DOI:
10.1039/C7SC02240A
(Edge Article)
Chem. Sci., 2017,
8, 7003-7008
Mechanistic analysis of a copper-catalyzed C–H oxidative cyclization of carboxylic acids†
Received
17th May 2017
, Accepted 16th August 2017
First published on 16th August 2017
Abstract
We recently reported that carboxylic acids can be oxidized to lactone products by potassium persulfate and catalytic copper acetate. Here, we unravel the mechanism for this C–H functionalization reaction using desorption electrospray ionization, online electrospray ionization, and tandem mass spectrometry. Our findings suggest that electron transfer from a transient benzylic radical intermediate reduces Cu(II) to Cu(I), which is then re-oxidized to Cu(II) in the catalytic cycle. The resulting benzylic carbocation is trapped by the pendant carboxylate group to give the lactone product. Formation of the putative benzylic carbocation is supported by Hammett analysis. The proposed mechanism for this copper-catalyzed oxidative cyclization process differs from earlier reports of analogous reactions, which posit a substrate carboxylate radical as the reactive oxidant.
Introduction
A detailed understanding of the mechanisms of reactions that promote C–H bond functionalization is important for the continued advancement of these technologies.1–5 We recently reported a copper-catalyzed oxidative cyclization of aliphatic and aromatic carboxylic acids using potassium persulfate (K2S2O8) as the terminal oxidant (Scheme 1).6 The reaction is performed open to air in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of acetic acid/water, uses low-cost copper acetate as a catalyst, and furnishes lactone products in modest to good yields for a variety of substrates. Results from kinetic isotope effect experiments indicate that copper ion has no influence on the C–H oxidation event.6 These findings coupled with other substrate selectivity data and the absence of detectable products of decarboxylation have led us to speculate that a sulfate radical anion functions as the active oxidant in this reaction. Our conclusion differs from prior literature reports7–11 for related processes, all of which have suggested that intramolecular C–H abstraction by a carboxylate radical initiates the cyclization event. In this study, we have leveraged the power of mass spectrometry to obtain evidence in support of our earlier proposal as well as additional mechanistic insights that reveal the complex nature of this process.
:1 mixture of acetic acid/water, uses low-cost copper acetate as a catalyst, and furnishes lactone products in modest to good yields for a variety of substrates. Results from kinetic isotope effect experiments indicate that copper ion has no influence on the C–H oxidation event.6 These findings coupled with other substrate selectivity data and the absence of detectable products of decarboxylation have led us to speculate that a sulfate radical anion functions as the active oxidant in this reaction. Our conclusion differs from prior literature reports7–11 for related processes, all of which have suggested that intramolecular C–H abstraction by a carboxylate radical initiates the cyclization event. In this study, we have leveraged the power of mass spectrometry to obtain evidence in support of our earlier proposal as well as additional mechanistic insights that reveal the complex nature of this process.
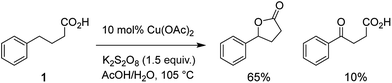 |
| | Scheme 1 Copper-catalyzed oxidative cyclization of a typical carboxylic acid. | |
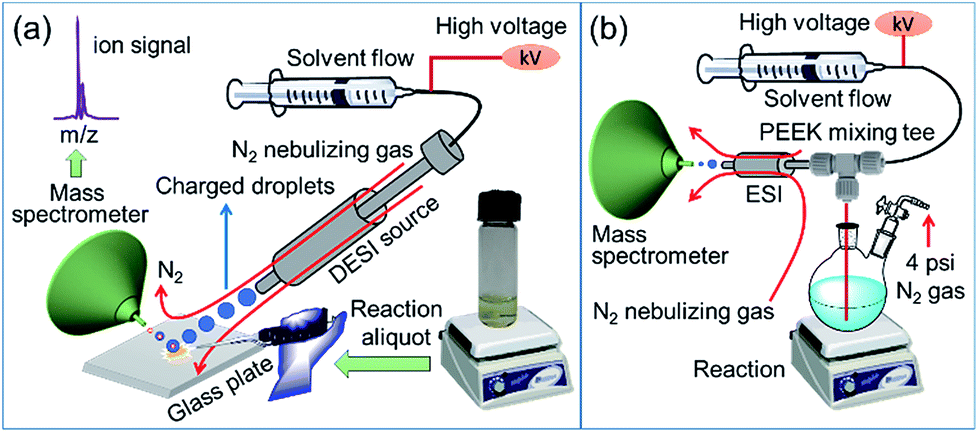
In order to elucidate the role of copper catalyst as well as to chart the transformation of substrate into product, we have investigated the mechanism of oxidative lactonization (Scheme 1) using a combination of ambient ionization mass spectrometric techniques and conventional physical-organic experiments. Desorption electrospray ionization mass spectrometry (DESI-MS; Fig. 1a)12–15 and online electrospray ionization mass spectrometry (ESI-MS; Fig. 1b)16–20 have enabled direct capture of transient intermediates derived from both substrate and copper catalyst, allowing us to posit a possible mechanistic scheme for this cyclization reaction. Our mechanistic hypothesis is corroborated by results from Hammett experiments as well as from studies using substrate probes. All told, these findings highlight the utility of mass spectrometry for the analysis of complex, multicomponent reactions, particularly those involving short-lived, redox-reactive species and kinetically labile transition metal salts.
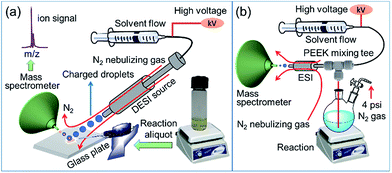 |
| | Fig. 1 Schematic diagram of the (a) DESI-MS and (b) online ESI-MS setup for intercepting species formed in the reaction mixture. | |
Fig. 1a shows the experimental set up of DESI-MS, which is used to monitor ion signals of intermediates formed during the progress of the reaction. In this DESI-MS experiment, a spray of charged microdroplets, generated in a 1:1 acetonitrile/dimethylformamide (ACN/DMF, v/v) solution, is directed toward a glass sample plate on which the reaction mixture is dispensed (Fig. S1,† see Experimental section for details). When the incident (primary) microdroplets impact the sample (reaction mixture), reactants, intermediate species, and products are extracted into secondary microdroplets. Subsequent solvent evaporation from these secondary microdroplets generates gas-phase analytes that are transferred into a high-resolution orbitrap mass spectrometer for mass-to-charge ratio (m/z) analysis. The high mass accuracy of this instrument allows for reliable identification of fleeting intermediates. Typically, the average lifetime of microdroplets in DESI or ESI is on the order of milliseconds.13,21 Accordingly, DESI-MS/ESI-MS can provide a ‘snapshot’ of reaction progress under ambient conditions in the form of ion signals of relevant species (substrate, catalyst, intermediate, and product).
Results and discussion
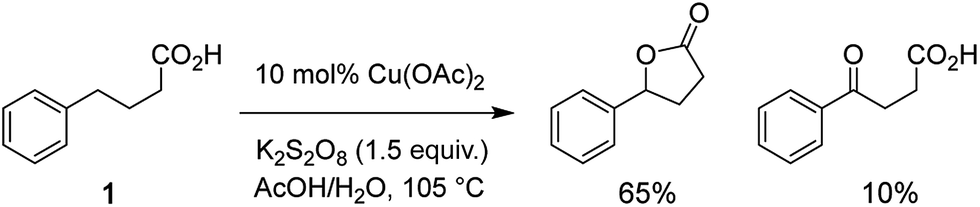
A. Mechanistic scheme
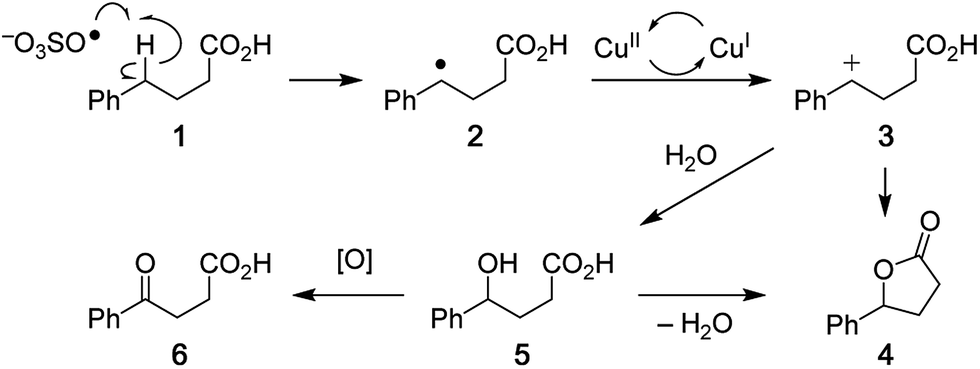
Analysis of the overall rate of product formation in the oxidative lactonization reaction (Fig. S2†) shows that the process is rapid, yielding maximal product within 5 min of initiation. Samples are thus taken from the reaction mixture at 2 min post initiation for analysis by DESI-MS. Results from these experiments (vide infra) and those in our earlier report6 have led us to propose Scheme 2 as one plausible mechanism for this Cu-catalyzed cyclization. Benzylic hydrogen abstraction in phenylbutyric acid 1 by SO4˙− affords a carbon radical intermediate 2. One-electron oxidation of 2 by the Cu(II) catalyst generates a resonance-stabilized carbocation 3 with concomitant formation of a Cu(I) species. Intermediate 3 can then cyclize directly to lactone product 4. If water traps the carbocation, alcohol 5 is formed, which can either close to lactone 4 or undergo further oxidation to ketone 6.6 In the presence of K2S2O8, Cu(I) is re-oxidized to Cu(II), thus allowing for catalytic turnover.22 We have detected ion signals corresponding to intermediates 2 (trapped by SO4˙−), 3, 5, products 4, 6, HSO4−, active oxidant SO4˙−, and Cu(I) species in our experiments (Fig. 2 and 3). In most cases, characterization of these species is further enabled by collision-induced dissociation (CID) (see Fig. S5, S11,† and 4).
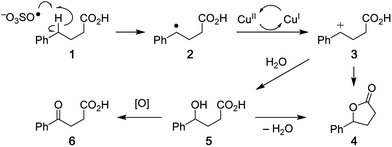 |
| | Scheme 2 Proposed mechanism for the copper-catalyzed oxidative cyclization of carboxylic acids to lactones. | |
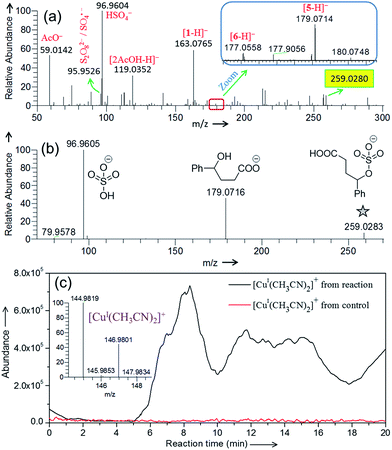 |
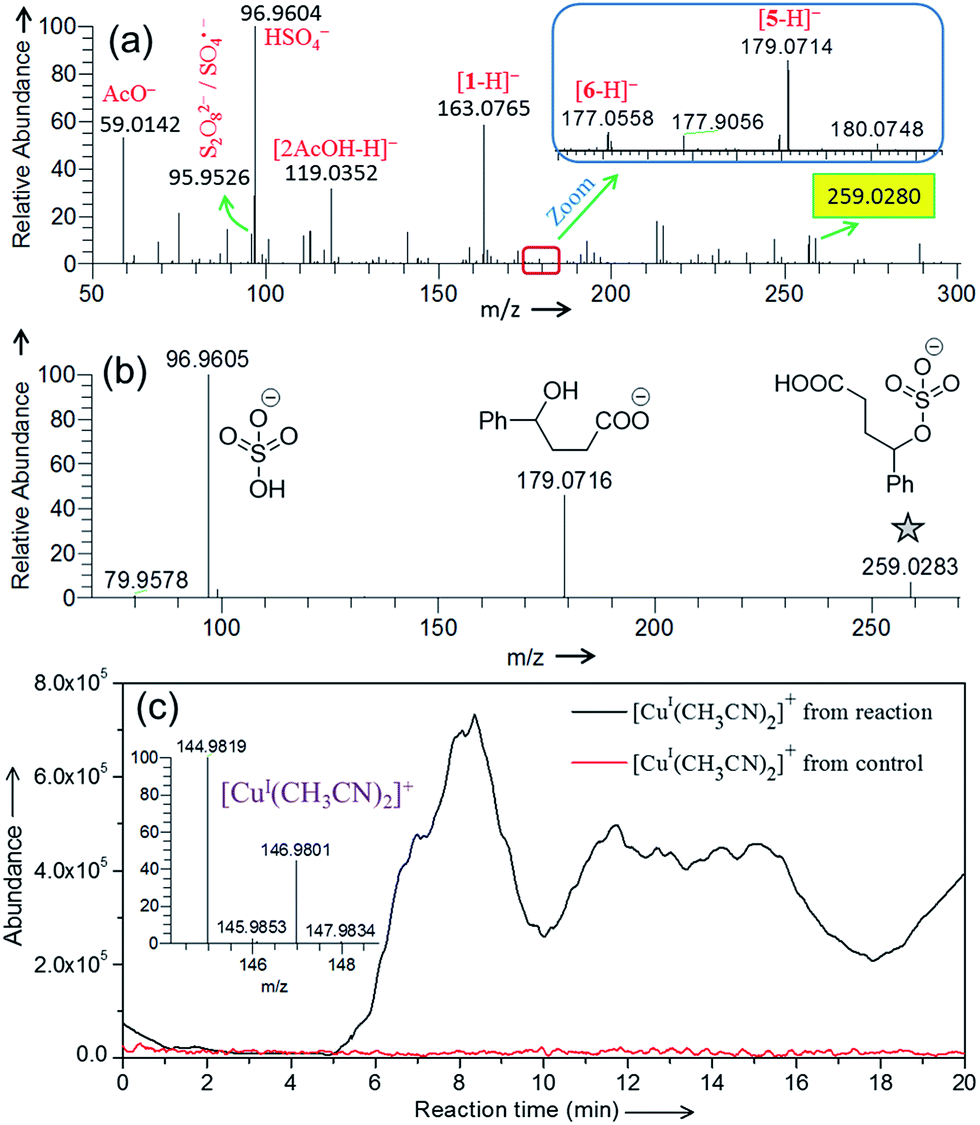
| | Fig. 2 (a) Negative ion mode DESI mass spectrum for the (a) Cu(II)-catalyzed oxidative cyclization of 4-phenylbutyric acid (Scheme 1). (b) CID-MS2 of the mass selected ion at m/z 259.0280 [highlighted in yellow in (a)]. See Table S1† for the accuracy of the m/z measurement. (c) Online ESI-MS monitoring of the formation of Cu(I) species in real time from the reaction mixture (containing 4-phenylbutyric acid substrate) and control (reaction mixture without substrate) at 75 °C. Inset shows the isotopic distribution of [CuI(CH3CN)2]+. See the Experimental section and Fig. S10 in ESI† for details. | |
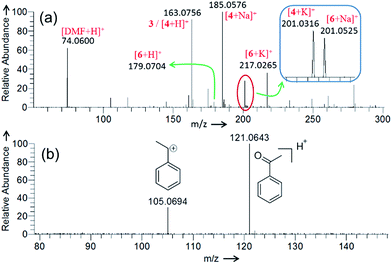 |
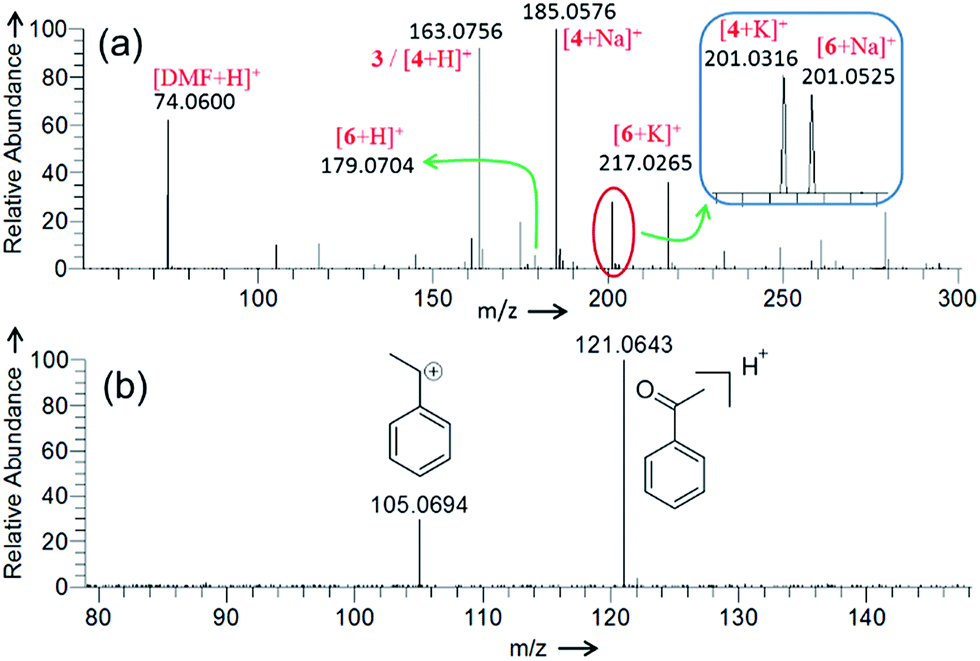
| | Fig. 3 Positive ion mode DESI mass spectrum for the (a) Cu(II)-catalyzed oxidative cyclization of 4-phenylbutyric acid (Scheme 1), and (b) Cu(II)-catalyzed oxidation of ethylbenzene. See the Experimental section in ESI for details and Table S1† for the accuracy of the m/z measurement. | |
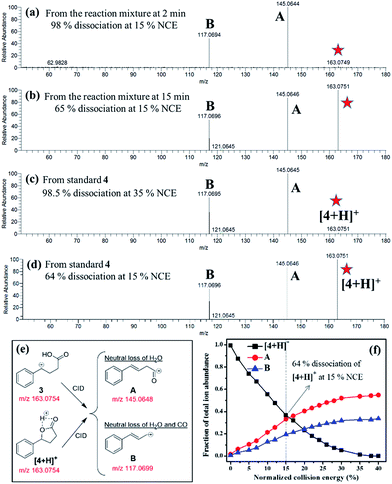 |
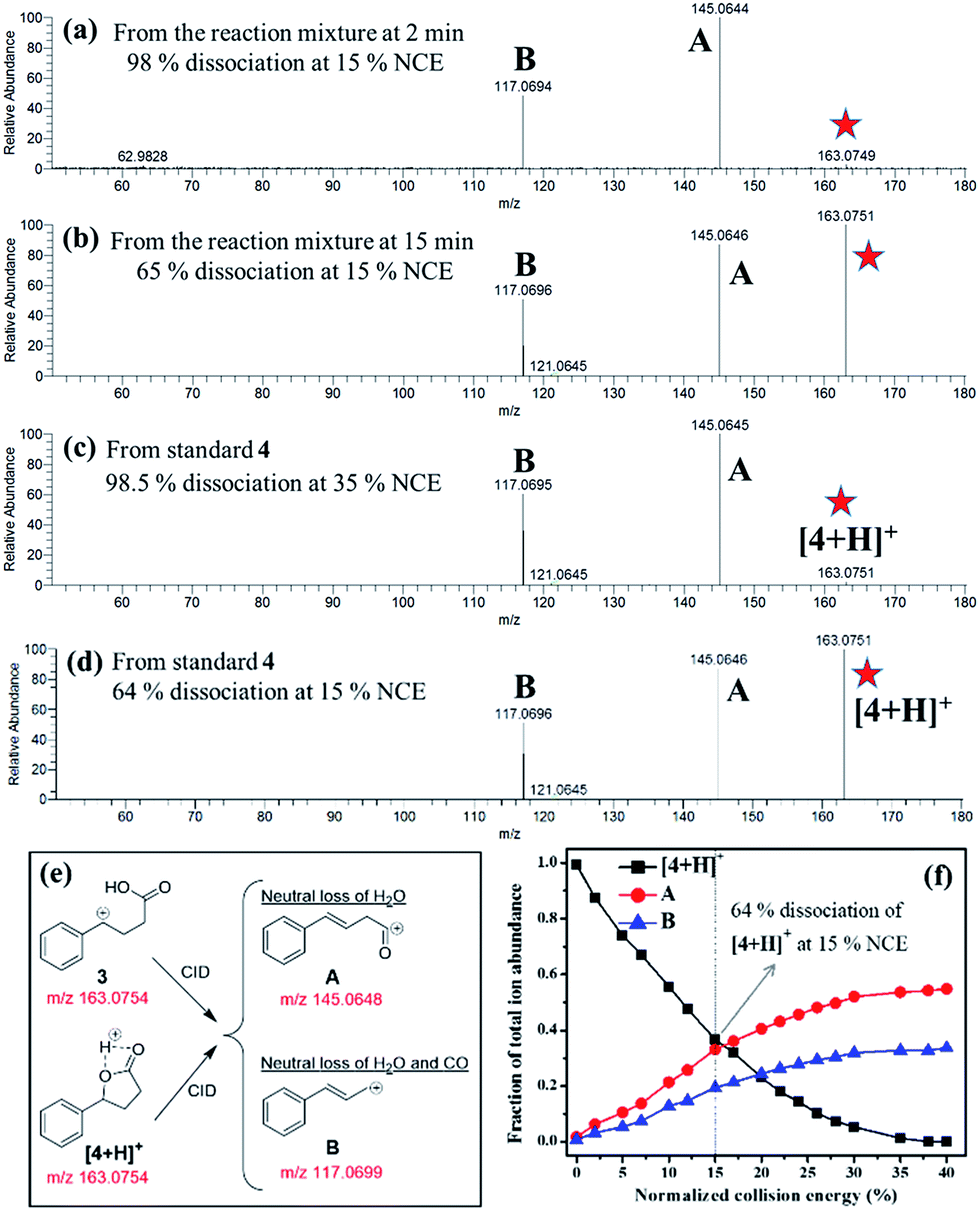
| | Fig. 4 CID-MS2 spectrum of the species at m/z 163.0756 detected from (a) the reaction mixture at 2 min (Fig. 2a), (b) the reaction mixture at 15 min, (c) the protonated 4 (standard) at 35% normalized collision energy (NCE28,29), and (d) the protonated 4 (standard) at 15% NCE. (e) The CID of 3 or [4 + H]+ produces the same product ions A and B, whose most plausible structures are postulated here. The theoretical m/z values of the corresponding species are presented in red. (f) A breakdown curve of standard [4 + H]+ species is also plotted to show its NCE-dependent dissociation profile. | |
B. Capturing intermediates by mass spectrometry
Negative ion mode DESI-MS (Fig. 2a) of the reaction mixture detects ion signals of hydrogen sulfate (HSO4−), sulfate radical anion (SO4˙−), and persulfate anion (S2O82−) generated from decomposition of K2S2O8. The detailed characterization of these species by their isotopic distribution is given in Fig. S3.† Please note that in Fig. 2 that the intensity of different ion signals cannot be simply related to concentrations of the corresponding species in bulk solution because of differences in ionization efficiencies. Careful evaluation of the mass spectra led us to identify a few among many detected signals which formed the basis of the proposed mechanism. Other unassigned signals mostly appear from impurities and background artifacts. Thermal homolysis of S2O82− can produce SO4˙−, which is capable of abstracting a hydrogen atom from 4-phenylbutyric acid to generate benzyl radical 2 and HSO4−.23 The ion signal at m/z 259.0280 (Fig. 2a) appears to correspond to the adduct of such a radical intermediate (2) with SO4˙−.24 This species has been characterized with high mass accuracy (0.8 ppm error) and closely matches the predicted isotopic distribution pattern (Fig. S4†) for the sulfated product.
The CID-MS2 of the m/z 259.0280 analyte shows two major fragments: HSO4− and 4-hydroxy-4-phenylbutanoate (Fig. 2b). Formation of 4-hydroxy-4-phenylbutanoate (m/z 179.0716, Fig. 2b) is verified by mass selection followed by CID-MS3 on this species (Fig. S5a†), which matches the CID-MS2 (Fig. S5b†) of standard 4-hydroxy-4-phenylbutanoate or [5-H]− in Fig. 2a. The structure of the analyte at m/z 259.0280 (Fig. 2a) has been confirmed by comparing its CID (Fig. 2b) with that of a synthetic standard of 3-carboxy-1-phenylpropyl sulfate (Fig. S6†). Generation of the sulfated product can be ascribed to reaction of the benzylic radical (i.e., 2) and K2S2O8. It is also possible that 3-carboxy-1-phenylpropyl sulfate (m/z 259.0280, Fig. 2a) forms from sulfate trapping of carbocation 3; however, in light of the poor nucleophilicity of sulfate anion,25 this reaction pathway is less likely.
We have demonstrated previously that cupric ion is required as a catalyst for optimal reaction performance.6 From DESI-MS, we have been unable to identify any type of organocopper complex derived from 1. A weak signal, however, has been detected in positive ion mode DESI-MS for Cu(I) complexed with acetonitrile ([CuI(CH3CN)2]+) at m/z 144.9827 (Fig. S7†). It is possible that this species results from reduction of Cu(II) ion in the spray ionization process.26,27 To determine if Cu(I) is generated under our reaction conditions, we have monitored the formation of [CuI(CH3CN)2]+ in real time by an on-line ESI-MS setup (Fig. 1b and S8†) in which the reaction mixture continuously flows through a capillary to the ESI source. Fig. 2c depicts the ion abundance of [CuI(CH3CN)2]+ as a function of reaction time. This ion chronogram reflects a clear increase in Cu(I) abundance over the course of the reaction with 4-phenylbutyric acid 1, as compared to a control experiment in which only 1 is excluded. A similar ion chronogram recording is obtained using an alternative substrate, 4-methylvaleric acid (Fig. S9†). Oxidative cyclization of this carboxylic acid yields the corresponding lactone, albeit less efficiently than for a substrate such as 1.
Fig. 3a presents the positive ion mode DESI mass spectrum for the Cu(II)-catalyzed cyclization of 4-phenylbutyric acid 1 (Scheme 1). The reaction products, lactone 4 and ketone 6, are detected as protonated, sodiated, and potassiated species, as expected.6 Fig. S11† and 4c present CID data for [6 + H]+ and [4 + H]+, respectively. The peak at m/z 163.0756 can be attributed to either protonated product [4 + H]+ or carbocation intermediate 3. These two analytes are isomeric and thus cannot be distinguished on the basis of m/z alone. Participation of the carbocation 3 as an active intermediate is well supported by the Hammett analysis (vide infra). However, in separate experiments employing ethylbenzene, DESI-MS analysis of an aliquot of the reaction mixture (2 min) unambiguously identifies the ion signal of the corresponding benzylic carbocation (Fig. 3b). These findings establish that carbocation formation can indeed occur under our reaction conditions and that such an intermediate (see Fig. S12† also) is detectable on the timescale of the DESI-MS experiment. From the reaction with 4-phenylbutyric acid 1, CID on m/z 163.0756 establishes that this species undergoes 98% dissociation in the gas phase at 15% normalized collision energy (NCE)28,29 (Fig. 4a). By contrast, analysis of a pure sample of lactone 4 shows that [4 + H]+ is 98% dissociated at 35% NCE (Fig. 4c and f). These data suggest that the ion at m/z 163.0756 (Fig. 3a) is likely a mixture of both benzylic carbocation 3 and protonated lactone [4 + H]+. Thus, we would expect the analyte at m/z 163.0756 to require less energy than [4 + H]+ to undergo the same extent of dissociation (Fig. 4) given the intrinsic reactivity of the benzylic carbocation.30 Notably, when the reaction mixture is analyzed at a later time point (15 min), the CID on m/z 163.0756 appears similar to that of a pure sample of 4 (Fig. 4).
C. Hammett analysis
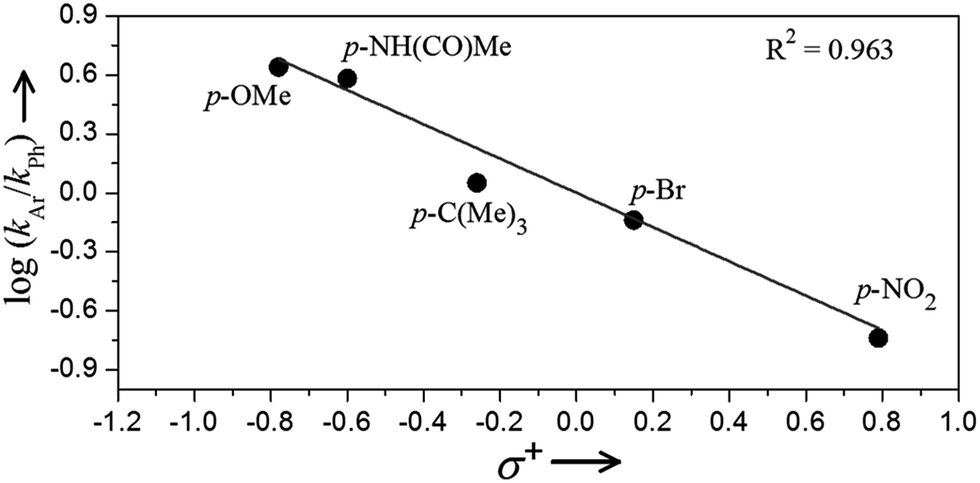
Further support for the proposed intermediacy of a benzylic carbocation intermediate 3 follows from Hammett analysis data.31 A series of competition experiments between 4-phenylbutyric acid and para-substituted 4-phenylbutyric acid derivatives shows a clear preference for oxidation of electron-rich arene substrates. Fig. 5 presents a plot of log(kAr/kPh) against the Hammett–Brown substituent constant σ+. The linear relationship between log(kAr/kPh) and σ+ yields a ρ+ value of −0.87 with an R2 of 0.96. Plots of this data against either the para-substituent constant σp or the Creary radical constant σc˙ do not give linear correlations (Fig. S13†).32 These results implicate a transition state with significant partial positive charge in the rate-determining step, consistent with the formation of intermediate 3.
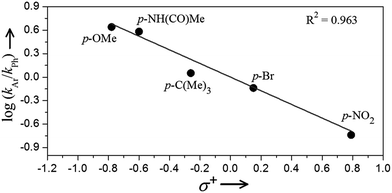 |
| | Fig. 5 Plot of log(kAr/kPh) vs. Hammett–Brown substituent constant σ+ from the competition experiments between 4-phenylbutyric acid and para-substituted 4-phenylbuytric acids for the oxidative cyclization reaction. | |
D. Mechanistic analysis using substrate probes
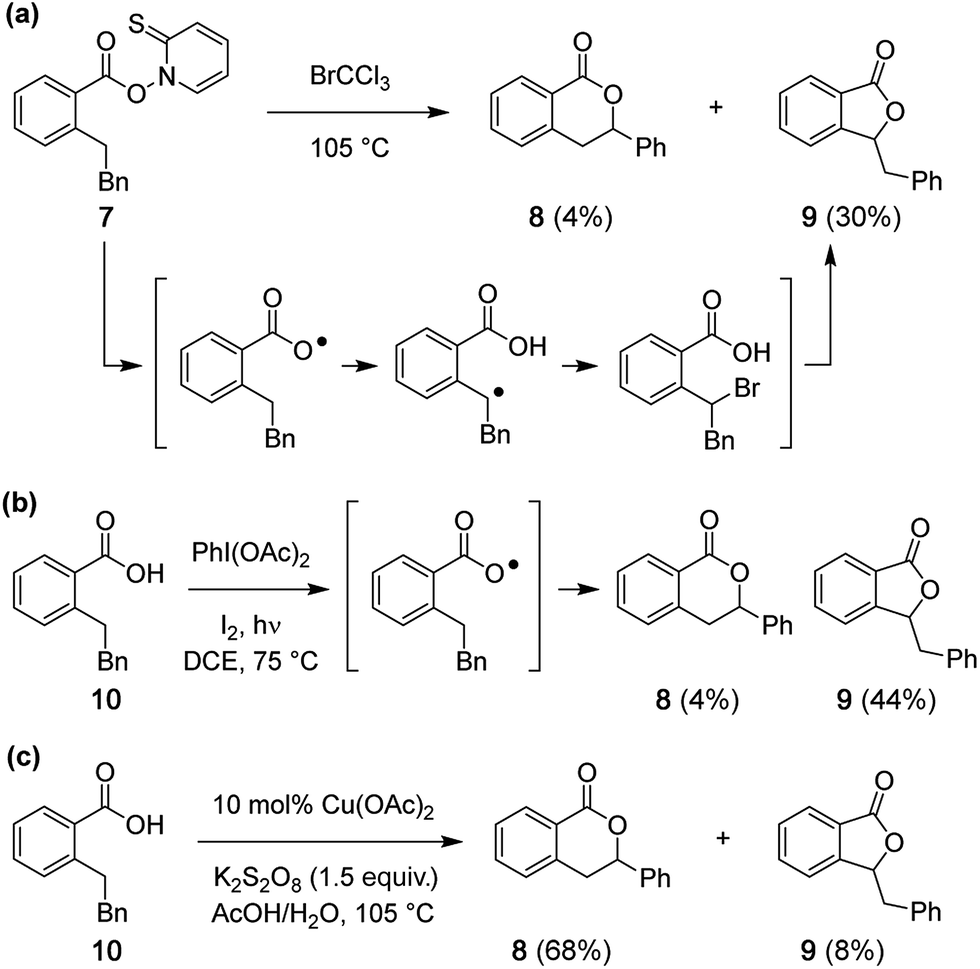
Reported studies of C–H oxidative lactonization reactions have posited reaction mechanisms that occur by way of carboxylate radical generation and subsequent 1,6-H-atom abstraction.7–10 In order to query if such a carboxylate radical is responsible for formation of 4 and 5, Barton ester 7 was prepared33,34 and heated for 2 h in BrCCl3 (Scheme 3a). Substrates of this type can fragment under thermal conditions to give transient radical species. Analysis of the reaction mixture of 7 with BrCCl3 (Fig. S14a†) shows the formation of 5-membered lactone 9 as a major product (30%) and 6-membered lactone 8 as a minor constituent (4%) (Scheme 3a). Similarly, when carboxylic acid 10 is treated with PhI(OAc)2 and I2, a reagent combination reported to promote lactone formation through an intermediate carboxylate radical,35,36 five-membered lactone 9 is again the major product (44%) (Scheme 3b and Fig. S14b†). In sharp contrast to these two results, the isomeric 6-membered lactone 8 is the predominant product under our Cu-catalyzed protocol (Scheme 3c and Fig. S14c†), confirming that the mechanism of this latter reaction does not operate through a carboxylate radical.
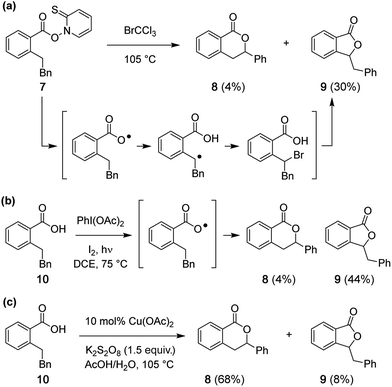 |
| | Scheme 3 (a) Fragmentation of Barton ester 7 produces five-membered lactone 9 as the major product. (b) Formation of 9 predominates under PhI(OAc)2/I2 conditions. (c) The Cu-catalyzed reaction favors formation of the 6-membered lactone 8. Note: Bn = CH2Ph. | |
Conclusions
We have investigated the mechanism of a Cu-catalyzed oxidative cyclization of carboxylic acids using a combination of DESI-MS, online ESI-MS, tandem mass spectrometry, Hammett analysis, and substrate probes. This mechanistic study is not based solely on mass spectrometry but also relies on evidence gathered from NMR analysis, which is not subject to possible artifacts from mass spectrometry. The evidence found from our mass spectrometric analysis is fully consistent with that from the NMR analysis. Although it is only possible to disprove a proposed mechanism and not prove it, the weight of all evidence supports a pathway for lactone formation that initiates through benzylic C–H abstraction by SO4˙− and proceeds through an intermediate carbocation, which is trapped by the pendant carboxylic acid (or by H2O). An alternative route (Scheme S1†) following through the intermediacy of an alkyl sulfate may account for lactone production in the absence of a Cu salt. High resolution mass spectrometry has allowed us to obtain a rather complete mechanistic picture of this reaction, including the role of Cu(II) as an outer-sphere oxidant. Using collision-induced dissociation, we were able to assign the structures of several of the observed species, which we believe are part of the catalytic cycle. Despite remarkable sensitivity, mass spectrometry remains an under-utilized tool for the analysis of reaction mechanisms.37,38 The present study highlights the power of mass spectrometric techniques when combined with more conventional physical organic experiments for elucidating complex catalytic processes.
Experimental
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO). HPLC grade solvents were purchased from Fisher Scientific (Nepean, ON, Canada). Reactions were performed using glassware that was oven-dried. Air- and moisture-sensitive liquids and solutions were transferred via syringe or stainless steel cannula. Organic solutions were concentrated under reduced pressure (∼15 Torr) by rotary evaporation. Solvents were purified by passage under 12 psi through activated alumina columns. Detailed procedures for synthesis of substrate probes and reactions for Hammett analysis are provided in the ESI.† DESI-MS studies were performed on a high-resolution mass spectrometer (Thermo Scientific LTQ Orbitrap XL Hybrid Ion Trap-Orbitrap mass spectrometer) using a homebuilt DESI source. The details of DESI-MS experiments are described in the ESI.† The online ESI-MS experiment for real time monitoring of the Cu(I) species was performed using the pressurized infusion method originally described by McIndoe and coworkers.20 See the ESI† for complete experimental details.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This work was supported by National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1205646) and Air Force Office of Scientific Research through Basic Research Initiative grant (AFOSR FA9550-16-1-0113).
Notes and references
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
-
A. Company, J. Lloret, L. Gómez and M. Costas, in Alkane C–H Activation by Single-Site Metal Catalysis, ed. P. J. Pérez, Springer, Netherlands, Dordrecht, 2012, pp. 143–228, DOI:10.1007/978-90-481-3698-8_5.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- M. C. White, Science, 2012, 335, 807–809 CrossRef CAS PubMed.
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
- S. Sathyamoorthi and J. Du Bois, Org. Lett., 2016, 18, 6308–6311 CrossRef CAS PubMed.
- N. O. Mahmoodi, K. Tabatabaeian, M. Kosari and S. Zarrabi, Chin. Chem. Lett., 2008, 19, 1431–1434 CrossRef CAS.
- N. O. Mahmoodi and M. Salehpour, J. Heterocycl. Chem., 2003, 40, 875–878 CrossRef CAS.
- N. O. Mahmoodi and M. Jazayri, Synth. Commun., 2001, 31, 1467–1475 CrossRef CAS.
- G. I. Nikishin, I. V. Svitanko and E. I. Troyansky, J. Chem. Soc., Perkin Trans. 2, 1983, 595–601, 10.1039/P29830000595.
- N. O. Mahmoodi and M. Salehpour, Russ. J. Org. Chem., 2003, 39, 1760–1763 CrossRef CAS.
- Z. Takáts, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef PubMed.
- R. H. Perry, M. Splendore, A. Chien, N. K. Davis and R. N. Zare, Angew. Chem., Int. Ed., 2011, 50, 250–254 CrossRef CAS PubMed.
- R. H. Perry, K. R. Brownell, K. Chingin, T. J. Cahill, R. M. Waymouth and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2246–2250 CrossRef CAS PubMed.
- Q. Zheng, Y. Liu, Q. Chen, M. Hu, R. Helmy, E. C. Sherer, C. J. Welch and H. Chen, J. Am. Chem. Soc., 2015, 137, 14035–14038 CrossRef CAS PubMed.
- X. Yan, E. Sokol, X. Li, G. Li, S. Xu and R. G. Cooks, Angew. Chem., 2014, 126, 6041–6045 CrossRef.
- A. J. Ingram, C. L. Boeser and R. N. Zare, Chem. Sci., 2016, 7, 39–55 RSC.
- A. J. Ingram, K. L. Walker, R. N. Zare and R. M. Waymouth, J. Am. Chem. Soc., 2015, 137, 13632–13646 CrossRef CAS PubMed.
- E. D. Lee, W. Mueck, J. D. Henion and T. R. Covey, J. Am. Chem. Soc., 1989, 111, 4600–4604 CrossRef CAS.
- K. L. Vikse, M. P. Woods and J. S. McIndoe, Organometallics, 2010, 29, 6615–6618 CrossRef CAS.
- A. Venter, P. E. Sojka and R. G. Cooks, Anal. Chem., 2006, 78, 8549–8555 CrossRef CAS PubMed.
- Potassium persulfate can re-oxidize Cu(I) to Cu(II) and, in this regard, we have found that cyclization of 1 occurs in 50% yield using 25 mol% CuCl as a catalyst.
-
H. Jakob, S. Leininger, T. Lehmann, S. Jacobi and S. Gutewort, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a19_177.pub2.
- G. Levey, G. Bida and J. O. Edwards, Int. J. Chem. Kinet., 1981, 13, 497–501 CrossRef CAS.
-
M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, 2007 Search PubMed.
- H. Lavanant, H. Virelizier and Y. Hoppilliard, J. Am. Soc. Mass Spectrom., 1998, 9, 1217–1221 CrossRef CAS.
- L. Gianelli, V. Amendola, L. Fabbrizzi, P. Pallavicini and G. G. Mellerio, Rapid Commun. Mass Spectrom., 2001, 15, 2347–2353 CrossRef CAS.
- Y. Zhang, S. B. Ficarro, S. Li and J. A. Marto, J. Am. Soc. Mass Spectrom., 2009, 20, 1425–1434 CrossRef CAS PubMed.
-
A. Mallet and S. Down, Dictionary of Mass Spectrometry, Wiley, 2010 Search PubMed.
- B. Paizs and S. Suhai, Mass Spectrom. Rev., 2005, 24, 508–548 CrossRef CAS PubMed.
-
E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science, 2006 Search PubMed.
- X. Creary, M. E. Mehrsheikh-Mohammadi and S. McDonald, J. Org. Chem., 1987, 52, 3254–3263 CrossRef CAS.
- D. H. R. Barton, D. Crich and W. B. Motherwell, Tetrahedron, 1985, 41, 3901–3924 CrossRef CAS.
- D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941, 10.1039/C39830000939.
- J. I. Concepcion, C. G. Francisco, R. Freire, R. Hernandez, J. A. Salazar and E. Suarez, J. Org. Chem., 1986, 51, 402–404 CrossRef CAS.
- T. Muraki, H. Togo and M. Yokoyama, J. Chem. Soc., Perkin Trans. 1, 1999, 1713–1716, 10.1039/A900791A.
- L. S. Santos, J. Braz. Chem. Soc., 2011, 22, 1827–1840 CrossRef CAS.
- M. N. Eberlin, Eur. J. Mass Spectrom., 2007, 13, 19–28 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02240a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Shyam
Sathyamoorthi‡
,
Shyam
Sathyamoorthi‡