
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule spectroelectrochemical cross-correlation during redox cycling in recessed dual ring electrode zero-mode waveguides†
Donghoon
Han‡
 a,
Garrison M.
Crouch‡
a,
Kaiyu
Fu
b,
Lawrence P.
Zaino III§
b and
Paul W
Bohn
*ab
a,
Garrison M.
Crouch‡
a,
Kaiyu
Fu
b,
Lawrence P.
Zaino III§
b and
Paul W
Bohn
*ab
aDepartment of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: pbohn@nd.edu; Fax: +1 574 631 8366; Tel: +1 574 631 1849
bDepartmemt of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA
First published on 19th June 2017
Abstract
The ability of zero-mode waveguides (ZMW) to guide light into subwavelength-diameter nanoapertures has been exploited for studying electron transfer dynamics in zeptoliter-volume nanopores under single-molecule occupancy conditions. In this work, we report the spectroelectrochemical detection of individual molecules of the redox-active, fluorogenic molecule flavin mononucleotide (FMN) freely diffusing in solution. Our approach is based on an array of nanopore-confined recessed dual ring electrodes, wherein repeated reduction and oxidation of a single molecule at two closely spaced annular working electrodes yields amplified electrochemical signals. We have articulated these structures with an optically transparent bottom, so that the nanopores are bifunctional, exhibiting both nanophotonic and nanoelectrochemical behaviors allowing the coupling between electron transfer and fluorescence dynamics to be studied under redox cycling conditions. We also investigated the electric field intensity in electrochemical ZMWs (E-ZMW) through finite-element simulations, and the amplification of fluorescence by redox cycling agrees well with predictions based on optical confinement effects inside the E-ZMW. Proof-of-principle experiments are conducted showing that electrochemical and fluorescence signals may be correlated to reveal single molecule fluctuations in the array population. Cross-correlation of single molecule fluctuations in amperometric response and single photon emission provides unequivocal evidence of single molecule sensitivity.
Introduction
The development of techniques to detect and manipulate individual molecules in solution has yielded unprecedented insights into molecular-scale processes, including macromolecular dynamics, conformational changes, and intermolecular interactions.1 Observing single molecules is critical, because the behavior of the ensemble may reflect either the broadly shared median behavior of the population, or the activity of a small number of highly active members, or a mixture of the two. The most widely-used single-molecule observation techniques rely on optical2,3 or force-transduction4 mechanisms, rather than complementary electrical or electrochemical measurements. Electrochemical detection of a single molecule in solution represents a formidable challenge, because the generated currents are at, or below, the noise floor of the measurement. To address this challenge, redox cycling (RC) is commonly employed to amplify electrochemical currents, especially in scanning electrochemical microscopy (SECM). RC has been used to detect short-lived intermediates in electrochemical reactions,5 image neurotransmitter release from living cells,6 implement stochastic sensing with nanogap transducers7,8 and investigate the catalytic properties of surfaces.9,10 It has also been used to detect small numbers of molecules in closed, nanometer-high cavities formed by impinging a wax-coated metal tip onto a conducting substrate11 or by immersing recessed nanoelectrodes in mercury.12Fluorescence microscopy is also sufficiently sensitive to interrogate individual molecules and complexes,13–17 giving it the capacity to discover and quantify the lifetimes and movements of novel species obscured in ensemble average studies of biological systems.18–25 However, single molecule fluorescence of biomolecules is constrained by the spatial resolution limitations imposed by the diffraction of light (∼250 nm for visible radiation).26–28 As a result, to attain single-molecule occupancy within the typical focal volume, ∼1 femtoliter (1 fL = 10−15 L), of a diffraction-limited laser spot, the concentration of fluorescent species is restricted to the low nanomolar range. That is low compared to the typical micromolar KD values characteristic of biomolecular interactions.29,30 A common scheme to overcome this “concentration barrier” in single-molecule fluorescence is to excite only a limited number of fluorescently-labeled molecules within the focal volume, while the majority of molecules outside the focal volume remain unexcited.29 This can be achieved by: (1) stochastic activation of photoactivatable/switchable fluorophores coupled to photoactivation, diffusion, and excitation;31–33 or (2) decreasing the focal volume,34 as in the case of total internal reflection microscopy, confocal, stimulated emission depletion microscopies, and light emission in zero-mode waveguides (ZMWs).35–41
ZMWs are subwavelength-diameter cylindrical nanoapertures clad in metal, e.g. gold, aluminum, which limit the incident excitation beam to a non-propagating zeroth-order mode, thus forming an evanescent field. The intensity of this field decays exponentially with distance in the nanoaperture, thus constraining the excitation volume to the order of 100 zL (1 zL = 10−21 L), ∼3 orders of magnitude smaller than that in diffraction-limited microscopy. Because of this small excitation volume, single-molecule fluorescence imaging using a ZMW allows for the detection of single, fluorescent molecules at bulk concentrations up to 10 μM, or even as high as mM with special techniques, such as ZMW FRET.42,43 In chemical sensing, ZMW-based technology is an attractive platform for single-molecule detection due to the potential for high parallelism and the tolerance of μM concentrations of fluorescent species.44–46
Previously, we exploited ZMWs to investigate homogeneous electron transfer dynamics of sarcosine oxidase, both with its nominal substrate, N-methylglycine (sarcosine), as well as a non-canonical substrate, L-proline.47 Subsequently, we extended these experiments to use the optical cladding layer of the ZMW as the working electrode in spectroelectrochemical studies of single electron transfer events in surface-functionalized flavin adenine dinucleotide48 and electrofluorogenic behavior of freely diffusing flavin mononucleotide (FMN) under single molecule occupancy conditions.49 Flavins, like FMN, contain an isoalloxazine chromopore, which is fluorescent in the oxidized state (FMN), while the reduced state (FMNH2) exhibits dramatically lower light emission, i.e. a dark state. Intriguingly, FMN has a relatively constant fluorescence emission efficiency over a large pH window, but the electrochemical behavior varies significantly with pH.50,51 These properties, coupled with the intrinsic biochemical relevance as important cofactors in flavoenzymes, make the flavins excellent candidates for monitoring single molecule fluorescence under active potential control in an electrochemical ZMW (E-ZMW).
Here we present the first experiments in which single-molecule fluorescence is cross-correlated with electron transfer events in freely diffusing FMN in an E-ZMW. The structure and principle of operation of the E-ZMW device is illustrated in Fig. 1(A). A recessed dual ring electrode (RDRE) E-ZMW array enables the detection of very low currents and the simultaneous optical monitoring of fluorescence emission, thus providing both optical and electrochemical signatures from the same population of single molecules. In the RDRE geometry, redox cycling occurs between two independently-biased ring electrodes that are separated by a thin (100 nm) insulating layer. Electrical currents generated at the bottom-ring electrode, ib, and top-ring electrode, it, are independently measured. Using the RDRE arrays as E-ZMWs, we are able to record the current produced by the redox cycling of FMN molecules at the same time as the photon flux generated by fluorescence of oxidized FMN molecules in the optically-active region enclosed by the bottom electrode, which simultaneously serves as the optical cladding layer in the nanophotonic E-ZMW structure. Constant potential (amperometric) experiments show strong correlation between Eappl and fluorescence intensity under single-molecule occupancy conditions, while potential step experiments are utilized to examine the coupling between molecular transport and electron transfer in the E-ZMW. Statistical fluctuations in array population are also characterized by cross-correlating the single-molecule fluctuations in amperometric response and single-photon emission from freely diffusing FMN. Cross-correlation analysis of the stochastic signals and analytical results for the distribution of residence times are in close agreement, supporting the interpretation that the current/fluorescence signal fluctuations represent single molecule population excursions.
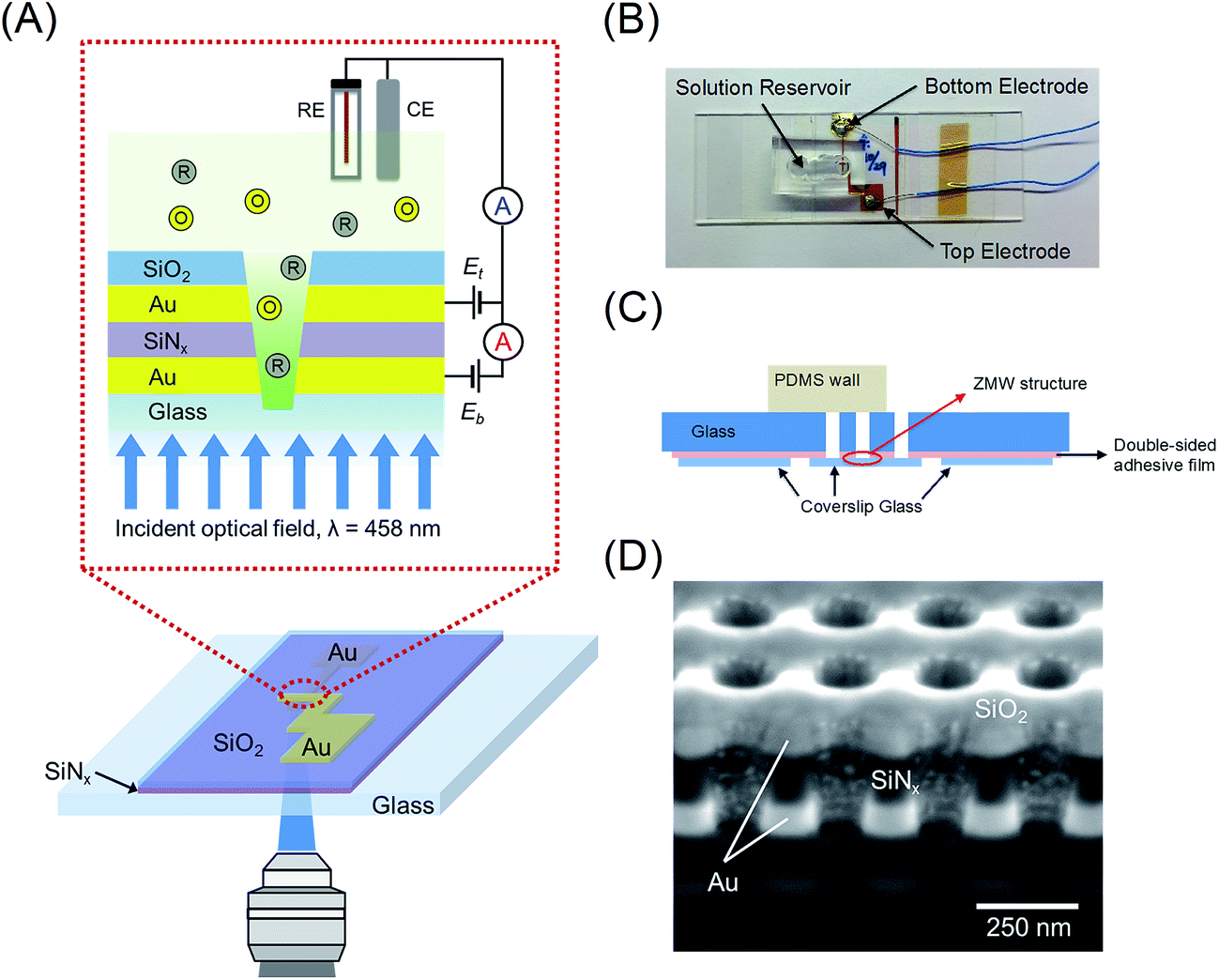 | ||
| Fig. 1 Electrochemical zero-mode waveguide (E-ZMW) device. (A) Schematic diagram of the experimental configuration. Freely diffusing redox-active molecules are repeatedly reduced and oxidized at recessed dual ring electrodes, leading to a measurable current. As focused light from a 458 nm laser penetrates into the E-ZMW, its intensity exponentially decreases to generate a zeptoliter-volume excitation volume confined near the bottom electrode. The E-ZMW can be used to observe single electron transfer events using simultaneous optical and electrochemical measurements. (B) Plan view photograph of the E-ZMW device at large scale. (C) Schematic cross-section of an E-ZMW device (not to scale). (D) Cross-section SEM image of 4 adjacent E-ZMW nanopores showing dual recessed Au ring electrodes separated vertically by ∼100 nm of SiNx. | ||
Experimental section
Chemicals and materials
Flavin mononucleotide, citric acid monohydrate, trisodium citrate dihydrate, sulfuric acid (95%), hydrogen peroxide (30%), acetone and Au etchant (KI–I2 solution) were obtained from Sigma-Aldrich. Photoresist (PR) AZ5214E (AZ Electronic Materials) and polydimethylsiloxane (PDMS) (Dow Corning) were purchased and used according to the manufacturers' specifications. All reagents were used as received without further purification, and the solutions were prepared using a Milli-Q Gradient water purification system (Millipore).Fabrication of E-ZMW devices
Similar to the procedure used for developing recessed dual ring electrode arrays,52 the nanopore arrays were fabricated via a combination of standard photolithography, layer-by-layer deposition, and focused ion beam (FIB) milling. The complete process flow is shown in Fig. S1.† A glass coverslip was cleaned in piranha solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 sulfuric acid (95%):hydrogen peroxide (30%) – Caution – Strong oxidizer, use with extreme care), rinsed with DI water, and dried at 110 °C. The cleaned glass coverslip was patterned by photolithography using AZ5214E PR to define a bottom electrode with a width of 100 μm. A 100 nm thick Au film was deposited by electron-beam evaporation (UNIVEX 450B, Oerlikon) using 10 nm Ti as an adhesion layer on the patterned glass coverslip. Then, an acetone lift-off step was used to remove the photoresist. A 100 nm thick SiNx layer was deposited by plasma-enhanced chemical vapor deposition (PECVD 790, Plasma-Therm), Fig. S1(A).† Using the same lithographic method, the top Au electrode was deposited perpendicular to the bottom electrode, creating an overlapping intersection area of ca. 200 μm × 200 μm containing two Au layers separated by a 100 nm-thick SiNx layer, Fig. S1(B).† Next, an additional 100 nm thick SiO2 layer was deposited after removal of the photoresist covering the entire substrate, Fig. S1(C).† A dual-source FIB instrument (Helios Nanolab 600, FEI Corp.) was used for milling and characterization. Nanopore arrays were patterned in a 20 μm × 20 μm square array with a lattice spacing of 250 nm, Fig. S1(D).† FIB milling was performed at 30 kV acceleration, 0.28 nA ion aperture, and 0.1 μs dwell time to produce the RDRE array. These FIB-milled pores exhibit a conical frustum shape with typical top diameter, dtop ∼ 100–120 nm, and bottom diameter, dbottom ∼ 60–70 nm. Immediately after milling the device, an ohmic background current was observed between the two electrodes, arising from a small bias (10 mV) observed between the top and bottom electrode in a dry environment. This is presumably caused by a combination of redeposition of partially etched Au along the nanopore walls and implanted Ga+, resulting in a partial short between the two electrodes. This conducting layer was removed by immersing the device in dilute Au etchant solution for 1–3 min. After cleaning the device with DI water, the top and bottom electrodes were well-insulated (resistance > 10 MΩ.).
:1 sulfuric acid (95%):hydrogen peroxide (30%) – Caution – Strong oxidizer, use with extreme care), rinsed with DI water, and dried at 110 °C. The cleaned glass coverslip was patterned by photolithography using AZ5214E PR to define a bottom electrode with a width of 100 μm. A 100 nm thick Au film was deposited by electron-beam evaporation (UNIVEX 450B, Oerlikon) using 10 nm Ti as an adhesion layer on the patterned glass coverslip. Then, an acetone lift-off step was used to remove the photoresist. A 100 nm thick SiNx layer was deposited by plasma-enhanced chemical vapor deposition (PECVD 790, Plasma-Therm), Fig. S1(A).† Using the same lithographic method, the top Au electrode was deposited perpendicular to the bottom electrode, creating an overlapping intersection area of ca. 200 μm × 200 μm containing two Au layers separated by a 100 nm-thick SiNx layer, Fig. S1(B).† Next, an additional 100 nm thick SiO2 layer was deposited after removal of the photoresist covering the entire substrate, Fig. S1(C).† A dual-source FIB instrument (Helios Nanolab 600, FEI Corp.) was used for milling and characterization. Nanopore arrays were patterned in a 20 μm × 20 μm square array with a lattice spacing of 250 nm, Fig. S1(D).† FIB milling was performed at 30 kV acceleration, 0.28 nA ion aperture, and 0.1 μs dwell time to produce the RDRE array. These FIB-milled pores exhibit a conical frustum shape with typical top diameter, dtop ∼ 100–120 nm, and bottom diameter, dbottom ∼ 60–70 nm. Immediately after milling the device, an ohmic background current was observed between the two electrodes, arising from a small bias (10 mV) observed between the top and bottom electrode in a dry environment. This is presumably caused by a combination of redeposition of partially etched Au along the nanopore walls and implanted Ga+, resulting in a partial short between the two electrodes. This conducting layer was removed by immersing the device in dilute Au etchant solution for 1–3 min. After cleaning the device with DI water, the top and bottom electrodes were well-insulated (resistance > 10 MΩ.).
The E-ZMW device was fabricated by attaching coverslip glass containing the nanopore array to a glass microscope slide. In brief, a slide (75 mm × 25 mm, 1 mm thick, Corning 2947) prepared to be the upper mounting layer was drilled to allow electrical and fluid contact with the nanopore array using a 2 mm-diameter diamond drill at 18000 rpm. Then, the drilled slide glass was cleaned with ethanol, acetone, and DI water and dried under a stream of N2. A double-sided adhesive film (no. 5603, Nitto) was used to attach to the nanopore array to the upper supporting slide, after appropriate openings were made for electrical contact with the nanopore array, forming an E-ZMW device, as shown in Fig. 1(B) and (C).
Modeling and calculations
Numerical calculations were performed with a finite element method using COMSOL Multiphysics version 5.2. The simulation was performed over a two-dimensional domain representing the geometry and dimensions of the zero-mode waveguides employed in our experiments. The zero-mode waveguide is represented by a single pore of the recessed dual ring electrode array, consisting of a recessed bottom-ring electrode, 100 nm insulating later, recessed top-ring electrode, and 100 nm top insulator layer. The geometry is adapted from scanning electron micrographs (SEMs) of FIB cross-sections. The modeling domain above the single pore was sufficiently large to avoid boundary artifacts, and the mesh was refined both within the pore and in the region just above the pore to provide sufficient numerical resolution. Details of the simulations are given in the ESI.†Electrochemical measurements
Electrochemical measurements were performed with a CH Instruments electrochemical analyzer (Model 750E) using a Pt wire and Ag/AgCl (RE-5B, BASi) as auxiliary and reference electrodes, respectively. The nanopore-confined top- and bottom-ring electrodes operate as separate working electrodes. All potentials are reported vs. Ag/AgCl reference at 300 K. Both the reference electrode and auxiliary electrode were immersed in a 100 μL PDMS reservoir in direct fluid contact with the E-ZMW device. Amperometric traces were acquired by fixing one electrode at an oxidizing potential and the other at a reducing potential, +0.3 V and −0.4 V, respectively.Fluorescence measurements
A custom-built confocal microscope was used to collect all fluorescence data. The instrument has a customized optical path depicted in Fig. S2(A).† 458 nm excitation radiation from a continuous wave laser (Sapphire 458 LP, Coherent) was passed through a spatial filter to select the TEM00 mode, then passed through a quarter wave plate to produce circularly polarized light to avoid polarization artifacts. A dichroic filter (Z488RDC, Chroma Technology Corporation) reflected the collimated beam into the back aperture of a 40×, 1.30 NA oil immersion objective (420260-9900-000, Carl Zeiss, Inc.) to produce a diffraction-limited spot with 40 μW power (2.5 × 104 W cm−2) at the focal plane. Fluorescence emission was collected by the same objective (epi-fluorescence) and passed through three emission filters (ET500lp, ET525/50m, ET500lp, Chroma Technology Corporation) before being focused through a 30 μm diameter confocal pinhole to reduce background radiation before detection by a single-photon avalanche photodiode (SPCM-AQR-16, PerkinElmer, Inc.). A hardware correlator (TimeHarp260 PICO Single, PicoQuant) was used to collect single molecule fluorescence data. The fluorescence optical path was augmented with a trans-illumination path to assist in locating the ZMW pores. To access the trans-illumination path, collimated radiation from a 632.8 nm laser was used to illuminate the back (upper) surface of the ZMW array. A mirror was selectively rotated into the detection path to allow monitoring of the transmitted radiation by a video-rate CCD camera (JE7462DC, Javelin Systems). Once located, the pore array was indexed to the motion of a piezoelectric stage (P-517.2CL, Physik Instrumente) to reliably center the x–y focal position of the objective on the ZMW pore of interest.Results and discussion
Characterization of E-ZMW device
A schematic illustration of the E-ZMW device is shown in Fig. 1(A). E-ZMWs with 250 nm inter-pore spacing were fabricated to form square arrays of annular insulator–electrode–insulator–electrode RDRE nanopore stacks, in which each electrode layer is individually addressable, i.e., the electrodes of the same layer in each nanopore are always at the same potential (see Experimental section for details). The design was chosen to optimize the probabilities of: (a) observing optical events with diffraction-limited microscope illumination, and (b) re-capturing molecules escaping from an individual nanopore in a neighboring nanopore. A plan-view photo and a schematic cross-section of the E-ZMW device are shown in Fig. 1(B) and (C), respectively. Fig. S1† illustrates the fabrication process using photolithography, layer-by-layer deposition and FIB milling to produce nanopore RDRE arrays. This simple direct-write approach enables direct formation of highly precise nanopore electrodes, but FIB direct writing produces pores with conical frustum shapes, rather than the cylindrical shapes obtained using electron beam53,54 or nanosphere lithography.55,56 Fig. S3† shows SEM images of the entire array with an inter-pore distance of 250 nm, as well as magnified images of the same array. Fig. 1(D) shows a cross-section of the typical pores produced by FIB milling.49 The overetched region below the Au/glass interface typically decreases with pore diameter, and well-controlled milling processes yield pores overetched by less than 50 nm. From bottom to top, the cross-section image in Fig. 1(D) shows the bottom Au electrode (bright), silicon nitride (dark grey), the top Au electrode (bright), and silicon dioxide (grey). The nanopores exhibit a conical shape with a slightly larger aperture at the top electrode than at the bottom electrode. Structures fabricated for this study generally had dtop ∼ 100–120 nm, and dbottom ∼ 60–70 nm with dtop:dbottom ratio ∼ 1.7. The E-ZMW RDRE arrays facilitates spectroelectrochemical experiments, because they allow optical access through the SiO2 floor to redox cycling of fluorigenic redox species.
For wavelengths above the cutoff wavelength of the nanoaperture, λc, where λc ∼ 1.7d and d is the pore diameter, the evanescent field decays exponentially with distance at a rate that depends on the radius and diameter. The optical intensity through a cylindrical ZMW can be expressed as
| I(z) = I0e−3z/Λ | (1) |
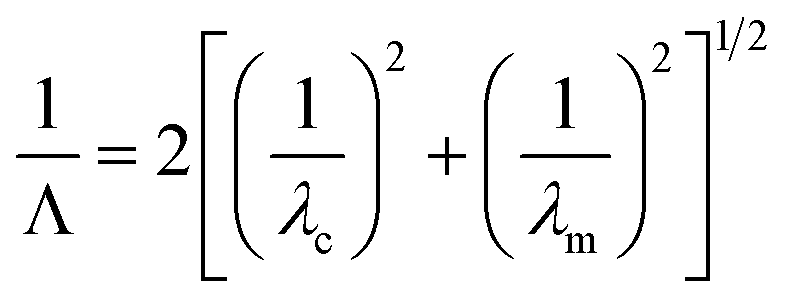 | (2) |
 | (3) |
To assess the behavior in the conical E-ZMW nanopores studied here, finite element simulations were performed at various conical frustum sizes and geometries. Fig. 2(A) and S4(A)–(E)† show the electric field in a series of conical frustum pores each having a ratio of top diameter, dtop, to bottom diameter, dbottom, dtop/dbottom = 2. As Fig. 2(A) and S4(A)–(E)† illustrate, the field decays exponentially, and the nanostructures provide excellent attenuation, primarily determined by dbottom. Fig. 2(B) shows that I(z) decays rapidly, limiting the optical observation volume to ca. the first 20 nm into waveguide, depending on the bottom diameter of the guide. Using attenuation in excess of 1/e2 as the threshold for an evanescent field strength unlikely to excite fluorescence, the exact position in the pore at which a fluorescent molecule can be excited varies with pore diameter and shape; however the effective volume, where the majority of excitation/emission events occur, is always constrained to the bottom-ring electrode area of the RDRE nanopore. Fig. S4(F)† shows the relative attenuation of energy density inside apertures. Fig. 2(C) shows how the effective optical volume, Veff/opt, depends on dbottom at dtop:dbottom values of 2.0. The effective volumes for different pore shapes deviate from each other only at large dbottom values. The concentration required to achieve a molecular occupancy expectation value of 1, i.e. 〈n〉 = 1, is also shown in Fig. 2(C) as a function of dbottom. Based on the simulations shown in Fig. 2(A) and taking into account the volume of the overetched region, the total effective volumes of the ZMWs used in this study are approximately 280 zL, assuming that the volumes of the metal-enclosed pore and the overetched region are roughly equal. At this volume, molecular occupancy of 〈n〉opt,pore = 1 is expected at 5.9 μM. In contrast, the effective volume for electrochemical reaction, Veff/elec, is shown as a function of dbottom in Fig. 2(D), as well as the corresponding concentration at which there is, on average, one molecule in the observation volume at any given time. The average number of molecules present in the electrochemically active region of a single nanopore is given by 〈n〉echem,pore = CVeff/elecNA, where C is the FMN concentration, NA is Avogadro's number, and Veff/elec is the volume of the active region for electrochemical reactions enclosed by the Au–SiNx–Au multilayer. For this volume, molecular occupancies of 〈n〉echem,pore = 1 is expected at 0.94 μM.
 | ||
| Fig. 2 (A) Cross-sectional heat map of evanescent field amplitudes in E-ZMW nanopore obtained by finite-element simulations, showing attenuation with distance from the aperture in the nanopore geometry. Note the color scale is logarithmic (dB). The structure exhibits dtop/dbottom = 2, and dbottom = 60 nm. (B) The energy intensity profile, I(z), for dtop/dbottom = 2 of 2.0 as a function of dbottom. (C) Effective volumes of the optical field (left ordinate) and concentration producing single molecule occupancy in the optical excitation volume (right ordinate). (D) Effective volumes for electrochemical reactions (left ordinate) and concentration for single molecule occupancy (right ordinate) in the electrical effective volume of a single nanopore. | ||
Dynamics of coupled fluorescence and electrochemistry
Fig. 3(A) shows the chronofluorometry of 1 μM FMN in 100 mM citrate buffer at pH 2.9 obtained from the 6561 nanopores E-ZMW RDRE device, conditions where the electrochemical occupancy per pore is 〈n〉echem,pore ∼ 1. For acidic aqueous environments with pH 3–5, FMN undergoes a 2e−/2H+ reduction to FMNH2.57 Analogous to the more familiar chronoamperometry experiment, chronofluorometry monitors the fluorescence, as opposed to the current, response of a redox system to alternating potential steps. Chronofluorometry was performed at the bottom working electrode, WEb, (leaving the top working electrode, WEt, to float) by stepping between oxidizing and reducing potentials vs. Ag/AgCl (Eappl = +0.3 V and −0.4 V) in order to produce conditions in which FMN alternates between fully oxidized FMN and fully reduced FMNH2, the latter being the fully protonated form of reduced FMN, appropriate to experiments at pH 2.9. Although both longer and shorter time windows were investigated, the 10 s potential steps used in Fig. 3(A) are sufficiently long to allow the system to achieve steady state at both oxidizing and reducing conditions, and it is clear that the fluorescence response is strongly correlated to the applied potential steps. We calculate that, under the diffraction-limited illumination conditions, the fluorescence signal is measured from ∼5 to 10 nanopores, each containing 〈n〉echem,pore ∼ 1, out of the 6561 nanopores in the E-ZMW array. | ||
| Fig. 3 (A) Experimental scheme and chronofluorometry of 1 μM FMN in 100 mM citrate buffer at pH 2.9. The potential of bottom electrode (Eb) is modulated at 10 s intervals between +0.3 V and −0.4 V, while the potential of top electrode (Et) floats. The temporal offset between optical and electrochemical data is caused by a trigger delay between the start of optical vs. electrochemical data acquisition. (B and C) Experimental schemes and current/fluorescence-time traces of 10 nM FMN in 100 mM citrate buffer at pH 2.9. Representative trajectories of the electrical and optical signals are given by the red and black solid lines, respectively. The insets show magnified views of the simultaneous electrical and optical signal excursions, which are highlighted in gray. (B) Eb = −0.4 V vs. Ag/AgCl, while Et floats. (C) Eb = +0.3 V while Et floats. | ||
Fig. 3(B) and (C) show current/fluorescence-time response measured at the much lower occupancy, with a per pore 〈n〉echem,pore ∼ 0.01 for a total array 〈n〉echem,array ∼ 65 obtained at 10 nM FMN. The amperograms were obtained by applying constant potential, Eb = −0.4 V (reducing potential, Fig. 3(B)) or +0.3 V (oxidizing potential, Fig. 3(C)), while the top electrode was disconnected (floating). The sign of current excursions at the bottom electrode are negative and positive, corresponding to reduction and oxidation, consistent with the applied reducing and oxidizing potentials to the electrodes. The presence of a FMN molecule in one of the pores in the array causes an observable electrochemical current due to self-induced redox cycling mediated by the floating electrode,58,59 as well as a transient change in the fluorescence intensity (gray highlights and insets in Fig. 3(B) and (C)). When a reducing potential is applied to the bottom electrode (Fig. 3(B)), oxidized species are able to redox cycle as the system approaches steady-state, causing a transient decrease in the fluorescence intensity. However, when an oxidizing potential is applied, the fluorescence signal increases transiently, since any redox cycling species are in the fluorescent oxidized state at the bottom electrode. Fig. S5 and S6† show control experiments performed in 100 mM citrate buffer at pH 2.9 as supporting electrolyte, exhibiting the background signal of current and fluorescence intensity at reducing, Fig. S5(A),† and oxidizing potentials, Fig. S5(B)† without redox cycling, and with redox cycling in Fig. S6.† It is noteworthy that the control experiments show background currents that are symmetric and display exceptionally low noise. Background optical signals are observed which are tentatively assigned to bulk fluorescence emission, scattering due to the sub-wavelength aperture array, and radiatively coupled surface plasmon modes, which are independent of applied potential.
Electrochemical and optical signal amplification by redox cycling
Encouraged by the subwavelength lateral field confinement provided by the E-ZMWs, we explored the enhancement of FMN molecules by redox cycling inside an E-ZMW, as well as the current amplification between generator and collector electrodes. Fig. 4 shows an example of simultaneous electrical and optical measurements obtained at 1 μM FMN, 〈n〉echem,pore ∼ 1, in the presence of redox cycling (GC mode) and without redox cycling as described previously (non-GC mode). The current/fluorescence-time traces in Fig. 4(A) were obtained by applying the oxidizing potential, Eb = +0.3 V vs. Ag/AgCl while WEt was held at Et = −0.4 V (GC mode) or floating (non-GC mode). Fig. 4(A) shows that the current and fluorescence intensity in GC mode (red line) are higher than those measured in non-GC mode (black line). The current and fluorescence amplification are attributed to redox cycling of FMN molecules between generator and collector electrodes. To further test this interpretation, we inverted the roles of the electrodes, making WEt oxidizing and WEb reducing, as shown in Fig. 4(B). The amperograms shown in Fig. 4(A) and (B) are very similar, reflecting nearly identical electrochemical amplification achieved by redox cycling, independent of which electrode is anode or cathode. Conversely, when WEb is set to a reducing potential of −0.4 V, Fig. 4(B), the fluorescence intensity in GC mode is lower than that in the non-GC mode, when the bottom electrode is allowed to float. This behavior is consistent with the reduction of fluorescent FMN to non-fluorescent FMNH2 at Eb = −0.4 V in combination with the optical field confinement in the E-ZMWs, illustrating the contribution of small excitation volume, spatial localization, and enhanced signal-to-background ratio in the spectroelectrochemical experiments.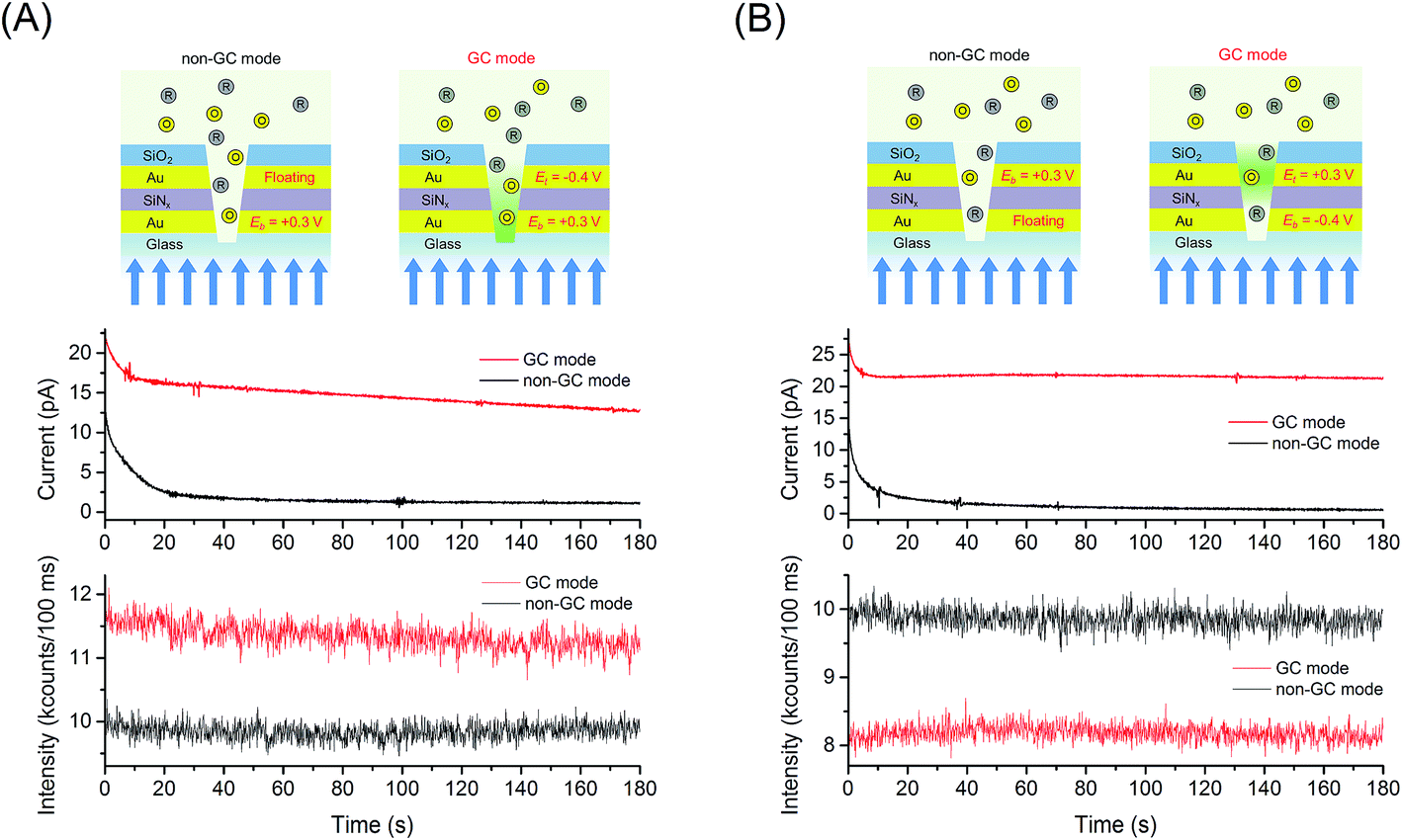 | ||
| Fig. 4 Experimental schemes for continuous electrical and optical measurements while switching the bias potentials of the electrodes. Data acquired from 1 μM FMN in 100 mM citrate buffer at pH 2.9 on an E-ZMW RDRE array with and without redox cycling. (A) Eb = +0.3 V vs. Ag/AgCl while Et floats (non-GC mode, black line) or held at −0.4 V (GC mode, red line). (B) Et = +0.3 V vs. Ag/AgCl (non-GC mode, black line) while Eb floats (non-GC mode, black line) or held at −0.4 V (GC mode, red line). Red and black lines in the optical signals correspond to GC mode and non-GC mode, respectively. | ||
Single molecule spectroelectrochemical behavior in GC mode
In a fluorigenic system, like FMN/FMNH2, electron transfer events may be translated to changes in fluorescence emission efficiency in the E-ZMW RDRE array. Here, we investigated electrofluorogenic behavior of freely diffusing FMN molecules under redox cycling conditions (Eb = −0.4 V, Et = +0.3 V). Fig. 5 shows a comparison of the current/fluorescence-time traces obtained at five different FMN concentrations. Fig. 5(A) shows the simultaneous electrical and optical control measurements performed in 100 mM citrate buffer at pH 2.9. Since no fluorigenic redox species are present in the system, the observed fluctuations (<±50 fA) correspond to the background noise level of the spectroelectrochemical measurement system. In contrast, Fig. 5(B) shows current/fluorescence-time traces measured at 1 nM FMN in GC-mode. First, it is evident from inspection that the generator and collector currents are mirror images, as expected. We attribute the top–bottom correlated current fluctuations to single molecule fluctuations in the array population, Δ〈n〉echem,array = ±1, which are reflected in the current at both electrodes due to high efficiency redox cycling. At 1 nM FMN, the average number of electrochemically accessible molecules in the entire E-ZMW RDRE array is given by (〈n〉echem,pore ∼ 0.001) × 6561 pores ∼ 6.5 = 〈n〉echem,array. | ||
| Fig. 5 Current/fluorescence-time traces and histograms for different FMN concentrations: (A) no FMN, (B) 1 nM, (C) 10 nM, (D) 100 nM, and (E) 1 μM. FMN concentrations of 1 nM, 10 nM, 100 nM and 1 μM, where the average molecular occupancy, 〈n〉opt,array ∼ 0.01, 0.1, 1, and 10, respectively. Each of the panels is organized as follows. Top: simultaneous time traces of electrochemical current (red and blue lines) and optical intensity (black line) in E-ZMWs under redox cycling conditions (Eb = −0.4 V, Et = +0.3 V). The currents measured at the bottom and top electrodes are represented by the red and blue solid lines, respectively. Simultaneous electrical and optical fluctuations of concurrent events are highlighted in gray (B and C). (B, inset) Magnified view of a single molecule spectroelectrochemical correlated event. Bottom: histograms of the fluorescence intensity distribution. Each histogram was created from data acquired over 180 s. | ||
In addition, sometimes correlated electrochemical and spectroscopic signal excursions were observed (highlight in Fig. 5(B)). Discrete features which we attribute to the entry or departure of a single FMN molecule from the optically probed region (10 pores) of the E-ZMW RDRE array. It is important to note that every observed spectroscopic excursion was correlated to a current excursion, although the converse was not true. This is because the electrochemical measurement probes all 6561 nanopores, while optical measurements only address a small subset of these, as the diffraction-limited focused laser spot only excites ∼10 pores, 〈n〉opt,pore ∼ 0.001 × 10 pores ∼ 0.01 = 〈n〉opt,array, at 1 nM. Thus, the number of optical detection events is expected to be a small fraction of the electrochemical events, as is observed.
For 10 nM FMN in Fig. 5(C), which corresponds to 〈n〉opt,array ∼ 0.1, discrete steps are still discernible in the electrochemical and optical measurements but occur more frequently. For higher concentration (Fig. 5(D) and (E)), changes in the instantaneous number of molecule become a smaller proportion of the overall signal and occur so frequently that they can no longer resolved with simultaneous electrical and optical measurements, although the fluctuations in the generator and collector currents are still correlated all the up to 1 μM.
Synchronization of the optical and electrochemical signal excursions was verified by cross-correlation analysis (see ESI† for details). Fig. 6 shows the analysis of current/fluorescence-time traces measured at 1 nM FMN in GC-mode for single-molecule redox cycling. The reduction and oxidation currents were fit to a model integrating responses from background, double layer reorganization, and Cottrell current, as shown in Fig. 6(A). As illustrated in Fig. 6(B), a normalized electrochemical signal was obtained by combining the two electrical signals from the separate amperometric traces to decrease random noise, then dividing by the standard deviation of the summed signal. The optical signal was fit to a linear model to remove drift, and then normalized by its standard deviation as well. As fluctuations occur much more frequently in the electrical signals, the overall cross-correlation of the electrical and optical signals shows little correlation; however, when the signals are examined in small windows of a few seconds, specific instances of correlation become apparent. Fig. 6(C) and (D) show cross-correlation of the optical and the associated electrochemical signals for the data shown in Fig. 6(B). In this particular data set, two times were identified with unequivocal evidence at t = 52 and 168 s – highlights in Fig. 6(C) and (D). The maximum lag is observed near −5 samples at 10 Hz sampling bandwidth. Such correlation relies on the interpretation that the optical deviation corresponds to a singular electrical signal, i.e. a low probability that two electrical deviation events occur within the same window, thus this method was only applied to the 1 nM FMN data. Fig. S7† shows the same analysis for optical and electrochemical data at t = 134 and 143 s, where no optical signal above the background is observed, from the data set shown in Fig. 6(B). The fitting and cross-correlation algorithms were checked for false positives by analyzing the traces from data collected with no FMN present, which showed no areas of correlation. Similar analysis of current/fluorescence-time traces measured at 10 nM FMN in non-GC mode also showed no correlation, as expected.
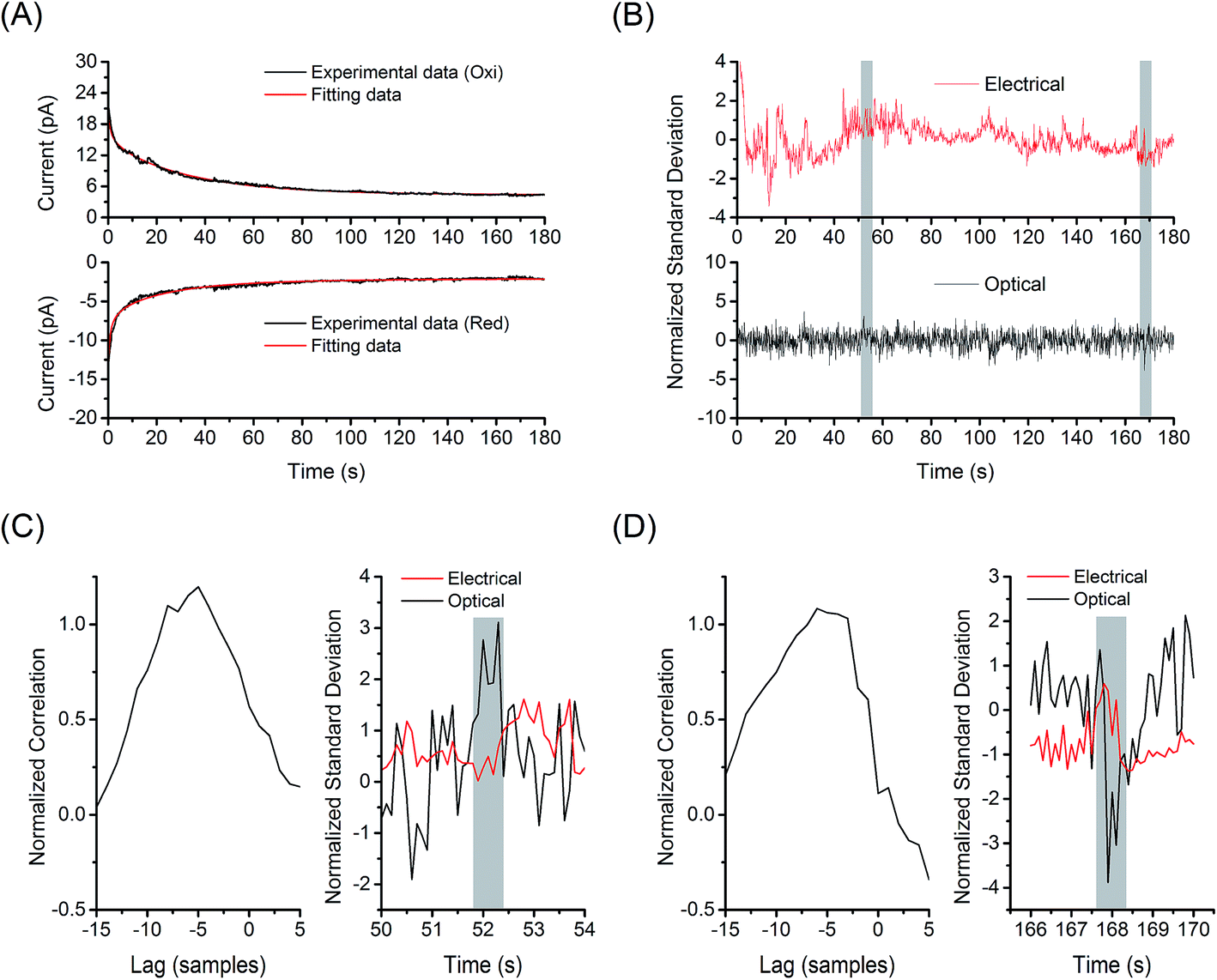 | ||
| Fig. 6 Cross-correlation analysis for single molecule detection. (A) Fitting the electrochemical signal from 1 nM FMN in the E-ZMW array under redox cycling condition to the model (red lines, see ESI† for details). The recorded amperometric traces (black lines) are given by the data set in Fig. 5(B). (B) Normalized electrochemical (red line, top) and fluorescence signal (black line, bottom) from the experimental data in Fig. 5(B). Cross-correlation of the optical events from E-ZMWs and the associated peaks in electrochemical current at (C) 52 s and (D) 168 s from the data set shown in (B). | ||
Conclusion
We demonstrate a new strategy for real-time observation of individual fluorigenic redox-active molecules as they freely diffuse into and out of zeptoliter-volume E-ZMW RDRE nanopores. In the RDRE geometry closely-spaced generator-collector electrodes are used to efficiently detect electrochemical response at pore occupancies in the range 〈n〉echem,pore ∼ 0.001–1, while the E-ZMW portion of the structure provides efficient confinement of the optical field, allowing investigation of the spectroelectrochemical behavior of FMN molecules both with and without redox cycling. The resulting coupled spectroelectrochemical events allowed observation of single molecule fluctuations in the FMN population on the array and, in some cases, detection of correlated electrochemical and spectroscopic events.These electrochemically-active zero-dimensional nanophotonic structures constitute a particularly advantageous platform for further studies because: (1) the average occupancy of individual pores may be controlled by solution concentration at facile concentration levels; and (2) the dual ring electrodes in each individual pore are held at the same potential as all the other pores through direct connection, thus the electrochemical signal is increased through integration over the entire array. We also note that the second ring electrode, instead of facilitating redox cycling, may be used to control solution conditions, e.g. pH, or synthesize short-lived reactive species which can then be captured in the nanopore and efficiently studied. Spectroscopic response can be examined either on a defined sub-array of E-ZMW nanopores, as was done here, or on the entire array using wide-field imaging. The central characteristic of the E-ZMWs in this study is that both single molecule electrochemical and optical fluctuations can be acquired simultaneously. We thus envision a wide range of applications integrated system for genomics, proteomics, and single-enzyme analysis where spectroelectrochemical detection may ultimately prove preferable to the currently established other detection techniques.
Acknowledgements
This work was supported by the National Science Foundation under grant 1404744 (DH, KF). GMC was supported by a NASA Space Technology Research Fellowship NNX16AM45H. Fabrication and structural characterization of the devices studied here were accomplished at the Notre Dame Nanofabrication Facility and the Notre Dame Integrated Imaging Facility, respectively. Their generous support is gratefully acknowledged.References
- N. G. Walter, C.-Y. Huang, A. J. Manzo and M. A. Sobhy, Nat. Methods, 2008, 5, 475–489 CrossRef CAS PubMed.
- W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12596–12602 CrossRef CAS PubMed.
- T. Schmidt, G. Schütz, W. Baumgartner, H. Gruber and H. Schindler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2926–2929 CrossRef CAS.
- K. C. Neuman and A. Nagy, Nat. Methods, 2008, 5, 491–505 CrossRef CAS PubMed.
- S. Bi, B. Liu, F.-R. F. Fan and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 3690–3691 CrossRef CAS PubMed.
- A. Schulte, M. Nebel and W. Schuhmann, Annu. Rev. Anal. Chem., 2010, 3, 299–318 CrossRef CAS PubMed.
- M. A. Zevenbergen, P. S. Singh, E. D. Goluch, B. L. Wolfrum and S. G. Lemay, Nano Lett., 2011, 11, 2881–2886 CrossRef CAS PubMed.
- S. Kang, A. F. Nieuwenhuis, K. Mathwig, D. Mampallil and S. G. Lemay, ACS Nano, 2013, 7, 10931–10937 CrossRef CAS PubMed.
- J. Rodríguez-López, M. A. Alpuche-Avilés and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 16985–16995 CrossRef PubMed.
- A. J. Bard, J. Am. Chem. Soc., 2010, 132, 7559–7567 CrossRef CAS PubMed.
- F.-R. F. Fan, J. Kwak and A. J. Bard, J. Am. Chem. Soc., 1996, 118, 9669–9675 CrossRef CAS.
- P. Sun and M. V. Mirkin, J. Am. Chem. Soc., 2008, 130, 8241–8250 CrossRef CAS PubMed.
- H. P. Lu, L. Xun and X. S. Xie, Science, 1998, 282, 1877–1882 CrossRef CAS PubMed.
- A. M. Van Oijen, Curr. Opin. Biotechnol., 2011, 22, 75–80 CrossRef CAS PubMed.
- S. Forth, M. Y. Sheinin, J. Inman and M. D. Wang, Annu. Rev. Biophys., 2013, 42, 583–604 CrossRef CAS PubMed.
- L. Bai, T. J. Santangelo and M. D. Wang, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 343–360 CrossRef CAS PubMed.
- J. Hilario and S. C. Kowalczykowski, Curr. Opin. Chem. Biol., 2010, 14, 15–22 CrossRef CAS PubMed.
- C. Joo, H. Balci, Y. Ishitsuka, C. Buranachai and T. Ha, Annu. Rev. Biochem., 2008, 77, 51–76 CrossRef CAS PubMed.
- T. Funatsu, Y. Harada, M. Tokunaga, K. Saito and T. Yanagida, Nature, 1995, 374, 555–559 CrossRef CAS PubMed.
- B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- X. Zhuang, L. E. Bartley, H. P. Babcock, R. Russell, T. Ha, D. Herschlag and S. Chu, Science, 2000, 288, 2048–2051 CrossRef CAS PubMed.
- W. Min, B. P. English, G. Luo, B. J. Cherayil, S. Kou and X. S. Xie, Acc. Chem. Res., 2005, 38, 923–931 CrossRef CAS PubMed.
- I. J. Finkelstein, M.-L. Visnapuu and E. C. Greene, Nature, 2010, 468, 983–987 CrossRef CAS PubMed.
- R. Roy, S. Hohng and T. Ha, Nat. Methods, 2008, 5, 507–516 CrossRef CAS PubMed.
- T. Ha and P. Tinnefeld, Annu. Rev. Phys. Chem., 2012, 63, 595–617 CrossRef CAS PubMed.
- F. Wei and Z. Liu, Nano Lett., 2010, 10, 2531–2536 CrossRef CAS PubMed.
- Z. Liu, H. Lee, Y. Xiong, C. Sun and X. Zhang, Science, 2007, 315, 1686 CrossRef CAS PubMed.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- P. Zhu and H. G. Craighead, Annu. Rev. Biophys., 2012, 41, 269–293 CrossRef CAS PubMed.
- J. M. Moran-Mirabal and H. G. Craighead, Methods, 2008, 46, 11–17 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- M. Bates, B. Huang and X. Zhuang, Curr. Opin. Chem. Biol., 2008, 12, 505–514 CrossRef CAS PubMed.
- B. Huang, W. Wang, M. Bates and X. Zhuang, Science, 2008, 319, 810–813 CrossRef CAS PubMed.
- Y. Zhao, S.-C. S. Lin, A. A. Nawaz, B. Kiraly, Q. Hao, Y. Liu and T. J. Huang, Opt. Express, 2010, 18, 23458–23465 CrossRef CAS PubMed.
- J. Ibach and S. Brakmann, Angew. Chem., Int. Ed., 2009, 48, 4683–4685 CrossRef CAS PubMed.
- M. J. Levene, J. Korlach, S. W. Turner, M. Foquet, H. G. Craighead and W. W. Webb, Science, 2003, 299, 682–686 CrossRef CAS PubMed.
- S. J. Sahl and W. Moerner, Curr. Opin. Struct. Biol., 2013, 23, 778–787 CrossRef CAS PubMed.
- P. Tinnefeld, Nat. Nanotechnol., 2013, 8, 480–482 CrossRef CAS PubMed.
- A. B. Loveland, S. Habuchi, J. C. Walter and A. M. Van Oijen, Nat. Methods, 2012, 9, 987–992 CrossRef CAS PubMed.
- G. Patterson, M. Davidson, S. Manley and J. Lippincott-Schwartz, Annu. Rev. Phys. Chem., 2010, 61, 345–367 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS PubMed.
- M. P. Goldschen-Ohm, V. A. Klenchin, D. S. White, J. B. Cowgill, Q. Cui, R. H. Goldsmith and B. Chanda, eLife, 2016, 5, e20797 Search PubMed.
- M. P. Goldschen-Ohm, D. S. White, V. A. Klenchin, B. Chanda and R. H. Goldsmith, Angew. Chem., Int. Ed., 2017, 56, 2399–2402 CrossRef CAS PubMed.
- J. Korlach, P. J. Marks, R. L. Cicero, J. J. Gray, D. L. Murphy, D. B. Roitman, T. T. Pham, G. A. Otto, M. Foquet and S. W. Turner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1176–1181 CrossRef CAS PubMed.
- K. Samiee, J. Moran-Mirabal, Y. Cheung and H. Craighead, Biophys. J., 2006, 90, 3288–3299 CrossRef CAS PubMed.
- D. Liao, P. Galajda, R. Riehn, R. Ilic, J. L. Puchalla, G. Y. Howard, H. G. Craighead and R. H. Austin, Opt. Express, 2008, 16, 10077–10090 CrossRef CAS PubMed.
- J. Zhao, S. P. Branagan and P. W. Bohn, Appl. Spectrosc., 2012, 66, 163–169 CrossRef CAS PubMed.
- J. Zhao, L. P. Zaino III and P. W. Bohn, Faraday Discuss., 2013, 164, 57–69 RSC.
- L. P. Zaino, D. A. Grismer, D. Han, G. M. Crouch and P. W. Bohn, Faraday Discuss., 2015, 184, 101–115 RSC.
- S. D. Islam, T. Susdorf, A. Penzkofer and P. Hegemann, Chem. Phys., 2003, 295, 137–149 CrossRef CAS.
- A. Sengupta, R. V. Khade and P. Hazra, J. Photochem. Photobiol., A, 2011, 221, 105–112 CrossRef CAS.
- D. Han, L. P. Zaino III, K. Fu and P. W. Bohn, J. Phys. Chem. C, 2016, 120, 20634–20641 CAS.
- K. Dawson, A. l. Wahl, R. Murphy and A. O'Riordan, J. Phys. Chem. C, 2012, 116, 14665–14673 CAS.
- S. E. Kleijn, A. I. Yanson and M. T. Koper, J. Electroanal. Chem., 2012, 666, 19–24 CrossRef CAS.
- C. Ma, N. M. Contento, L. R. Gibson and P. W. Bohn, ACS Nano, 2013, 7, 5483–5490 CrossRef CAS PubMed.
- A. Valsesia, P. Lisboa, P. Colpo and F. Rossi, Anal. Chem., 2006, 78, 7588–7591 CrossRef CAS PubMed.
- S. L. Tan, J. M. Kan and R. D. Webster, J. Phys. Chem. B, 2013, 117, 13755–13766 CrossRef CAS PubMed.
- C. X. Ma, N. M. Contento and P. W. Bohn, J. Am. Chem. Soc., 2014, 136, 7225–7228 CrossRef CAS PubMed.
- C. X. Ma, L. P. Zaino and P. W. Bohn, Chem. Sci., 2015, 6, 3173–3179 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02250f |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA. |
| This journal is © The Royal Society of Chemistry 2017 |