Int. Ed., 2016, 55, 2022 CrossRef CAS PubMed.
- (a) T. Wang, Z. Yu, D. L. Hoon, C. Y. Phee, Y. Lan and Y. Lu, J. Am. Chem. Soc., 2016, 138, 265 CrossRef CAS PubMed; (b) X. Han, H. Ni, W.-L. Chan, X. Gai, Y. Wang and Y. Lu, Org. Biomol. Chem., 2016, 14, 5059 RSC; (c) H. Ni, W. Yao and Y. Lu, Beilstein J. Org. Chem., 2016, 12, 343 CrossRef CAS PubMed; (d) T. Wang, Z. Yu, D. L. Hoon, K.-W. Huang, Y. Lan and Y. Lu, Chem. Sci., 2015, 6, 4912 RSC; (e) W. Yao, X. Dou and Y. Lu, J. Am. Chem. Soc., 2015, 137, 54 CrossRef CAS PubMed; (f) F. Zhong, G.-Y. Chen, X. Han, W. Yao and Y. Lu, Org. Lett., 2012, 14, 3764 CrossRef CAS PubMed; (g) X. Han, S.-X. Wang, F. Zhong and Y. Lu, Synthesis, 2011, 2011, 1859 CrossRef.
- (a) M. B. Isman and P. Proksch, Phytochemistry, 1985, 24, 1949 CrossRef CAS; (b) O. Kayser, A. F. Kiderlen, U. Folkens and H. Kolodziej, Planta Med., 1999, 65, 316 CrossRef CAS PubMed; (c) T. Nakayama, J. Biosci. Bioeng., 2002, 94, 487 CrossRef CAS PubMed; (d) C. Zwergel, S. Valente, A. Salvato, Z. Xu, O. Talhi, A. Mai, A. Silva, L. Altuccice and G. Kirsch, Med. Chem. Commun., 2013, 4, 1571 RSC.
- For selected examples, see: (a) Y. Kim, J. Kim and S. B. Park, Org. Lett., 2009, 11, 17 CrossRef CAS PubMed; (b) C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232 CrossRef CAS PubMed; (c) M. Wang, Z.-Q. Rong and Y. Zhao, Chem. Commun., 2014, 50, 15309 RSC; (d) F. Wang, C. Luo, Y.-Y. Shen, Z.-D. Wang, X. Li and J.-P. Cheng, Org. Lett., 2015, 17, 338 CrossRef CAS PubMed; (e) Z.-Q. Liang, Z.-H. Gao, W.-Q. Jia and S. Ye, Chem.–Eur. J., 2015, 21, 1868 CrossRef CAS PubMed.
- (a) X. Han, W.-L. Chan, W. Yao, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2016, 55, 6492 CrossRef CAS PubMed; (b) H. Ni, W. Yao, A. Waheed, N. Ullah and Y. Lu, Org. Lett., 2016, 18, 2138 CrossRef CAS PubMed; (c) T. Wang, D. L. Hoon and Y. Lu, Chem. Commun., 2015, 51, 10186 RSC; (d) T. Wang, W. Yao, F. Zhong, G. H. Pang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 2964 CrossRef CAS PubMed; (e) X. Han, W. Yao, T. Wang, Y. R. Tan, Z. Yan, J. Kwiatkowski and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 5643 CrossRef CAS PubMed; (f) F. Zhong, X. Dou, X. Han, W. Yao, Q. Zhu, Y. Meng and Y. Lu, Angew. Chem., Int. Ed., 2013, 52, 943 CrossRef CAS PubMed; (g) F. Zhong, J. Luo, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2012, 134, 10222 CrossRef CAS PubMed; (h) X. Han, F. Zhong, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 767 CrossRef CAS PubMed; (i) F. Zhong, Y. Wang, X. Han, K.-W. Huang and Y. Lu, Org. Lett., 2011, 13, 1310 CrossRef CAS PubMed; (j) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 123, 7983 CrossRef.
- CCDC 1517706 contains the supplementary crystallographic data for this compound.†.
- CCDC 1517707 contains the supplementary crystallographic data for this compound.†.
- (a) Y.-M. Yan, X.-L. Wang, Q. Luo, L.-P. Jiang, C.-P. Yang, B. Hou, Z.-L. Zuo, Y.-B. Chen and Y.-X. Cheng, Phytochemistry, 2015, 114, 155 CrossRef CAS PubMed; (b) Q.-M. Li, J.-G. Luo, Y.-M. Zhang, Z.-R. Li, X.-B. Wang, M.-H. Yang, J. Luo, H.-B. Sun, Y.-J. Chen and L.-Y. Kong, Chem.–Eur. J., 2015, 21, 13206 CrossRef CAS PubMed; (c) M. Braun, A. Hessamian-Alinejad, B. F. Lacroix, B. H. Alvarez and G. Fischer, Molecules, 2008, 13, 995 CrossRef CAS PubMed; (d) Q. Luo, L. Di, W.-F. Dai, Q. Lu, Y.-M. Yan, Z.-L. Yang, R.-T. Li and Y.-X. Cheng, Org. Lett., 2015, 17, 1110 CrossRef CAS PubMed.
- See the ESI† for full details of DFT calculations.
- (a) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 10646 CrossRef CAS PubMed; (b) A. E. Hayden and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 4048 Search PubMed; (c) A. G. Green, P. Liu, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 4575 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1517706 and 1517707. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02176c |
| This journal is © The Royal Society of Chemistry 2017 |